Organic Chemistry - Final Exam Flashcards
1/196
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
197 Terms
Atomic Orbitals
An electron orbital is: 3 dimensuibal: these are probabilities: they are most likely in x location
Some important reminders:
maximum allowed capacitu of 2 electrons! this rule can never be broken
Low energy shells are preferred, but not always.
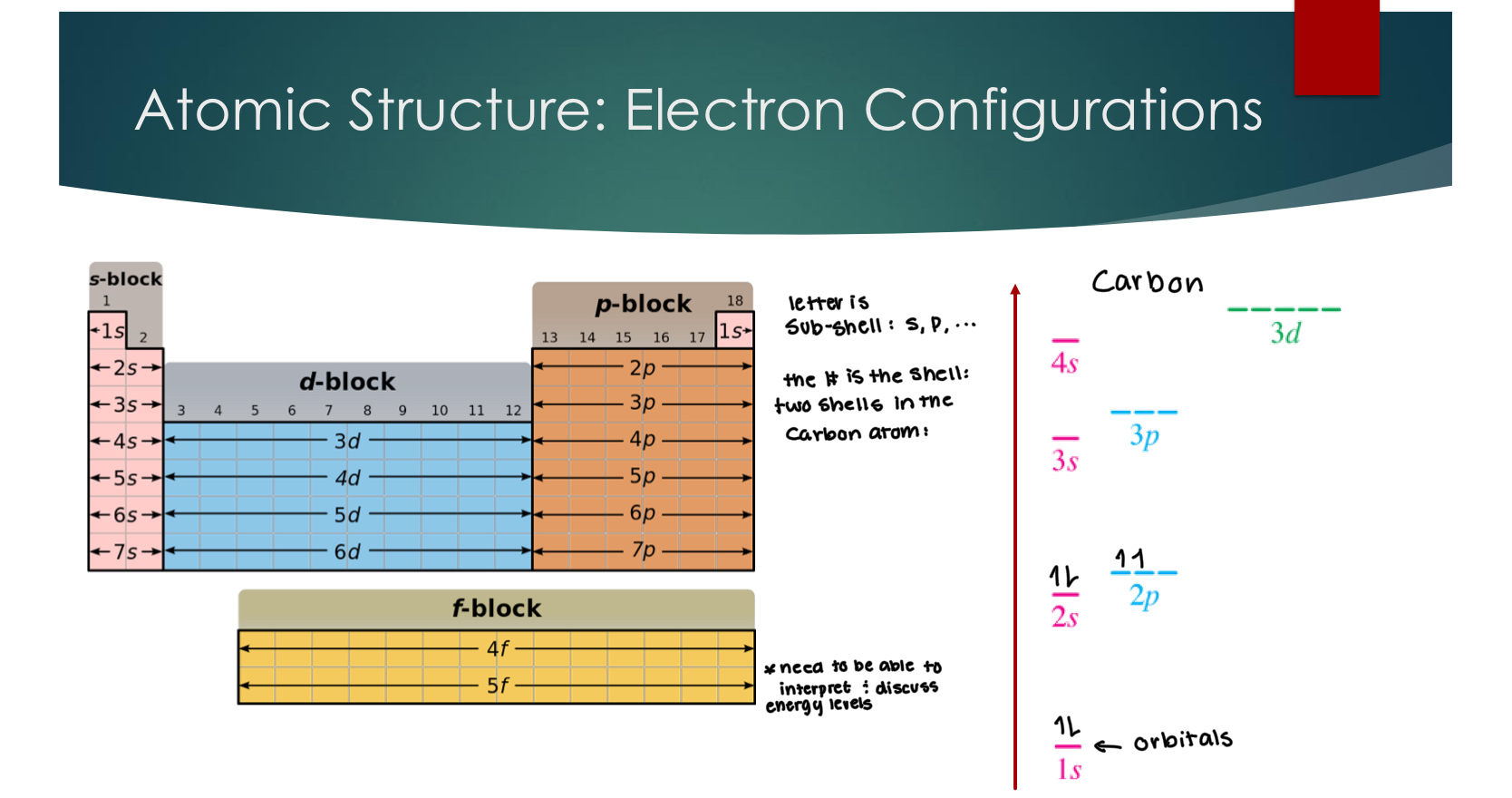
Atomic Structure: Electron Configurations
The Valence electrons are the ones contained in the highest shell, in this example, carbon has 4 valence electrons in the 2 shell.
Drawing Atomic Orbitals: Orgo Edition
A periodoic trick for determining valence electrons

Electronegativity of Atoms
The ability of an atom to attract electrons through a covalent bond
F > O > N ~ Cl > Br > I ~ S > C > H
Molecular Structure: Bonded Atoms
Key Principle: Completely filled orbitals are very stable → the octet rule
Ionic:
typically between a metal and nonmetal (because they usually have a large EN diff. between atoms) : its a transfer of electrons. DO NOT DRAW LINES ON LEWIS STRUCTURES FOR IONIC COMPOUNDS
Covalent:
Less EN diff. between atoms. generally, nonmetals bonded with other nonmetals. the share electrons.
Molecular Structure: Covalent Bonds
Key Principle: Bigger difference in atom electronegativity → more polar bond Non-
Polar Covalent
When there is unequal EN levels, arrow points toward the more EN atom.
We can rank how polar bond are because of the EN diff. between atoms.
Nonpolar Covalent
e- are equally shared. Only occurs when the two participating in the bond have the exact same electronegativity, usually its the same atom.
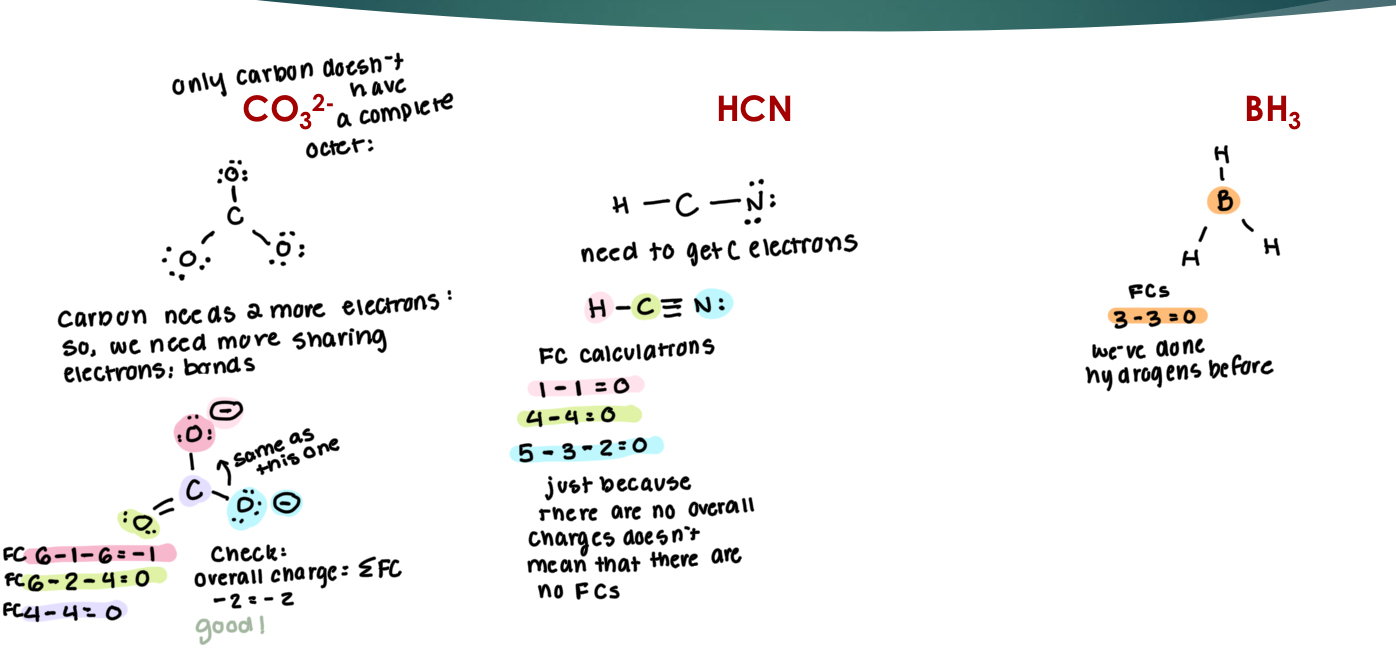
Molecular Structure: Lewis Structures
Represents the total number of valence electrons in a molecule → The best Lewis structure will have: Filled octets Fewest formal charges (-) on more EN atoms (+) on less EN atoms
FC = VE - dots - sticks
Overall charge vs formal charge (FC)
Step 1: Count valence electrons & connect atoms using single bonds with least EN atom in center
Step 2: Add remaining e- as lone pairs to outer atoms first then central atoms until you have accounted for total valence e-
Step 3: Consider multiple bonds & octet rule exceptions to draw the best Lewis Structure with all formal charges shown
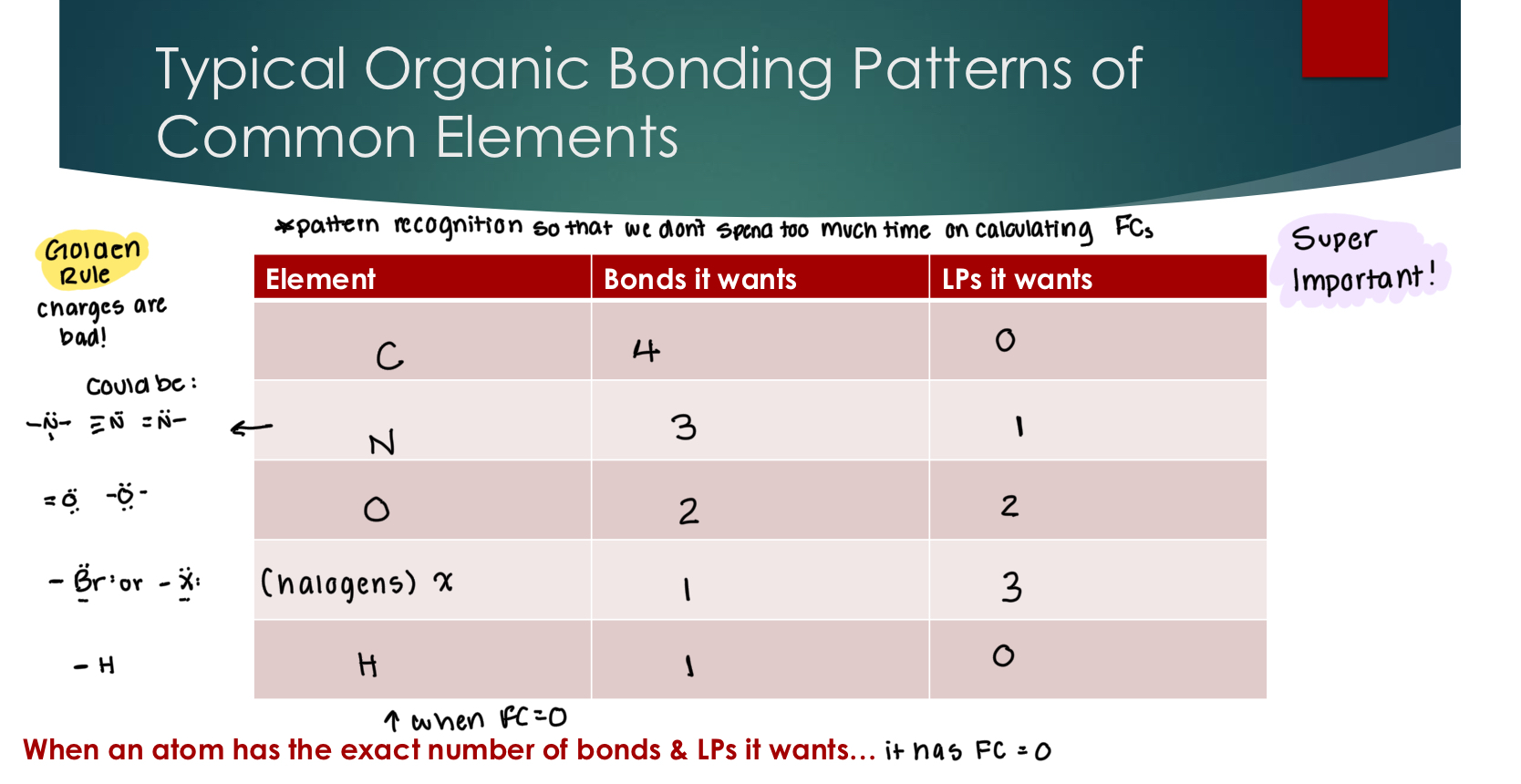
Typical Organic Bonding Patterns of Common Elements
Can a carbon atom ever have more than 8 electrons?
Carbon’s ideal bonding pattern starts with 4 bonds, meaning all bonding electrons are being covalently shared.
