Nitrogen Metabolism/ Urea Cycle- Ellis
1/54
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
55 Terms
How do humans ingest most nitrogen?
predominantly in the form of proteins into the digestive tract.
How will proteins be broken down within the digestive tract?
the stomach's low pH and proteolytic enzymes will break down the proteins and the small intestine will convert proteins into amino acids
What are 3 things that amino acids will be used for?
1. converted into proteins (hemoglobin)
2. nitrogen containing compounds
ammonia (converted to urea)
3. carbon skeletons ( energy production through glucose and ketone bodies)
Can someone live without carbohydrates?
yes
Since it is implied that carbohydrates are not needed from the diet where do carbohydrates come from?
human bodies can make them from scratch
Is proteins required for the diet?
yes
What are the 10 amino acids that cannot be synthesized by the human body?
1. phenylalaine
2. valine
3. Threonine
4. Tryptophan
5. isolucine
6. methionine
7. histidine
8. arginine *
9. lysine
10. leucine
What is the one essential amino acids is needed in children but not in adults ?
arginine
What are amino acids used for in the liver during a fed state?
protein and other nitrogen containing compounds
Is the Km high or low for protein and nitrogen containing compound synthesis in the liver in a fed state?
low Km because these are anabolic processes and amino acids are used for these processes even when the concentration is relatively low
At higher concentration for enzymes what is the pathway for amino acids in the liver in a fed state?
The amino acids can enter degradative pathways for wither energy production or storage of fatty acids
What is gluconeogenesis?
synthesizing glucose from scratch
What is the most protein rich organ in the body?
muscle
How is carbon obtained for gluconeogenesis in a prolonged fast?
by breaking down muscle
in a prolonged fast when amino acids are sent out of the muscle and converted into alanine what is it then converted into in the liver?
pyruvate
in a prolonged fast when amino acids are sent out of the muscle and converted into glutamine where are two places that glutamine can go to next?
the intestine or kidney where it will be converted into alanine
Why is glutamine being sent to the kidney important during acidosis?
Acidosis ph too acidic
Glutamine will be sent to the kidney and be converted into ammonia (NH₃) and pick up a protron to make NH₄⁺ which will be excreted through the urine and increase the pH
What is the enzyme that will allow for glutamine to be converted into ammonia in the kidney?
glutaminase
When alanine is converted into pyruvate in the liver what can it be used for?
gluconeogenesis and ketone bodies
What kind of reaction does alanine undergo to become glutamic acid?
transamidation reaction
What vitamin is pyridoxal phosphate from?
vitamin B6
What is the corresponding keto acid to alanine?
pyruvate
In order for alanine to become pyruvate it must give up a amino group, where does the amino group go to?
the amino group gets transferred to α-ketoglutarate to replace the carbonyl group and make glutamic acid
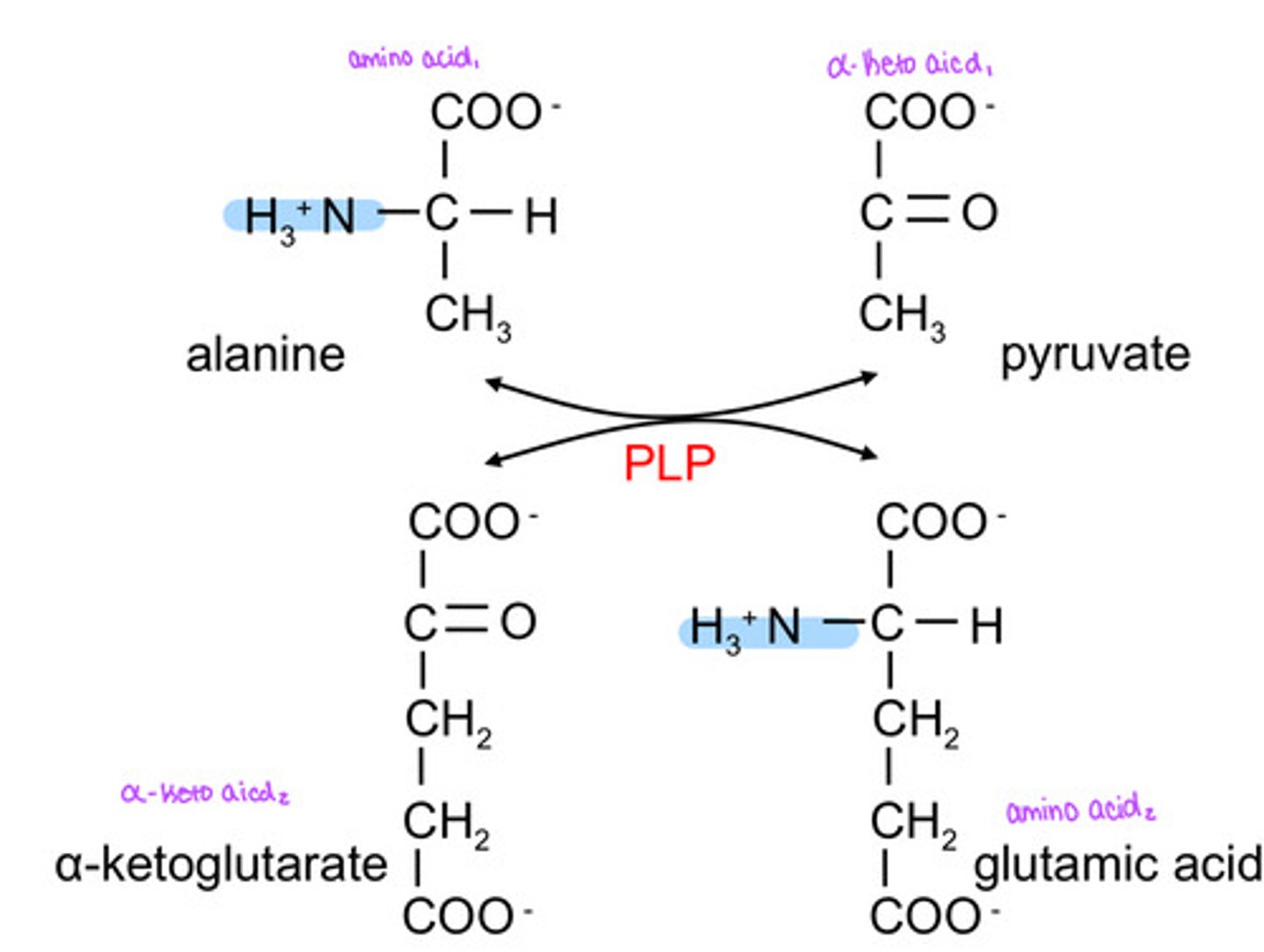
What is the enzyme that will convert alanine to glutamic acid?