Removing a bond results in: taking a negative away: results in a positive charge and an incomplete octet
Replacing a bond with a lone pair results in: adds a negative charge but still is a complete octet
Can any of the other common elements have more than 8 electrons? any elements that are greater than or equal to 3 on the periodic table
From the ideal bonding pattern, replacing a lone pair with a bond → decreasing electron density, which results in a positive charge.
From the ideal bonding pattern, replacing a bond with a lone pair → increasing electron density
Note NOT changing the # of electrons in these examples.
Valence Shell Electron Pair Repulsion
Key Principle: e- groups repel each other → dictates molecular shape
What counts as an electron group?

Electron Geometry & Line Angle Structure
Key Features of Line Angle Representation
1. Carbon (C) atoms are shown as bends & ends of lines
2. Like Lewis structures, line represent covalent bonds (2e- per line)
3. Bonds between C atoms and Hydrogen (H) atoms are not shown explicitly
4. Bonds between heteroatoms (anything not C) and H atoms are shown but can be abbreviated
5. Formal charges are always included
6. Lone pairs are sometimes included
Bond angles need to be somewhat accurate
The way line angle structures are drawn corresponds to the electronic geometry
(based on VSEPR) of each central atom
Line angle structures should include reasonably accurate bond angles that correspond to the correct geometry
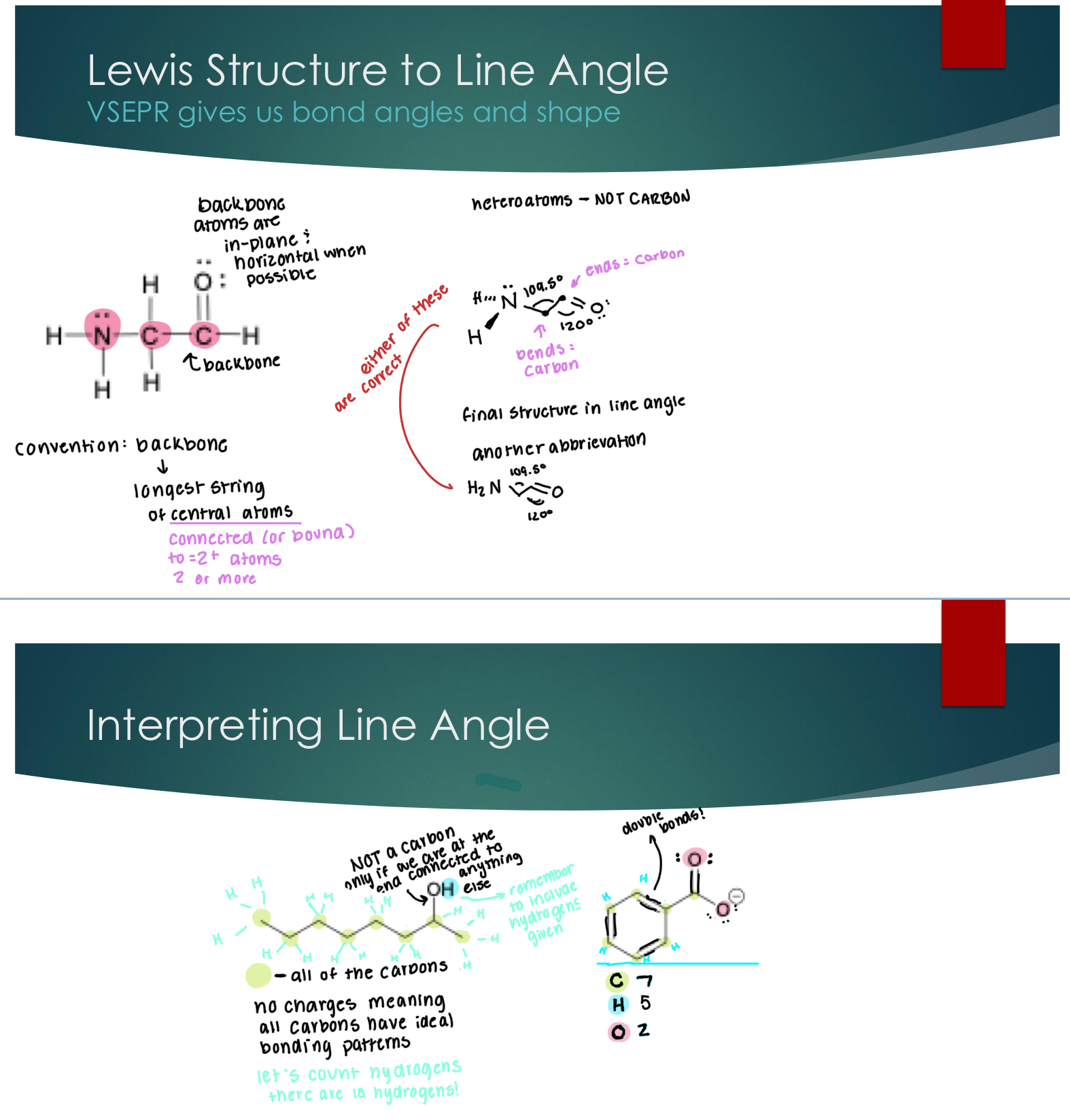
Valence Bond Theory (VBT)
Electrons exist in orbitals
Bonding occurs when electron orbitals overlap:
✓ Half filled + Half filled
✓ Empty + Full
x Full + Full
When bonding, electrons exist in different types of orbitals compared to isolated atoms (atomic orbitals)
Localized method: only the bonding (valence) electrons have a change in orbital
type
The new orbitals are formed through Hybridization
Hybridization
1. Hybrid orbitals do not exist in isolated atoms, they are only formed in
covalently bonded atoms
2. Hybrid orbitals have unique shapes and energies compared to atomic
orbitals (AOs)
3. Hybrid orbitals are created through the combination of AOs
4. The number of AOs combined = the number of hybrid orbitals created
5. Hybrid orbitals in the same set (created from the same AOs) have the
same shape and energy
6. The type of hybrid orbitals that are formed depends on its electron-pair
geometry (VSEPR)
H-atoms cannot hybridize.
NOT CHANGING # OF ORBITALS
Hybrid Orbitals in Organic Chemistry
Hybrid orbitals don’t really “exist” this is a model that is used to explain bonding and reactivity
This model works very well for C bonding and therefore for most organic molecules which is why we learn and use it in this class
A more accurate and detailed model: Molecular Orbital (MO) Theory (Next Class!) # of e- groups e- geometry hybrid orbital

Hybrid Orbitals: sp3
Created by the combination of 1 s AO + 3 p AOs
Any atom that is sp3 we can then say that it will have 4sp3 orbitals in the valence shell.
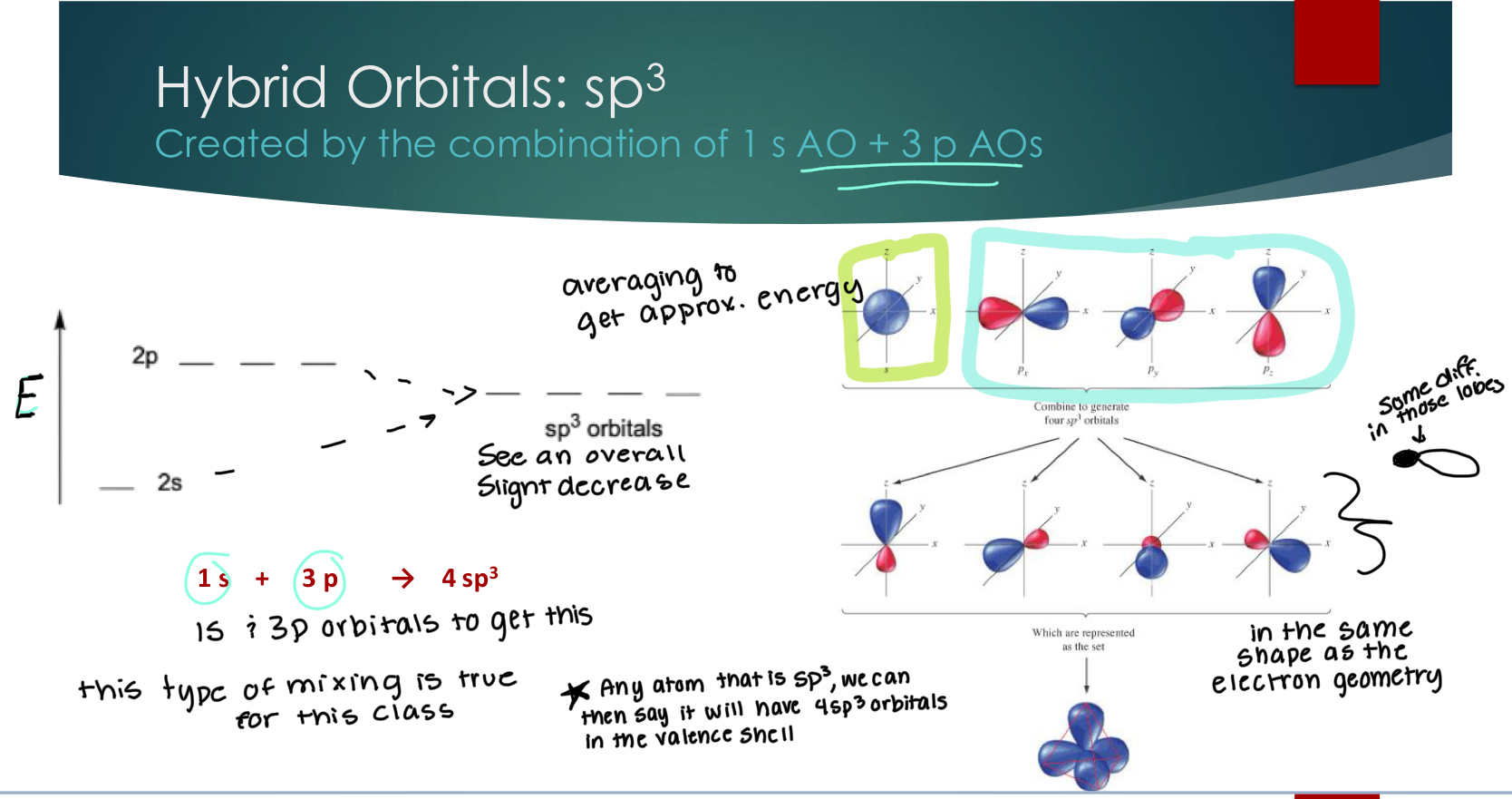
Hybrid Orbitals: sp2
Created by the combination of 1 s AO + 2 p AOs
Little less energy than the sp3 bc missing a p orbital. In vakence shells, we should have a total of 4 orbitals, even if they’re not all hybridized.
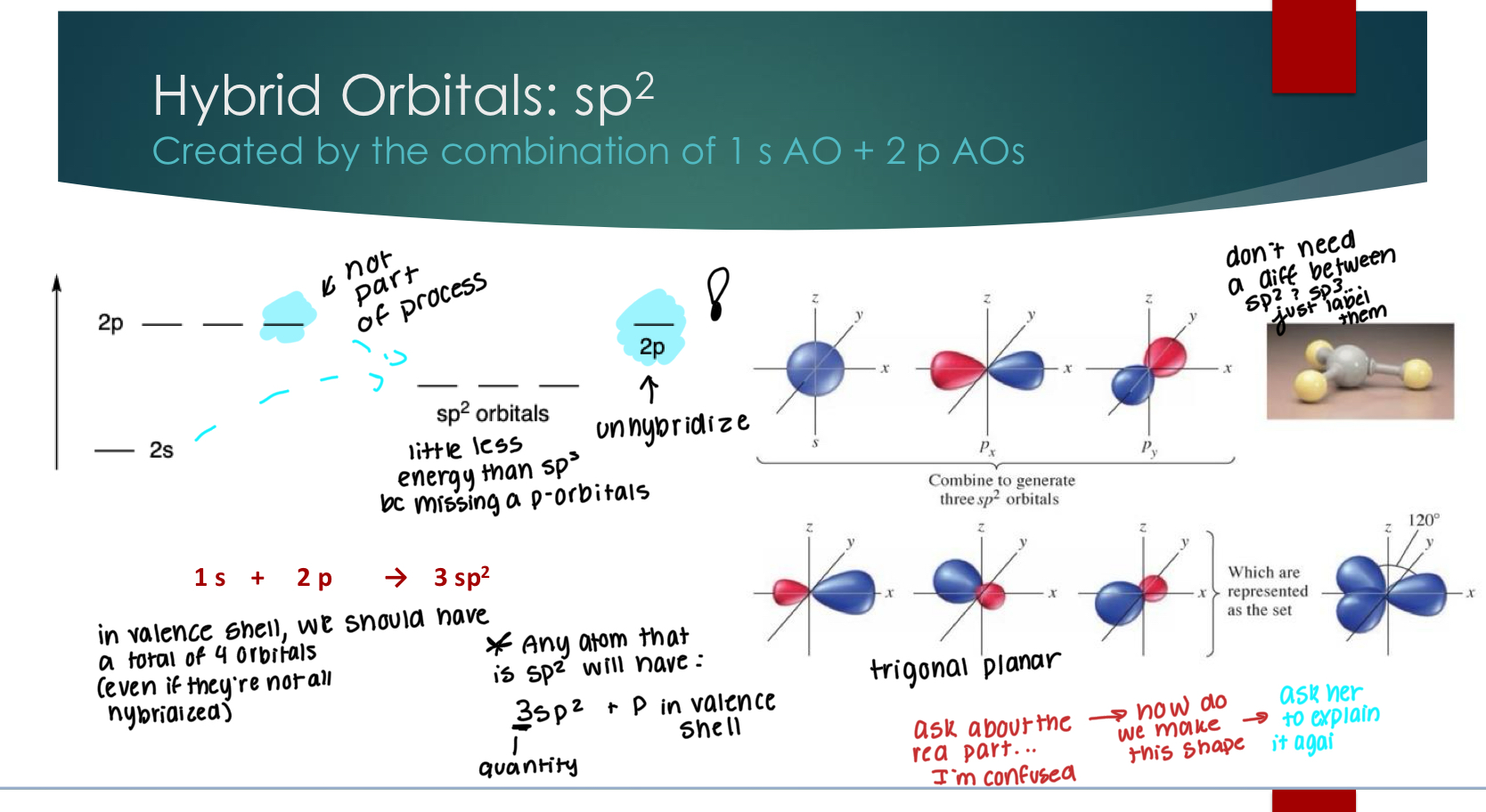
Hybrid Orbitals: sp
Created by the combination of 1 s AO + 1 p AO
all orbitals are perpendicular.