ALT= alanine transaminase
What is ALT- alanine transaminase cofactor?
PLP- pyridoxal phosphate
What is the enzyme that will convert aspartic acid to glutamic acid?
AST= aspartate transaminase
In addition to the reaction in the kidney to counter act acidosis what else will glutaminase convert glutamine into?
glutamic acid
If glutamine comes into the kidney and gets converted to glutamate what how will this push the transamidase reaction?
towards α- ketoglutarate in the process generate alaine and now alaine will exit the tissue through the liver
During metabolic acidosis, how do glutaminase and alanine transaminase (ALT) in the kidney help regulate acid–base balance, and what happens to the alanine formed?
In the kidney, glutaminase converts glutamine → glutamate + NH₃; the ammonia binds H⁺ to form NH₄⁺, which is excreted to remove acid. ALT then transfers an amino group from glutamate to pyruvate, forming alanine and regenerating α-ketoglutarate. The alanine travels to the liver, where ALT reverses the reaction to produce pyruvate for gluconeogenesis and glutamate for urea formation.
In a extended fasted state, what will alanine be converted into for gluconeogenesis?
pyruvate (uses ALT)
α-ketoglutarate is a collection of ___________ atoms
nitrogen
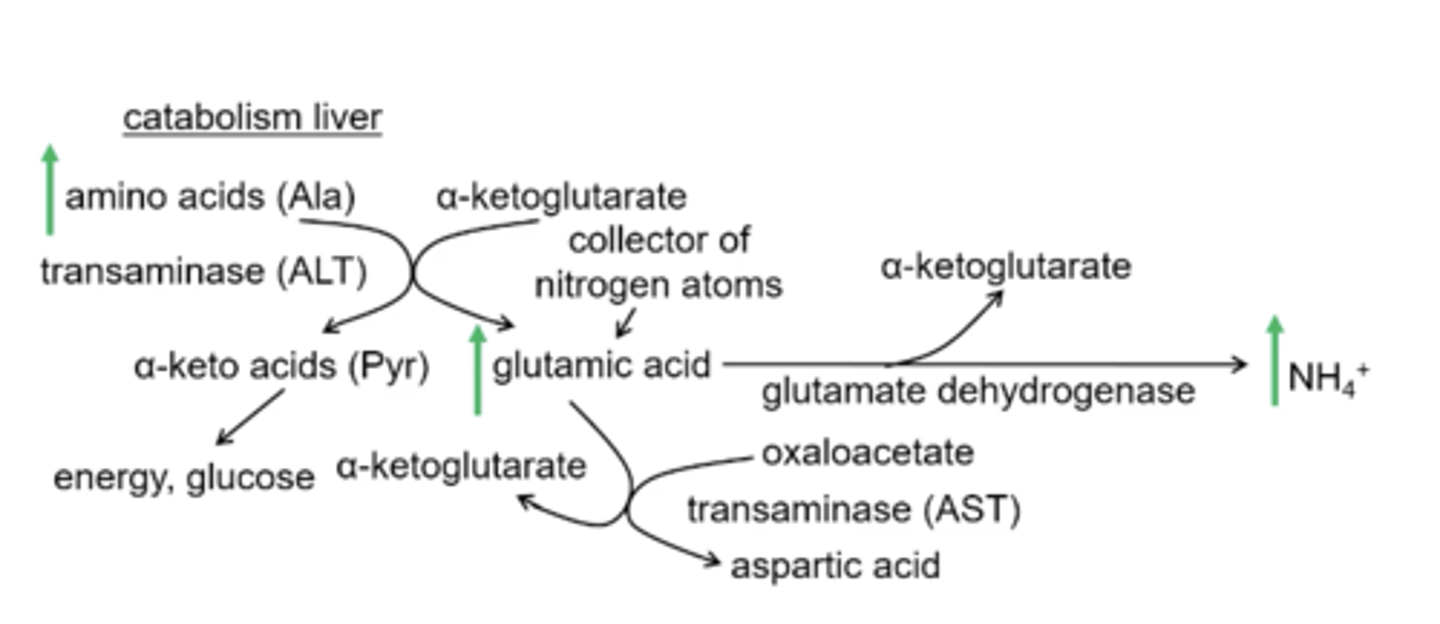
After α-ketoglutarate is converted into glutamic acid glutamate dehydrogenase produces what products?