Comparing Hybrid Orbitals
s-character & electronegativity
increasing s-character increases electronegativity because there is more area for the nucleus to attract in a sphere than the other shape,

Drawing & Shading Orbitals
Electrons have wave-like behavior
shading indicates phase (describing motion for wave-like patterns).
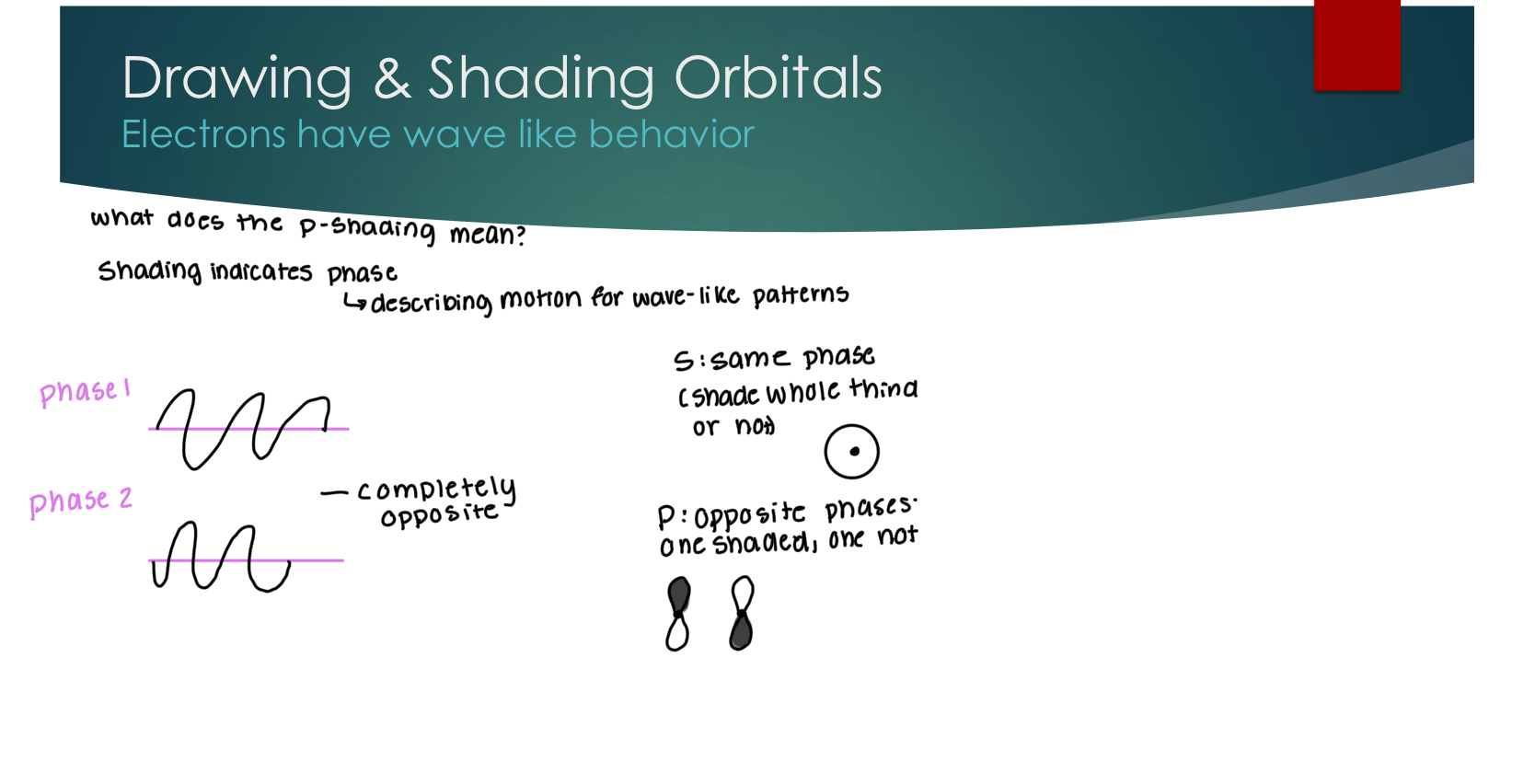
Constructive vs Destructive Interactions
Only constructive orbital overlap only leads to bonding & more overlap = better bonding
constructuve wave (in phase overlap) is additive
destrictive wave (out of phase overlap) cancels.
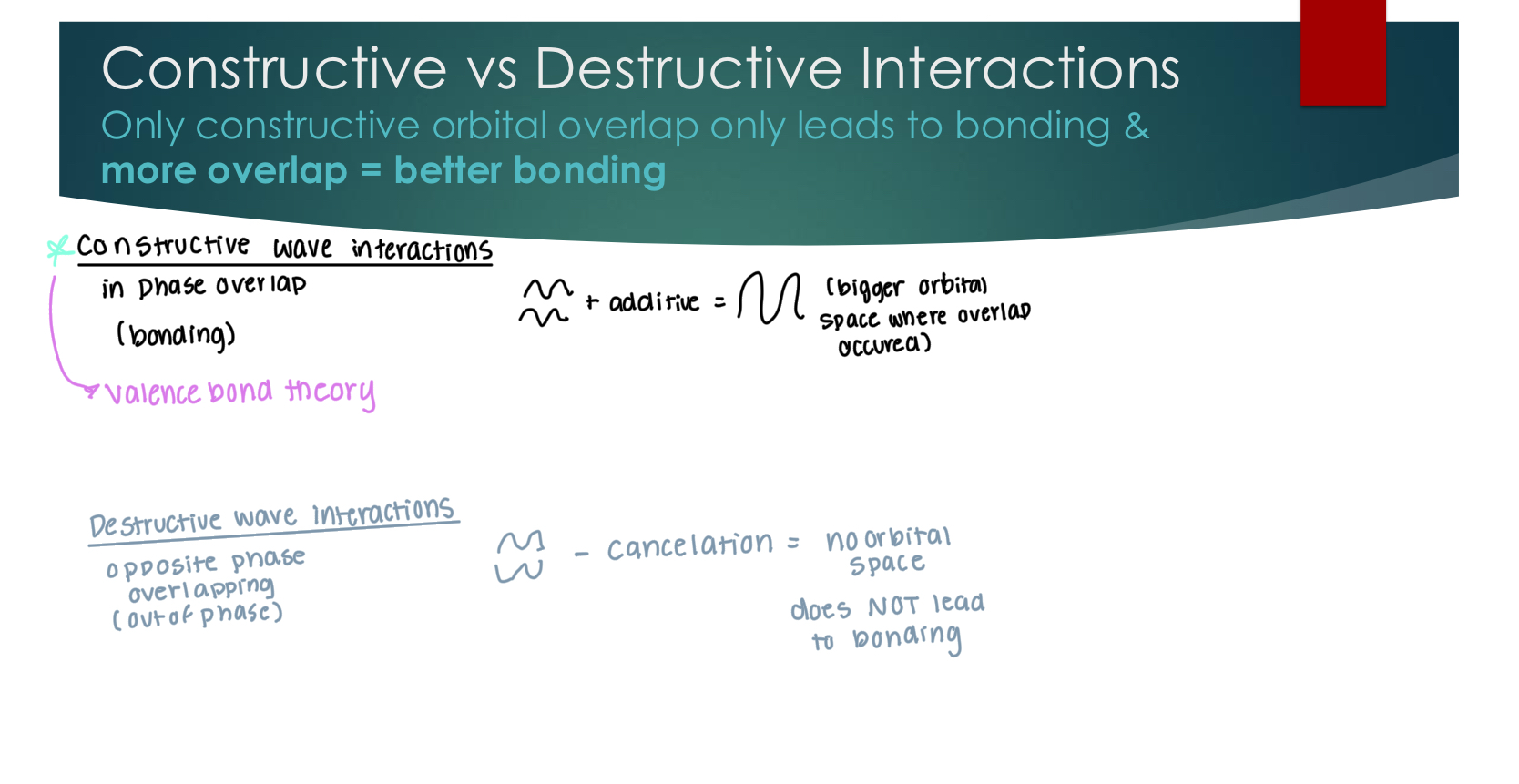
Classifying Bond Types with VBT
1. Un-hybridized s orbital (H) + Un-hybridized s orbital (H)
only exists w/ H-H (sigma bond)
2. Un-hybridized p orbital + Un-hybridized p orbital
phases are aligned on the same axis. pi bond. in VBT, p orbitals can only overlap with other p-orbitals.
3. Un-hybridized s orbital (H) + any hybridized orbital
Lots of possibilities, but we want best overlap. Larger lobe of hybrid in the same phase.
4. Any hybridized orbital + any hybridized orbital
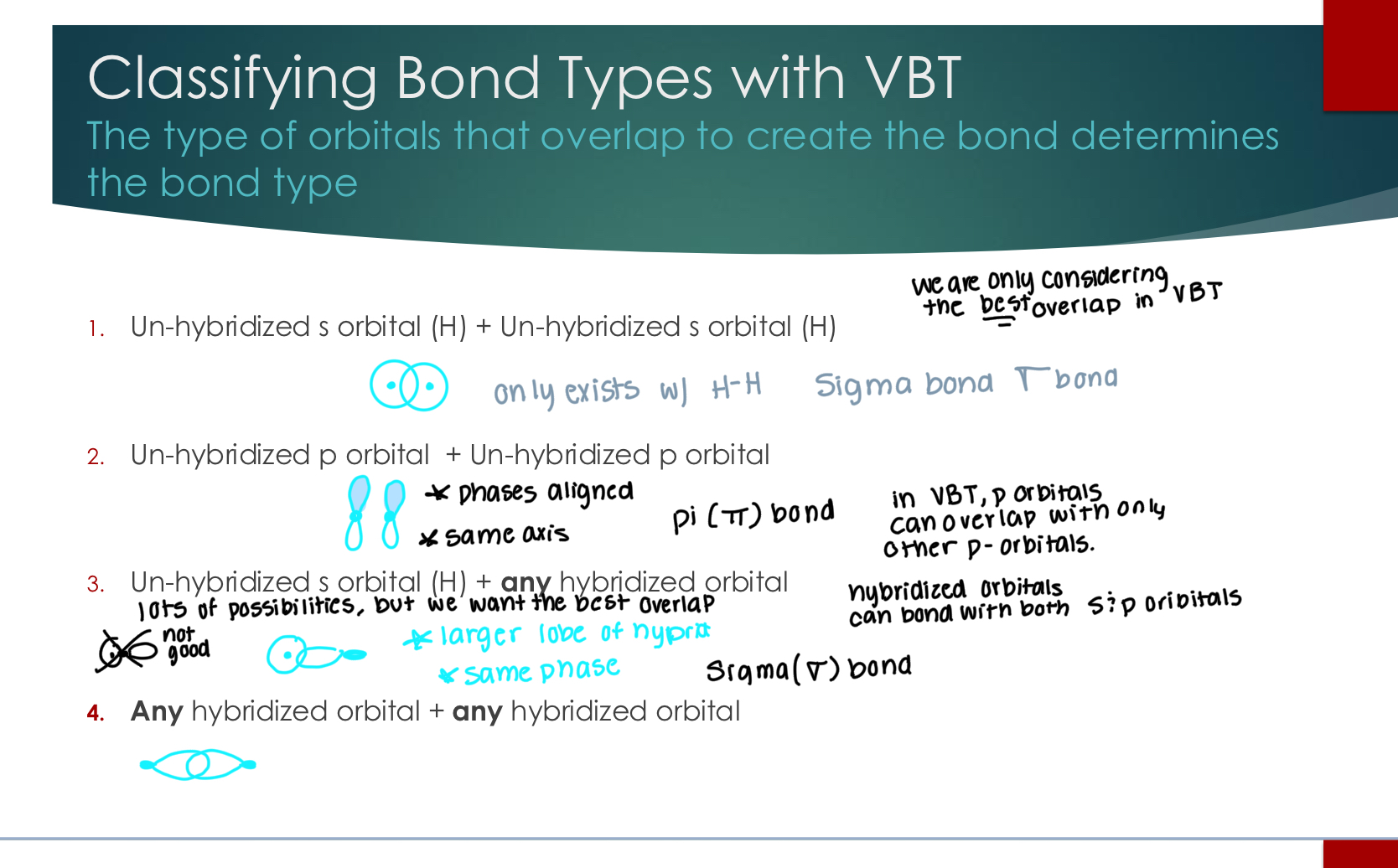
Drawing Sigma Bonding Schemes
Showing overlap between all hybrid & s type orbitals
We only draw valence shell electrons with letters, empty orbitals must contain the lone pairs. If there is overlap, that is a sigma bons.
Drawing Pi Bonding Schemes
Showing overlap between all p type orbitals
*We have to remember orientation: if p-orbitals are overlapping, they must have the same axis and aligned phases.
*P-orbitals must only overlap w/ other p orbitals based on other diagram, must be on a diff axis. Except if they’re bonded.
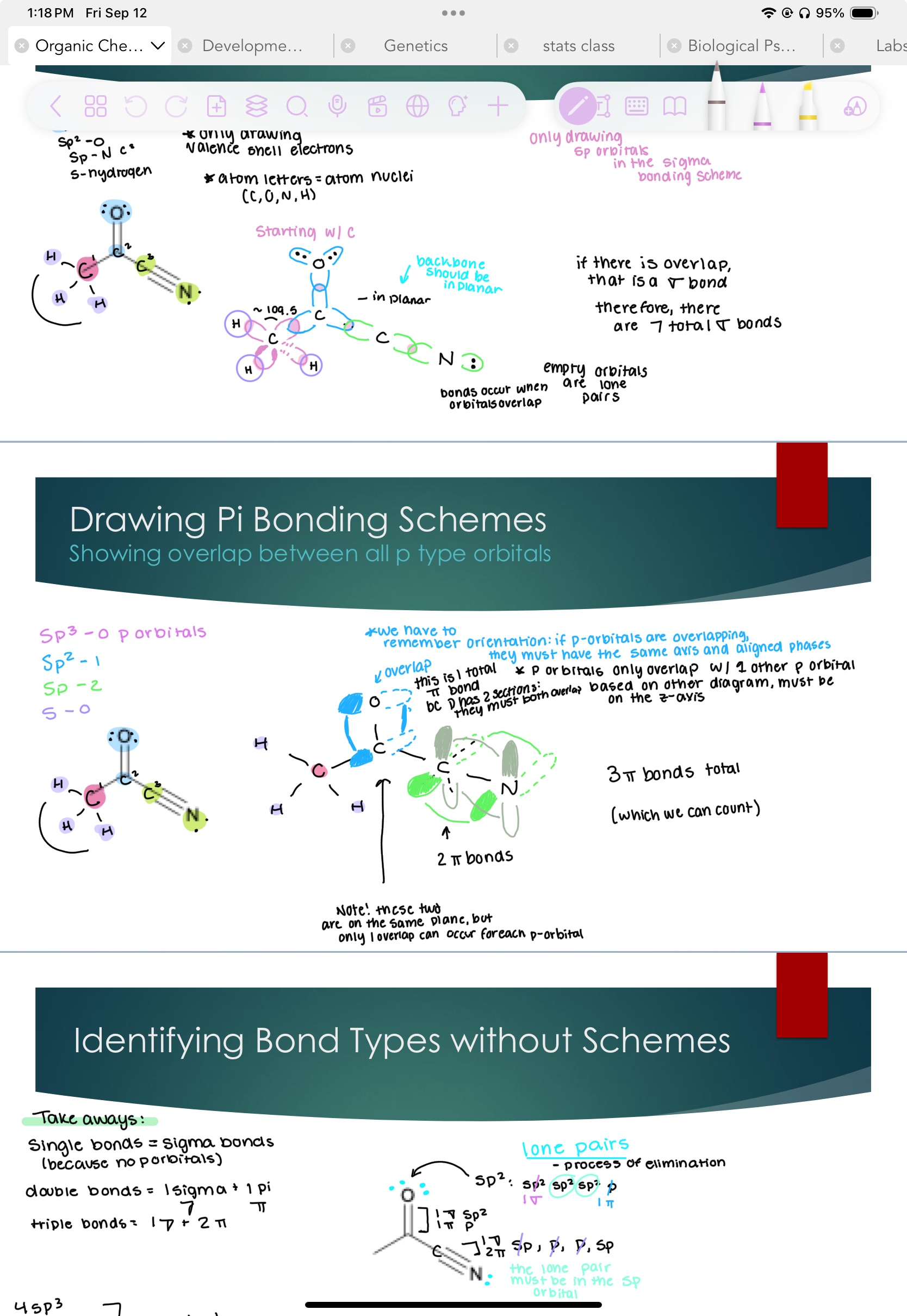
Identifying Bond Types without Schemes
Takeaways:
Single bonds= sigma bonds (because no p orbitals)
Double bonds = 1 sigma and 1 pi bond
Triple bonds = 1 sigma bond and 2 pi bonds
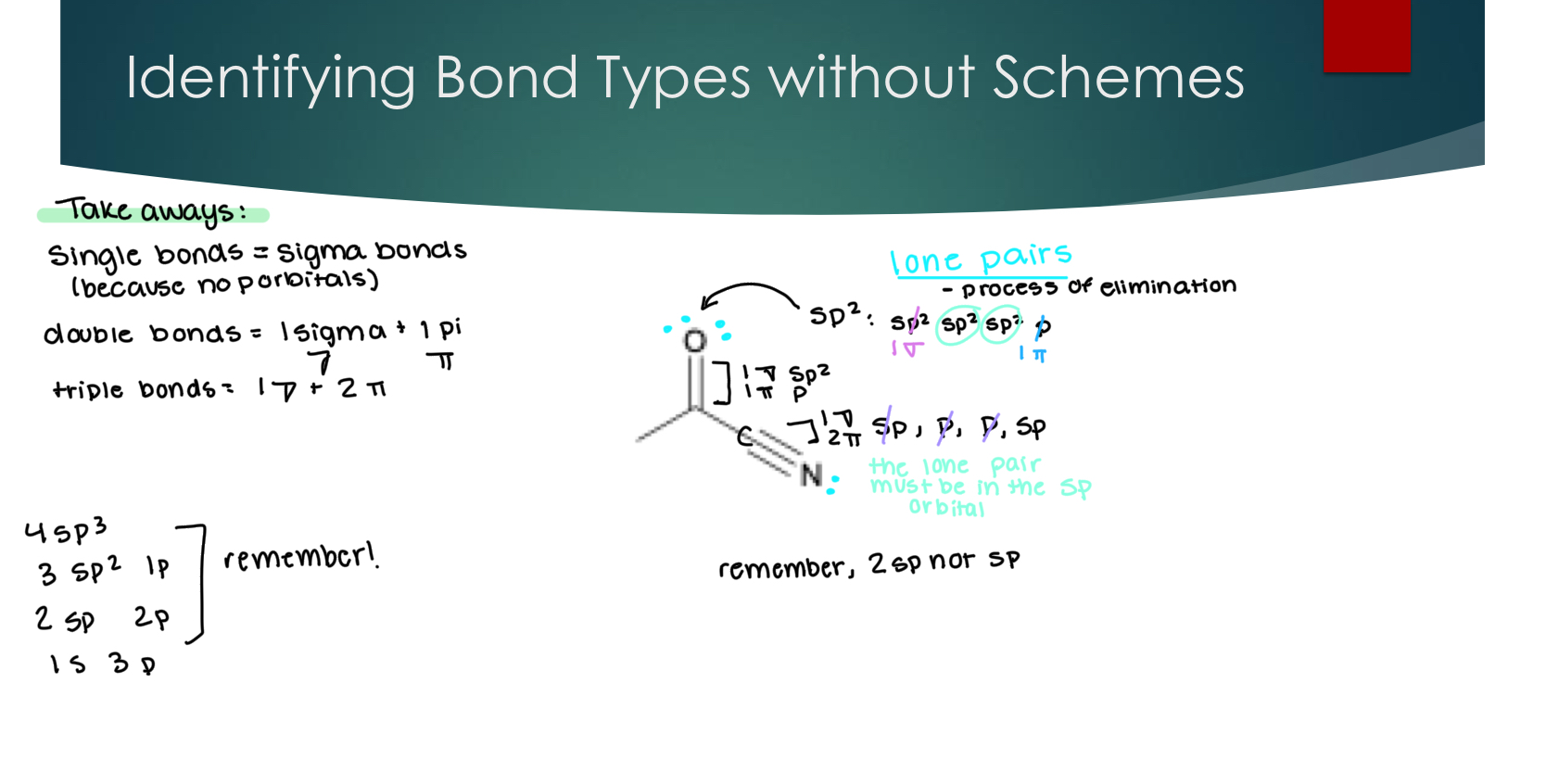
A unique structure: Allene (and its orbitals)
While we draw sigma & pi orbitals in separate schemes, these orbitals exist together at the same time
The reason the h’s are in different planes is because the p bonds have to be in the same plane.
Molecular Orbital Theory
Like VBT: the orbitals that hold electrons will differ between isolated atoms and bonded molecules
Unlike VBT: when atoms form covalent bonds, all orbitals holding electrons undergo a change in energy and shape
Linear combination of atomic orbitals (LCAO): mathematical process of combining atomic orbitals (AOs) to form new molecular orbitals (MOs)