α-ketoglutarate and ammonia
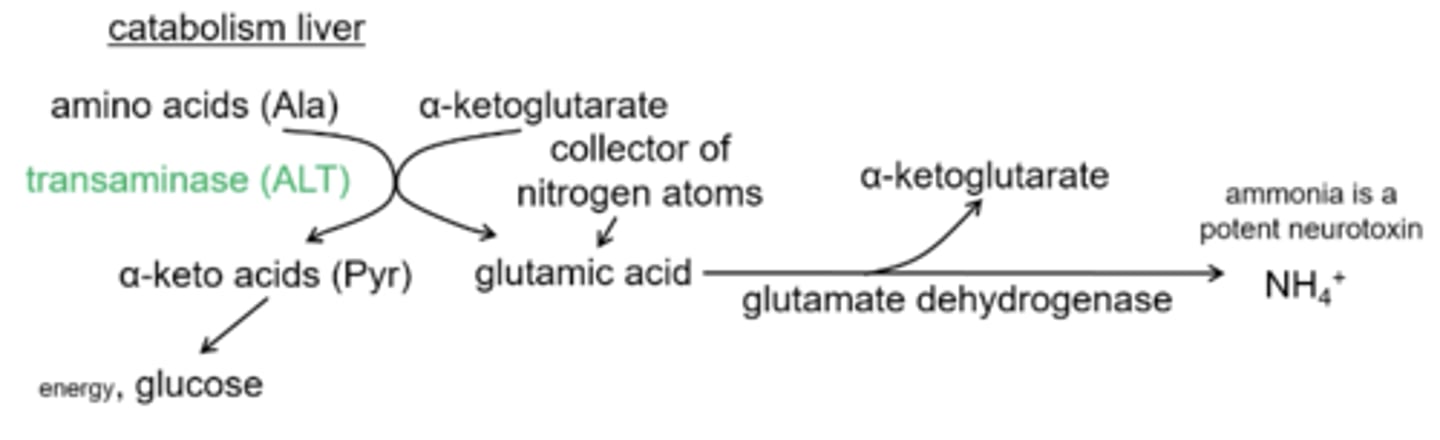
Why does ammonia need to be fixed into the urea cycle?
because it is a very potent neurotoxin
Other than ammonia getting detoxified in the urea cycle what is the other way that nitrogen will enter the urea cycle?
ALT - alanine transaminase- will give up a amino group from glutamic acid to oxaloacetate and form aspartic acid which feeds into the urea cycle
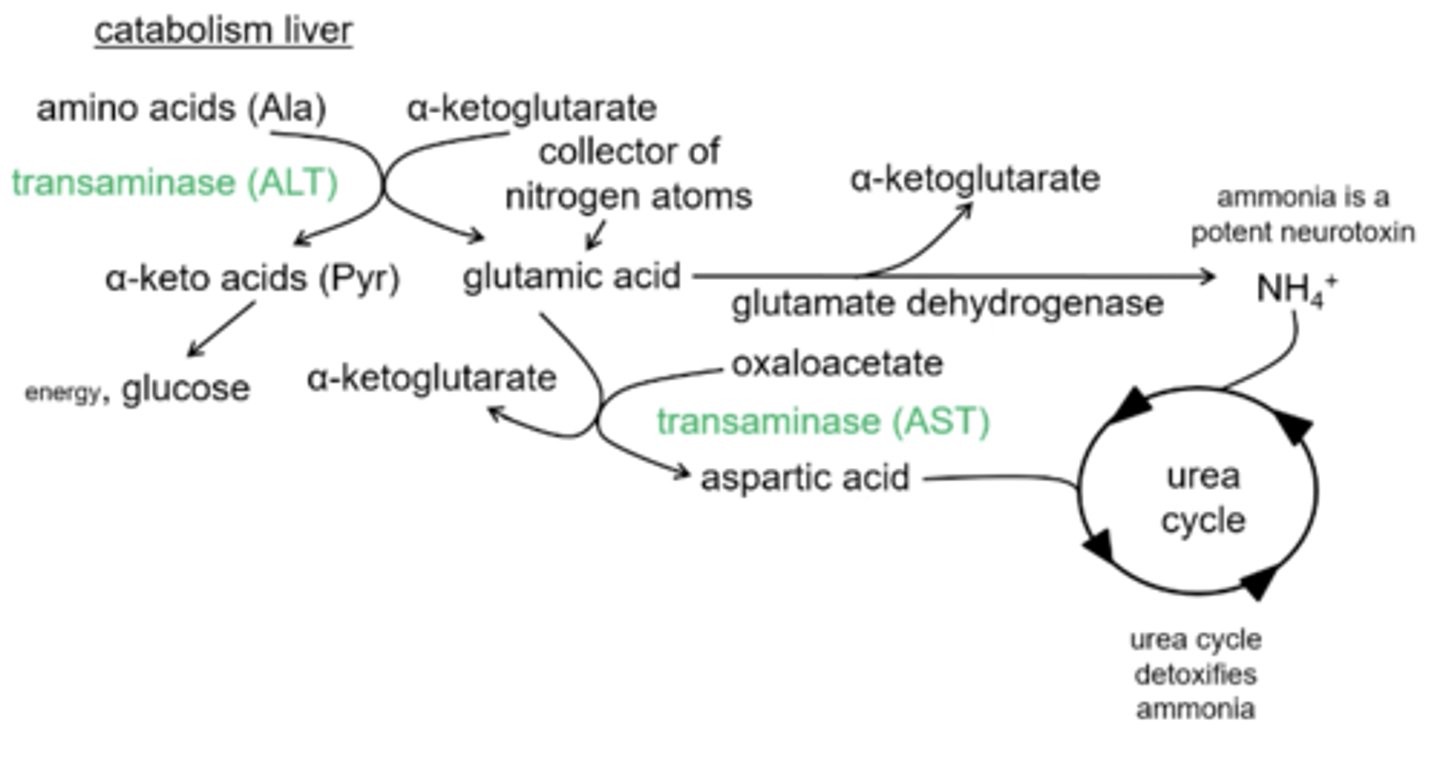
What will convert oxaloacetate to aspartic acid?
AST= aspartic acid transaminase
What two enzymes are expressed in high concentrations in the liver?
AST= aspartic acid transaminase
ALT= alanine transaminase
Compare:
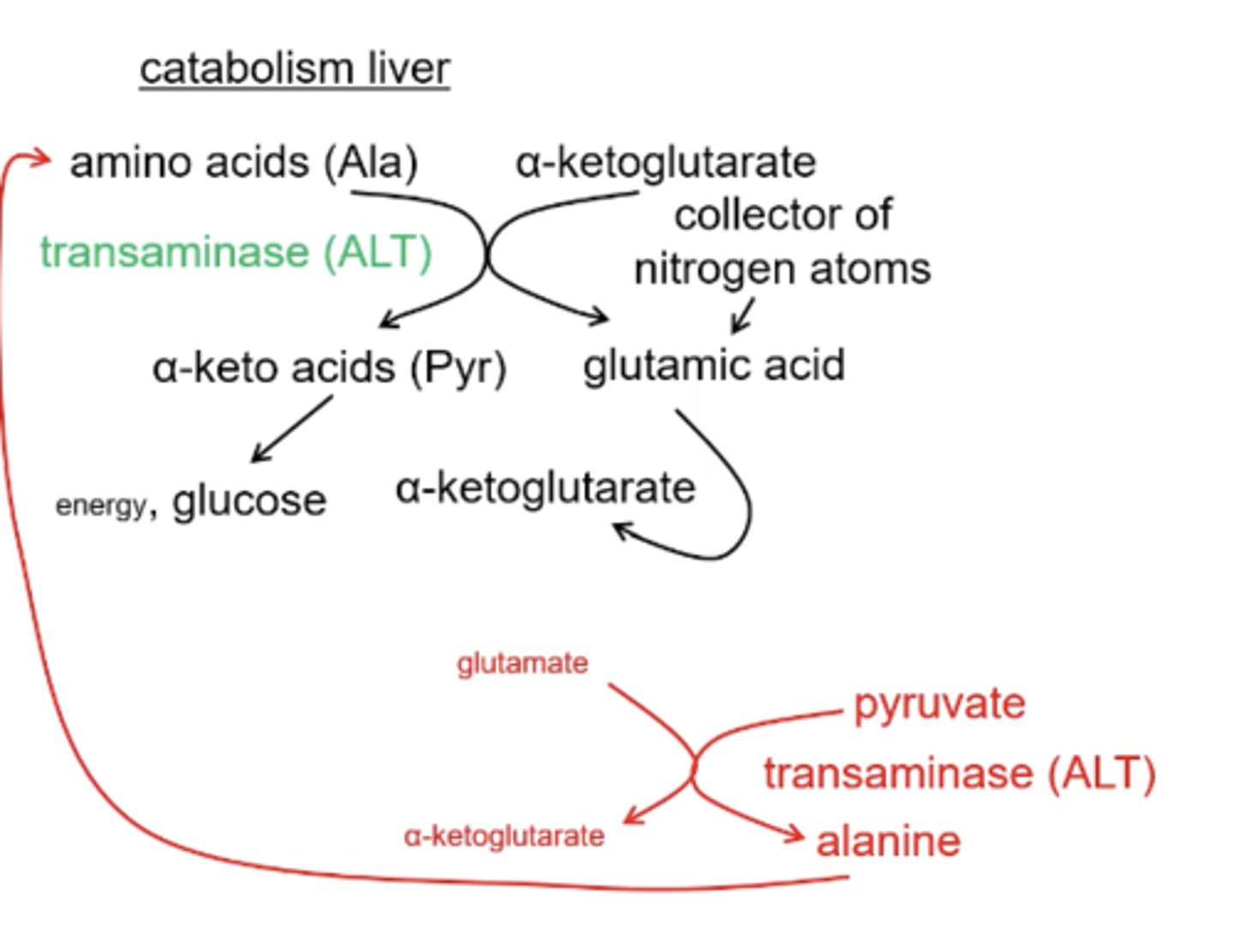
In the muscle cell transaminase ALT converts ___________ into __________
In the liver transaminase ALT converts __________ into ________
alanine; pyruvate
pyruvate; alanine
What are the five amino acids broken down to pyruvate?
Cys: cystine
Gly: glycine
Ser: serine
Thr: threonine
Trp: tryptophan
What is the point of the anplerotic reactions that add intermediates to the TCA?