# of AOs combined = # of MOs formed
MOs can be classified as: bonding, anti-bonding, non-bonding
Bonding MOs from s orbital overlap
Key Principle: electron density is located between the atoms
In-phase overlap of AOs and places the majority of electron density in the
internuclear region
One possibility is that they’re in-phase, other is that they’re out of phase

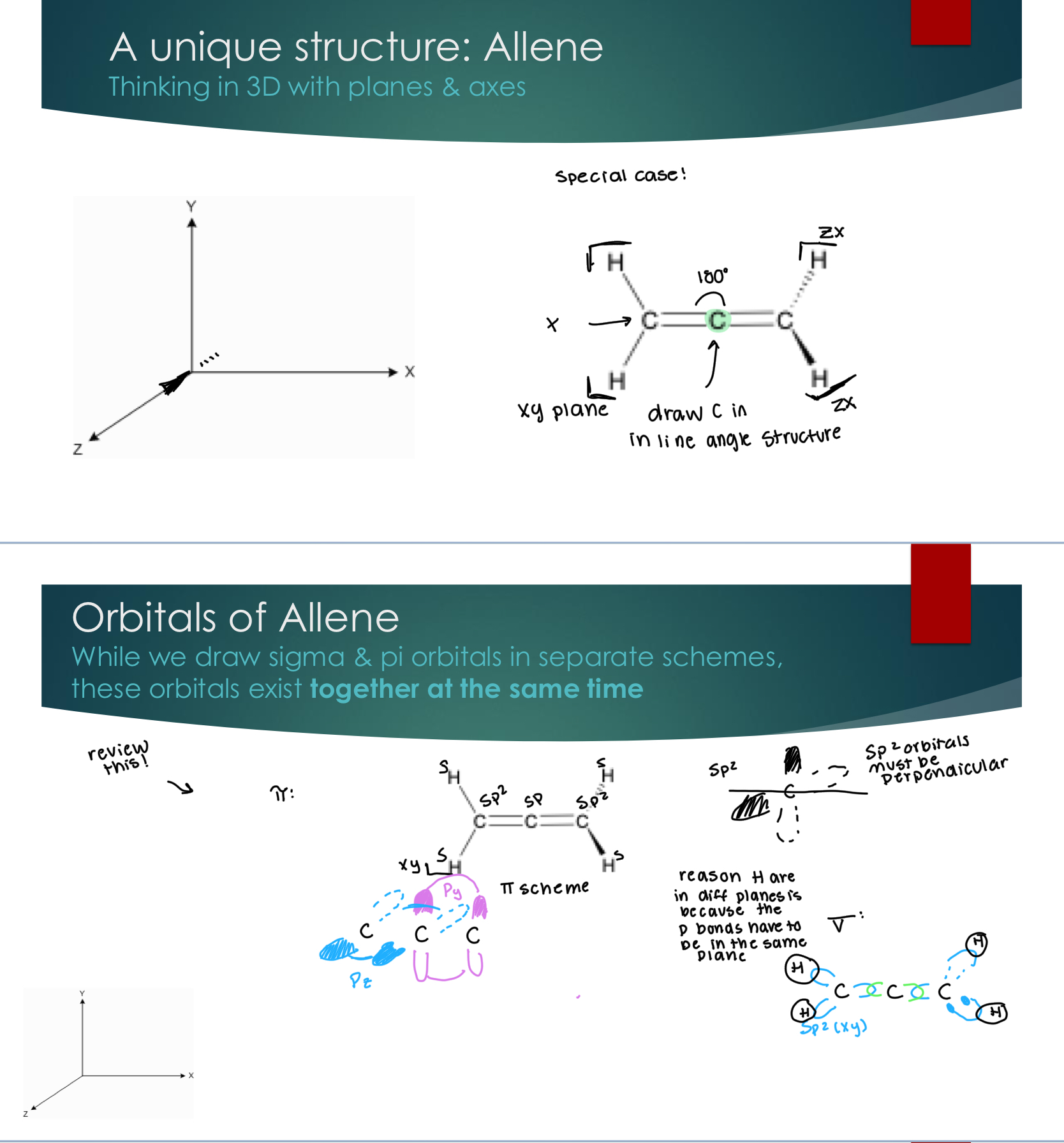
Anti-Bonding MOs from s orbital overlap
Key Principle: electron density is located not between the atoms
Out-of-phase overlap of AOs and places the majority of electron density outside of the internuclear region
Stil an S1 Sigma Molecular bond
Non-Bonding MOs
Key Principle: electron density is equally between and outside of the atoms
As molecular complexity increases, the amount of in-phase and out-of-
phase overlap can be very similar which results in a non-bonding MO
Electron density is about equal inside and outside the internuclear region for these MOs and the have the same Energy as the AOs used to create it!
Bond Order & HOMO / LUMO Identification
Bond Order = (# e- in bonding MOs - # e- in anti-bonding MOs) / 2
Higher bond order = more electrons in the internuclear region = more stable
HOMO: highest occupied molecular orbital : highest energy MO that contsins electrons
LUMO: lowest unoccupied molecular orbital : lowest energy empty MO (no e-)
Which is the most stable molecule : H2 or He2
Atoms give the highest energy electrons so donors → HOMO and acceptors → LOMO
The higher the bond order, the more stable the bond!
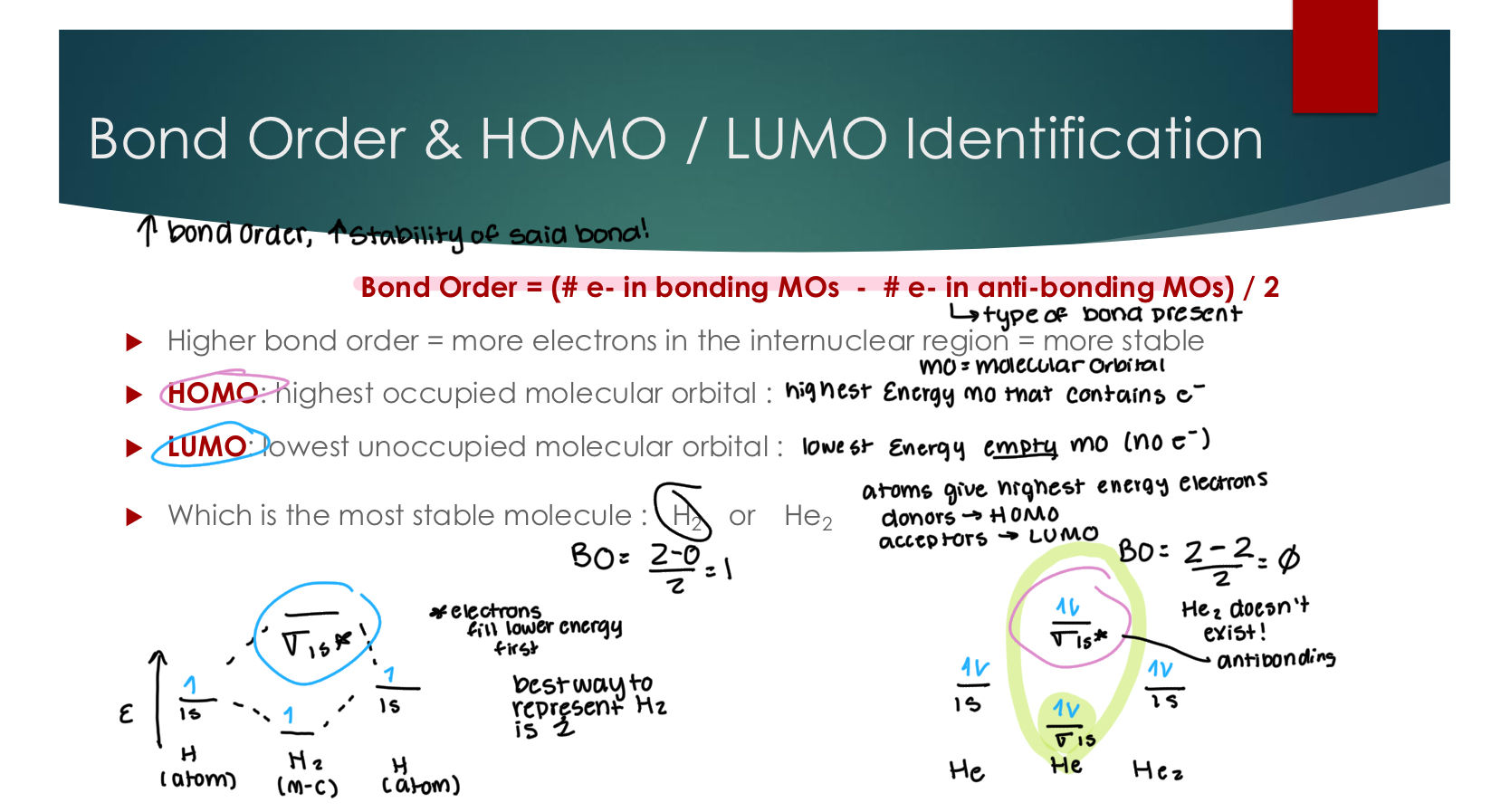
The Combination of P Orbitals and LCAO: p orbitals
Key takeaway: o-orbitals appear in pairs, meaning there are 2 lines.
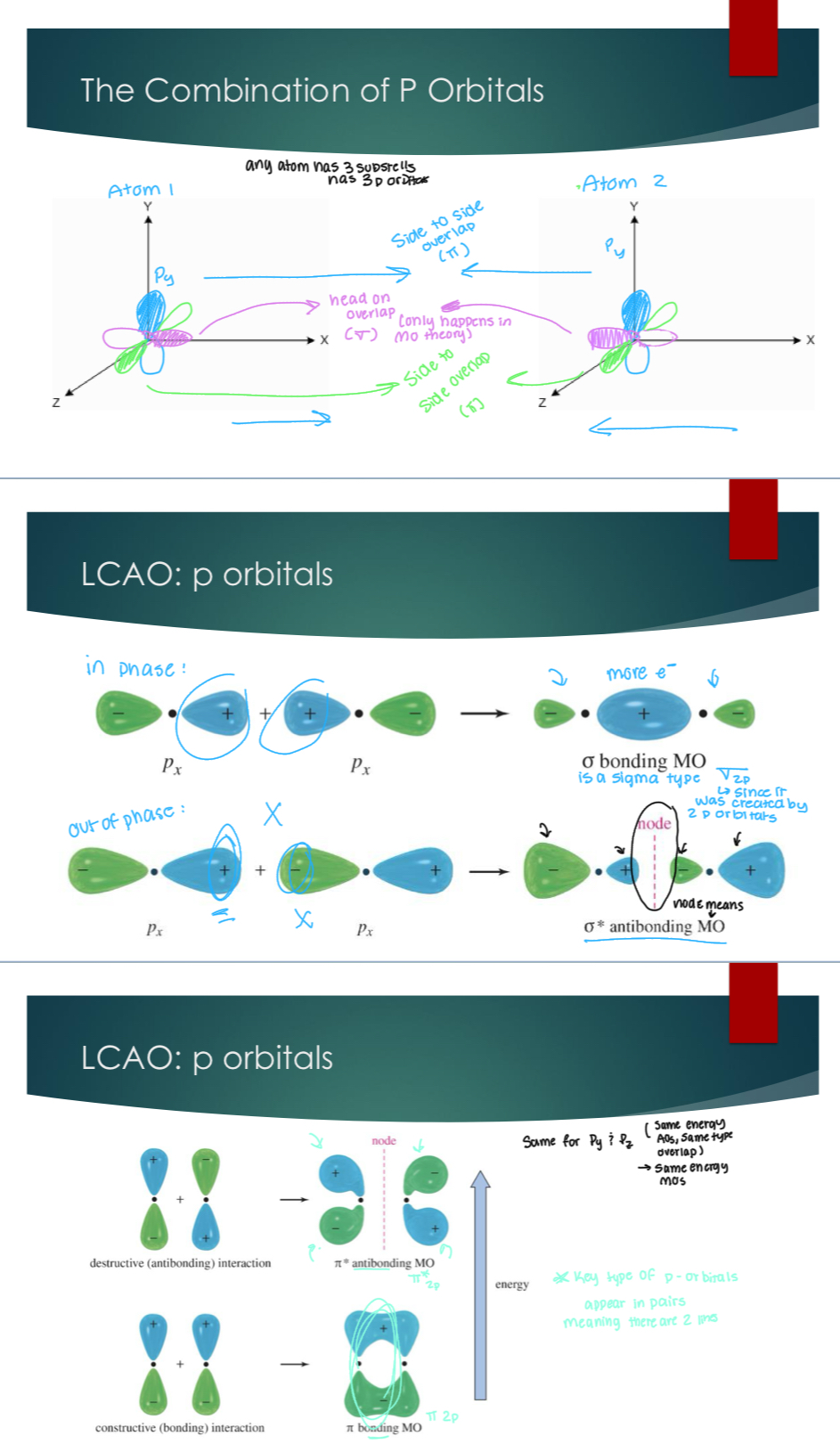
Energy of MOs from p-orbitals
Bonding Theories: Key Takeaways
Electrons move around a lot and orbitals are 3D areas where they are most likely to be found
Bonding >>> “Bonds”
Better overlap = more bonding electrons = more stable
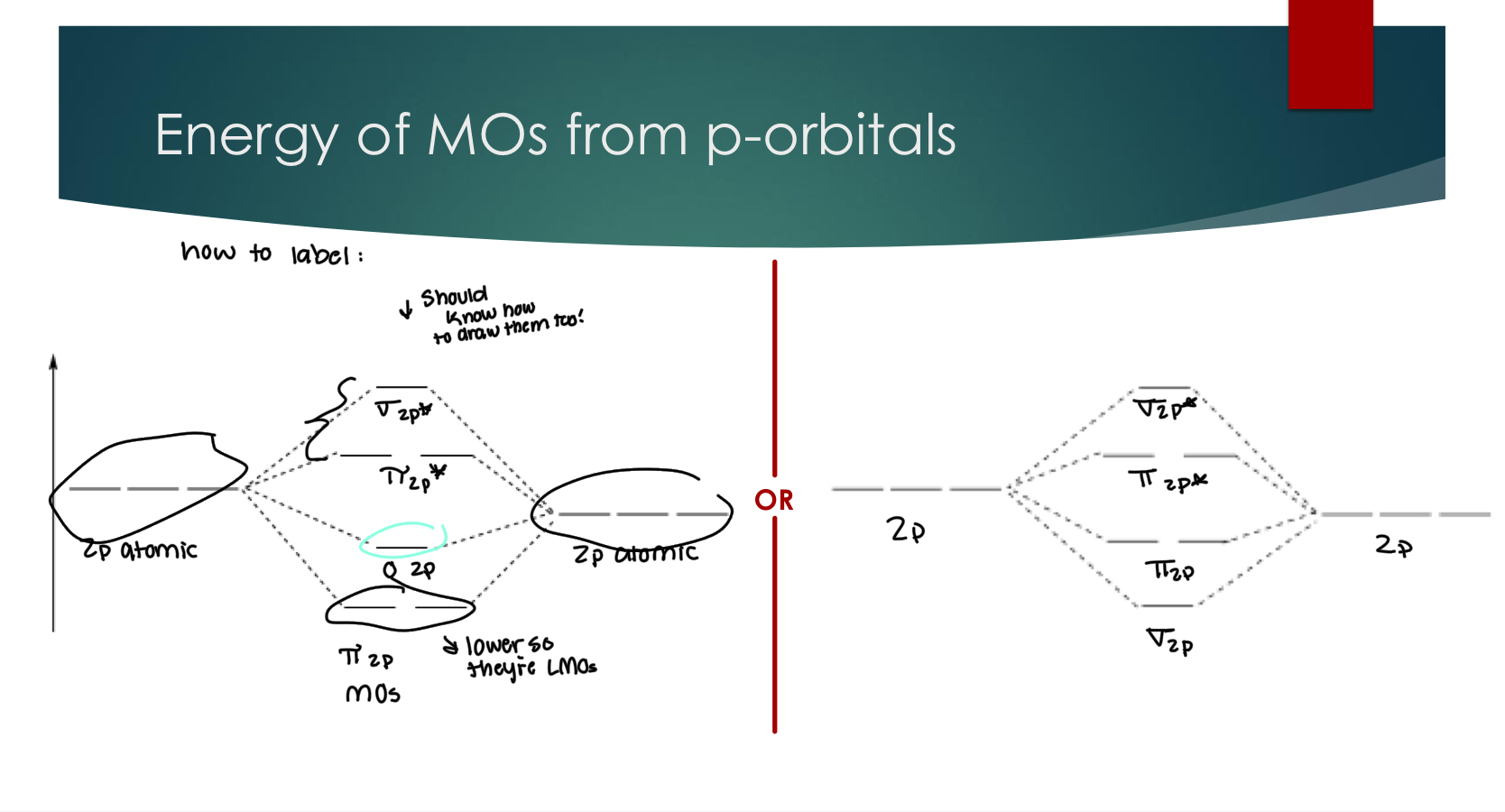
MO review question
Fill out the provided MO diagram for O2+ and determine the
Bond Order for the molecule. The identify & sketch the HOMO & LUMO
NOTE:
HOMO- highest order that contains electrons,
LUMO - lowest unoccupied order
When these two are the same orbital it becomes SOMO:
A singularly occupied MO
What is Resonance?
Electrons like to move around, once they are in hybrid / molecular orbitals they want to move around if possible
Electrons in single bonds (sigma bonds / hybrid orbitals) These are not able to move via resonance
These are close to nuclei, meaning the negative electrons are close to the positive nucleus, meaning e- are tightly held, meaning they are more stabilized.
Electrons in multiple bonds & lone pairs (pi bonds / p orbitals)
These are far from nuclei, less stable, and less tightly held: which means that they can move via resonance
When we think about resonance, we only need to think of p-orbitals and π type overlap.
Resonance Structures
Resonance is the movement of electrons through adjacent p-orbitals and resonance structures help us think about and depict the different locations the electrons could be
Resonance structures are not different molecules, they are just a different “snapshot” of possible electron locations
Not all resonance structures are equal ... some locations of electrons may be more stable and therefore the electrons would prefer to be in that position more of the time