All the anaplerotic reactions will add intermediates that will exit the TCA after one turn and get converted to pyruvate and then to alanine
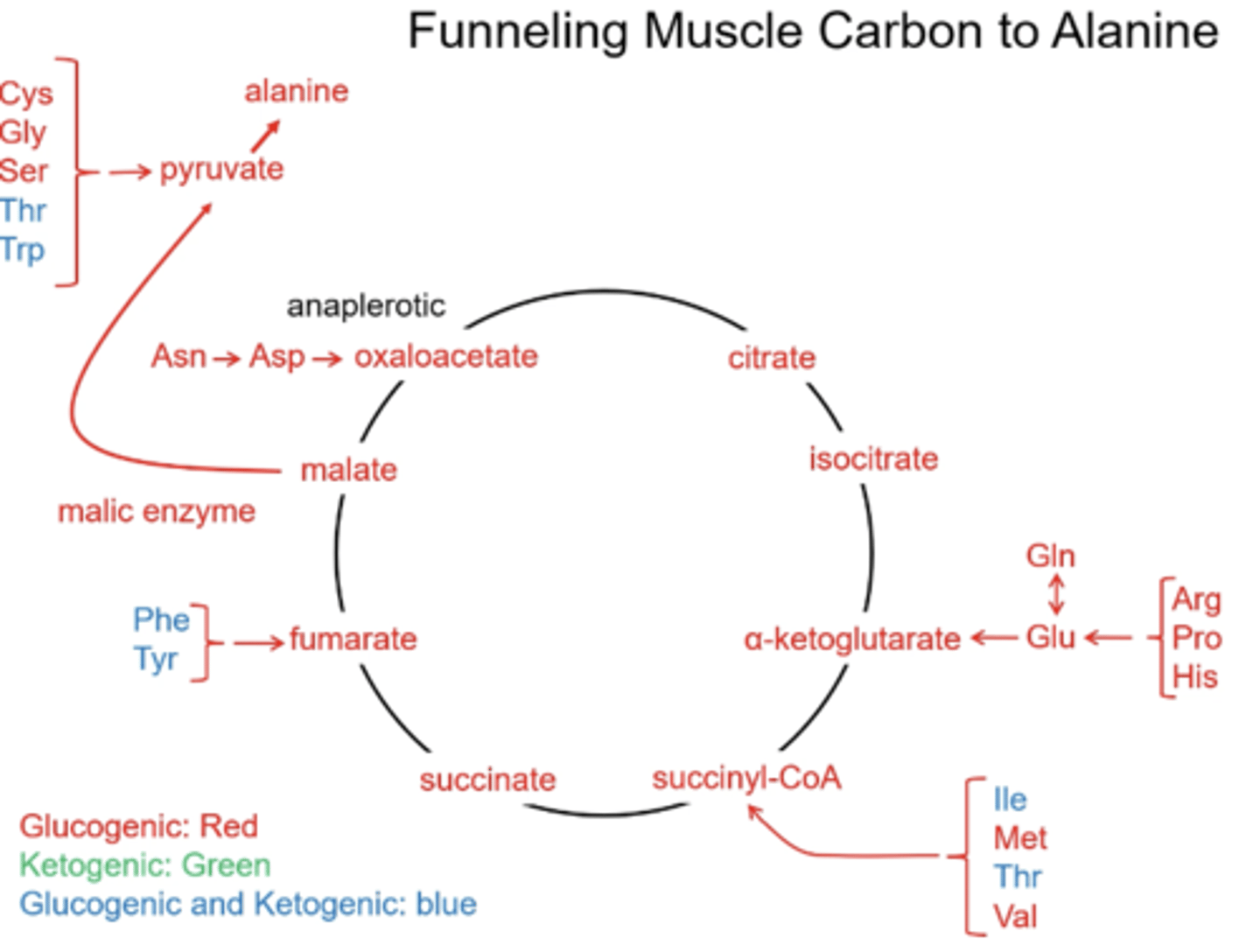
What are the two amino acids that are exclusively ketogenic?
Leu: leucine
Lys: lysine
What is the only product(s) that leucine and lysine will generate?
acetoacetate and Acetyl-CoA
Can the carbons of Acetyl- CoA be used for glucose synthesis?
no
Why isn't acetyl-CoA being added to the TCA anaplerotically?
There are no net intermediates being added
Explain the steps of the Urea Cycle
1. Carbamoyl phosphate synthetase I; CO₂ and NH₄⁺ are used to make carbamoyl phosphate
2. ornithine transcabomolase; carbamoyl phosphate + ornithine = citrulline
3. argininosuccinate synthetase; citrulline (shuttled out of the mitochondria) + aspartic acid= argininosuccinate
4. argininosuccinase; argininosuccinate will release fumarate to go into the TCA and will be left with arginine
5. arginase; arginine will be converted to ornithine with the addition of a carbonyl and allow for the completion of the cycle
What is the CPS1 deficiency?
The urea cycle can not start because there is no ammonia to feed into the cycle so there is no is no detoxification that can happen. Ammonia will spike in step one of the urea cycle
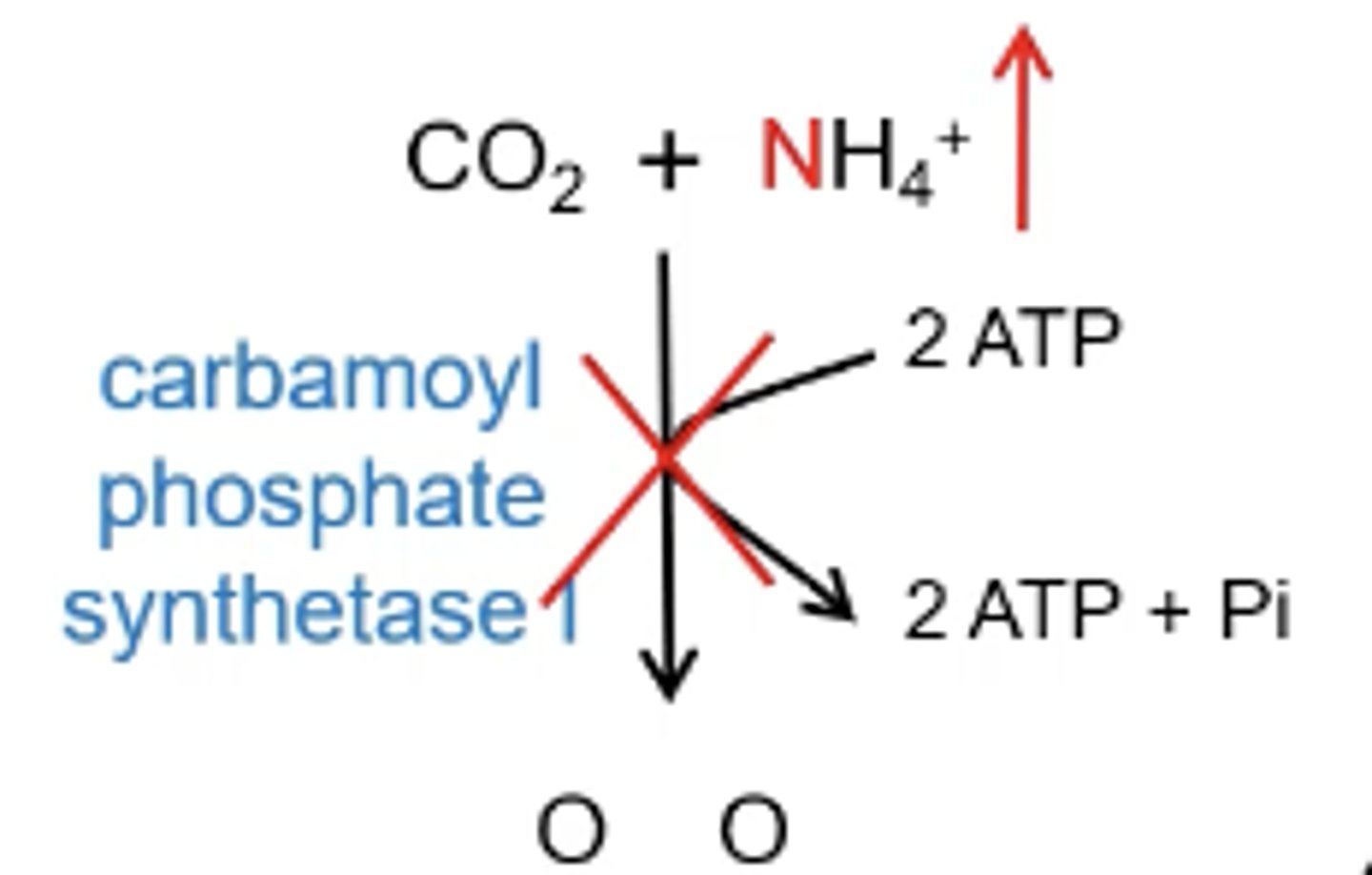
What is OT deficiency?
ornithine transcarbamolase will be inhibited causing a build up of carbamoyl phosphate leading to elevated ammonia
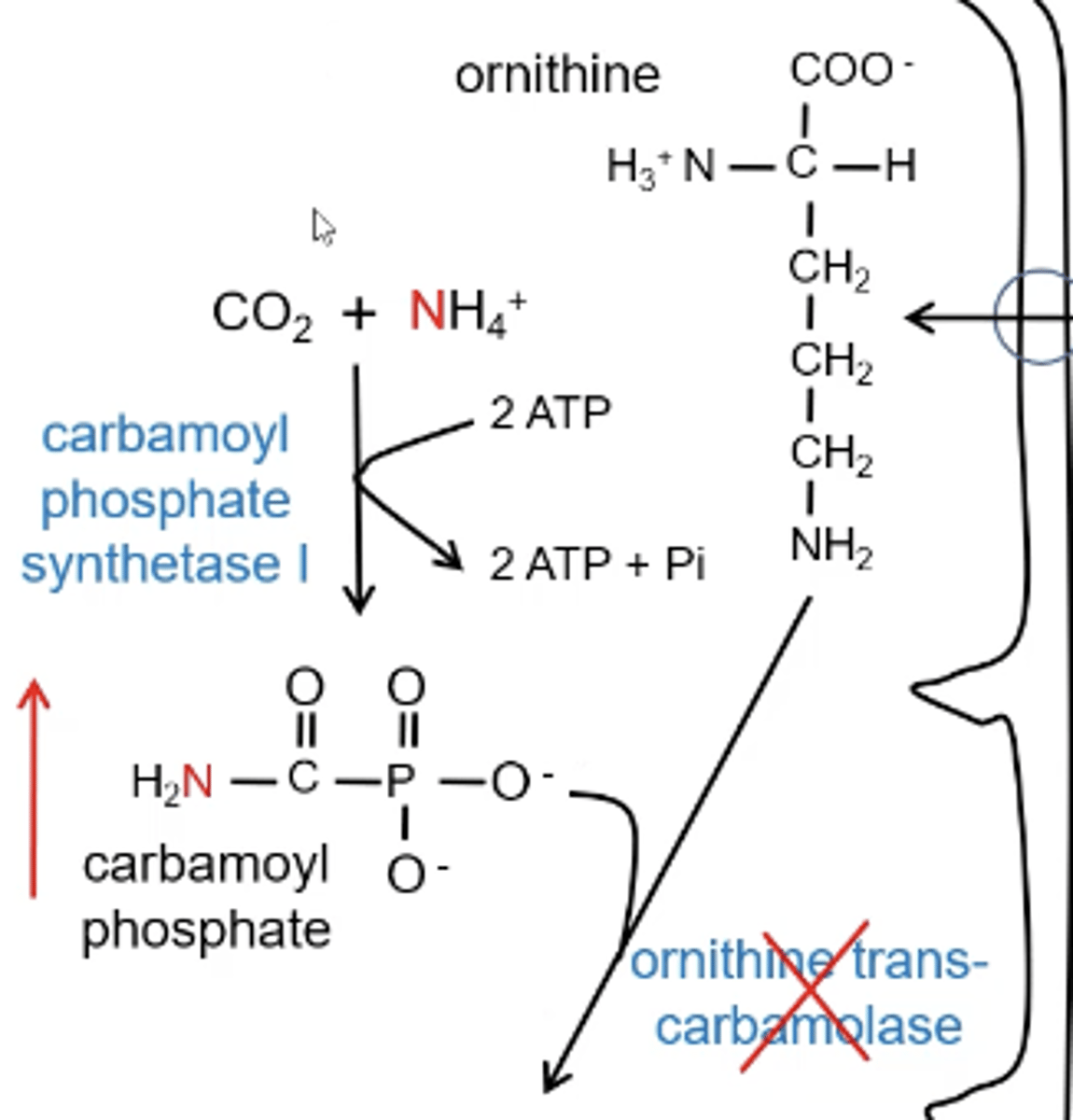
Why does a neonatal catastrophe happen with CPS1 and OT definecny?
As soon as the baby ingest proteins their ammonia will go through the roof and they will get sick and even more sick when they eat so they stop eating. Now the baby is in a fasted state and breaking down all the proteins that are building up producing more ammonia causing a very high blood ammonia
Why would glutamic acid and alanine build up?
if there is no urea cycle and ammonia builds up then the glutamate dehydrogenase reaction will build up its product (ammonia) and substrate (glutamic acid) and when glutamic acid builds up the transaminase reaction will go towards alaine production
What is the difference in orotic acid levels with a CPS1 deficiency and OT deficiency?
CPS1 deficiency has normal orotic acid levels
OT has elevated orotic acid levels
Why would it be important to identify is it is a OT deficiency or a CPS1 deficiency even though they both result in neonatal catastrophe?
OT deficiency is X-linked
Why does OT deficiency result in elevated levels of orotic acid?
Carbamoyl phosphate will build up with a OT deficiency and will spill out of the mitochondria and into the cytoplasm and elevates carbamoyl phosphate in the cytoplasm which results in spiked levels of orotic acid
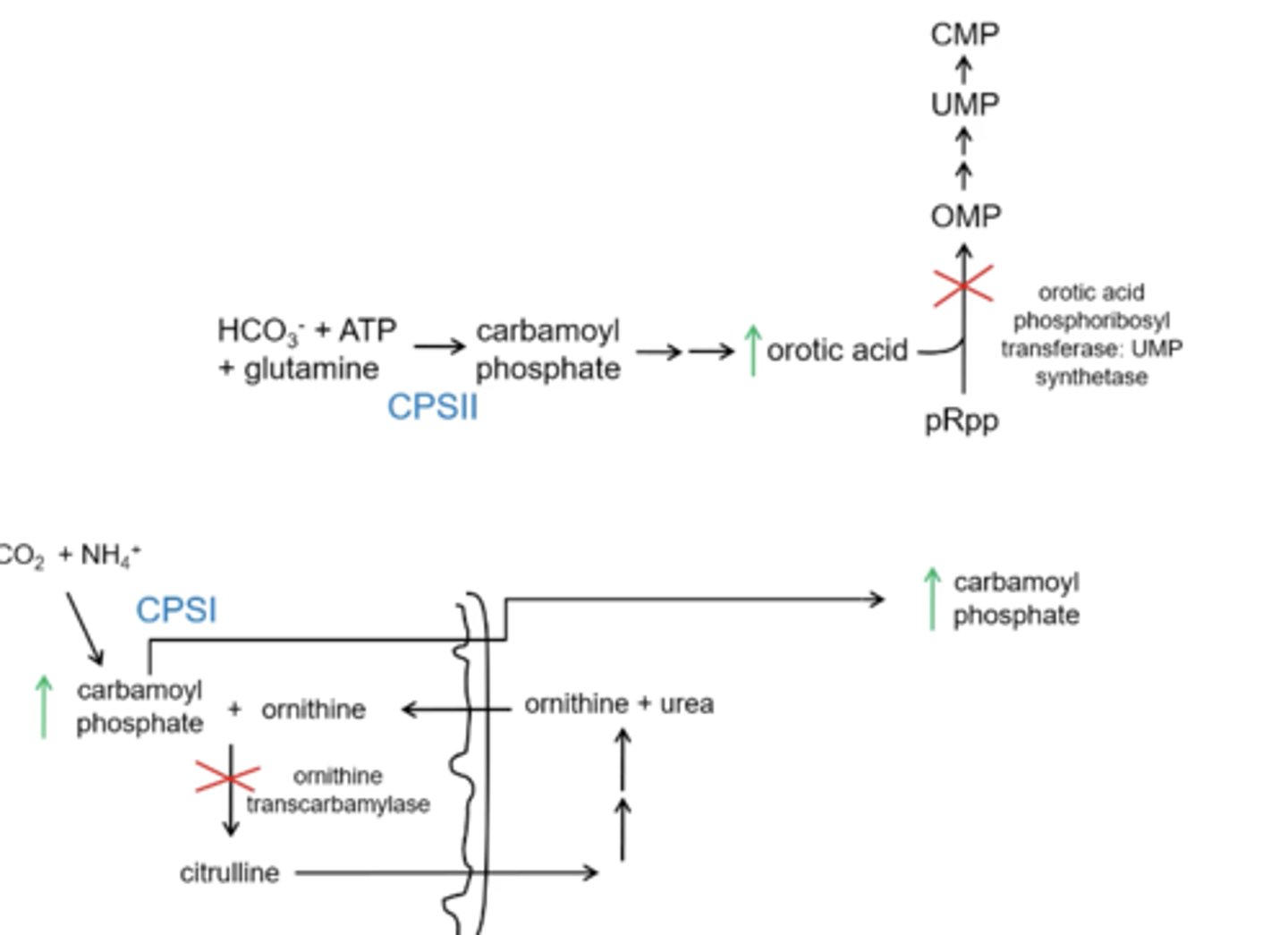
Why would there not be a increase in orotic acid levels with a CPS1 deficiency?
the production of carbamoyl phosphate will be inhibited and will not spill out to the cytoplasm
In addition to controlling diet what are two other ways to treat for a urea cycle defect?
1. Benzoic acid + glycine --> hippruric acid: this leads to renal excretion resulting in glycine getting excreted and depleting glutamic acid (which is a good. thing since it is a nitrogen reservoir)
2. Phenylacetate+ glutamine --> phenylacetyl glutamine: will result in renal excretion and glutamine will have to be replaced with the reserve of the glutamic acid using up the nitrogen reservoir
Why are Citrullinemia and Argininosuccinaturia deficiency not as severe?
Citrulline and argininosuccinate will build up but be excreted in the urine so there will be minimal nitrogen build up
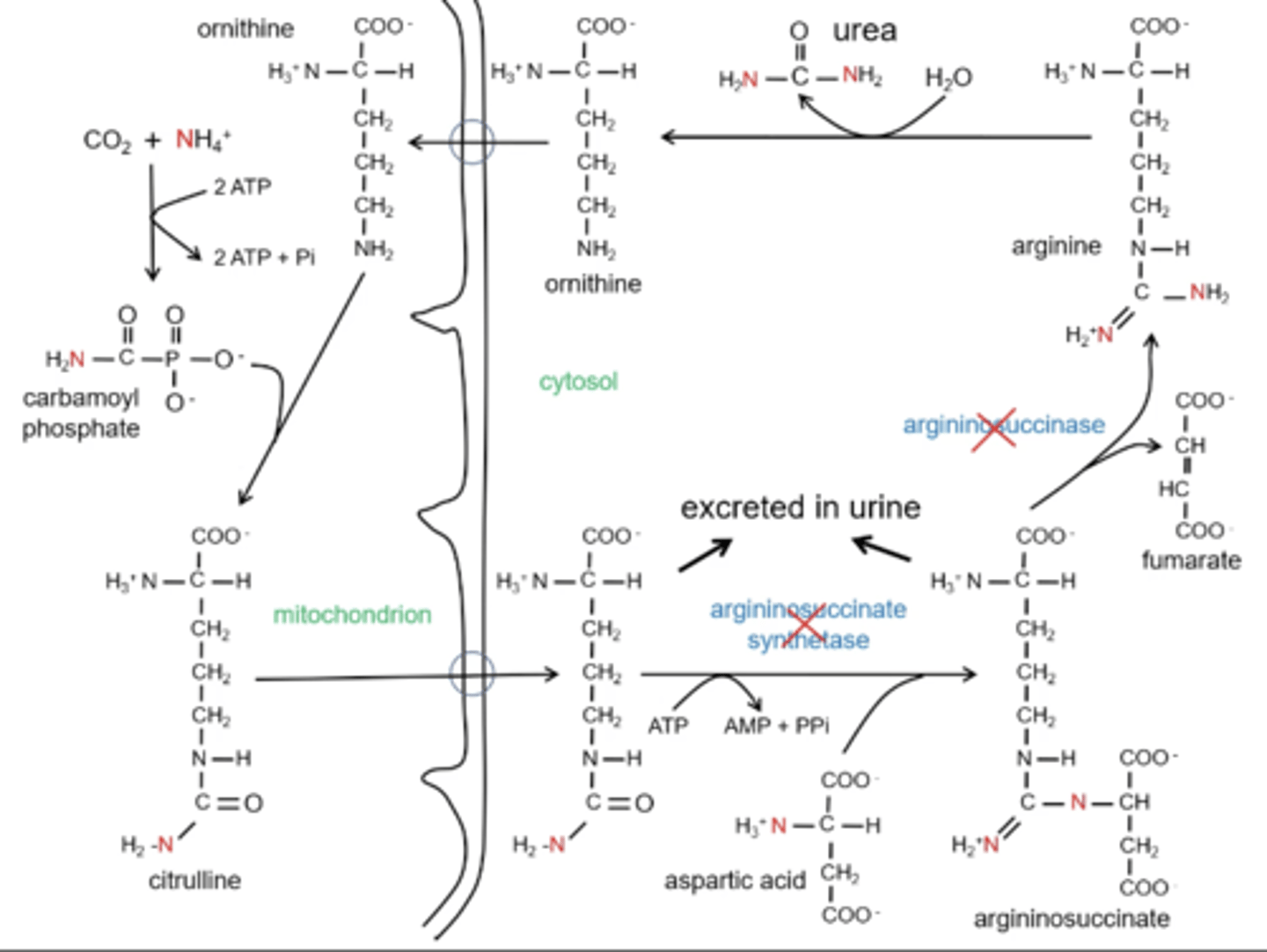
Why would someone need to supplement with arginine with Citrullinemia and Argininosuccinaturia deficiency?
If citrulline and argininosuccinase are be excreted there is not completion of the cycle. If the cycle is not completed then ornithine will not be regenerated and carbamoyl phosphate will not be picked causing this to eventually look like a early disorder.
Arginine is supplemented and ornithine is regenerated.