The limitations is that when they’re moving, they cannot break or form single bonds because they have to keep the established connectivity.
Ex. People move a lot in chairs because the movement is more comfortable – although they may stay in one spot longer if it’s super comfortable. However, you can't just take off your arm is your arm is still, so the connectivity has to stay the same.
The Resonance Hybrid
Basically, if we could average all structures and draw them: it’s the best pictured model
Because electrons are constantly moving, the molecule never looks like a single resonance structure
The resonance hybrid is a picture of the molecule that averages together all the possible resonance structures: this is the best representation of electron location in a molecule
Since the electrons prefer more stable resonance structures and spend more time in these locations, more stable resonance structures have a bigger influence on the resonance hybrid: “major contributors” (greater than 3 adjacent p-orbitals and more stable).
Electrons will spend less time in less stable resonance structures so these will have less influence on the resonance hybrid: “minor contributors” (less than three adjacent p-orbitals and less stable)
Hybridization & Resonance
How lone pairs can participate in resonance
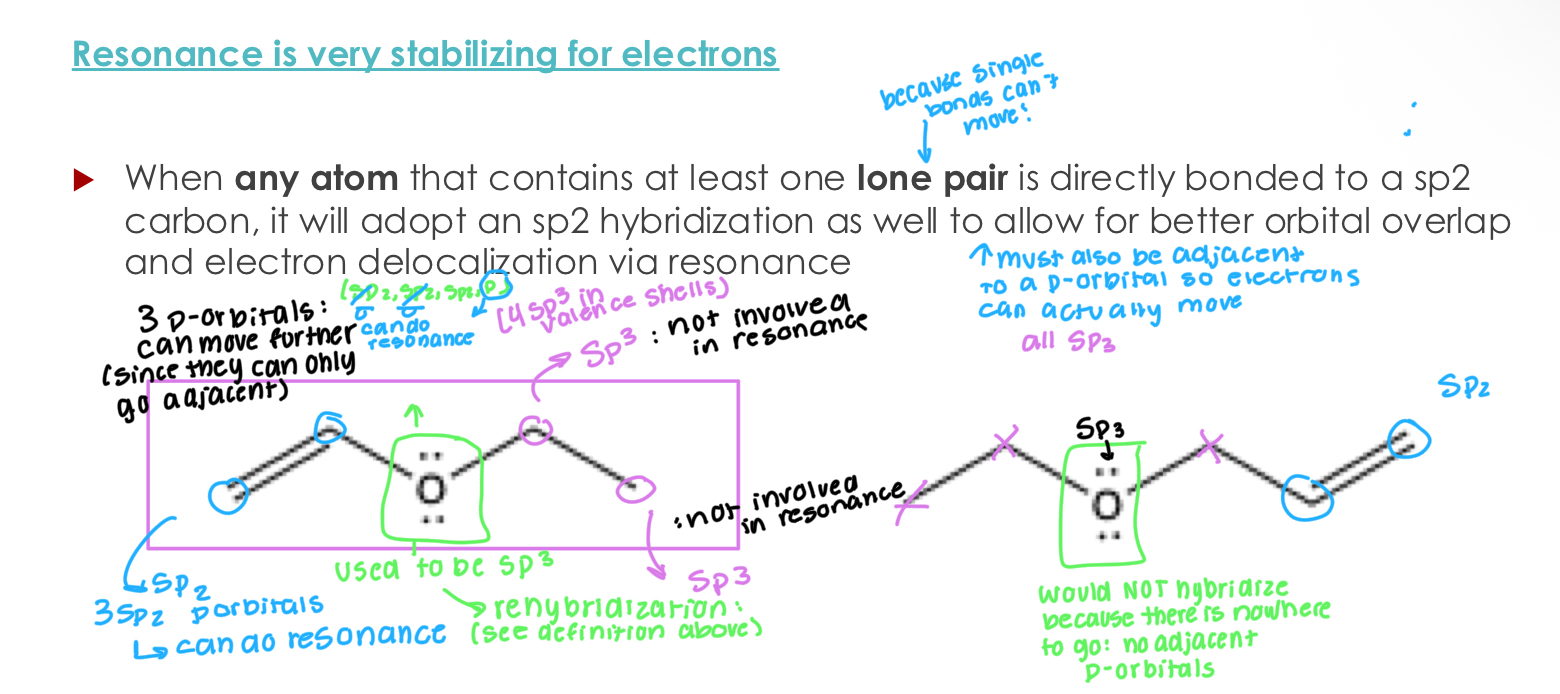
Resonance is very stabilizing for electrons
When any atom that contains at least one lone pair is directly bonded to a sp2 carbon, it will adopt an sp2 hybridization as well to allow for better orbital overlap and electron delocalization via resonance
Manner of thinking:
Is it hybridized?
Does it have a p-orbital?
Does it have adjacent p-orbitals?
Then, it has resonance if yes to all.
Yes, you can do resonance with empty p-orbitals.
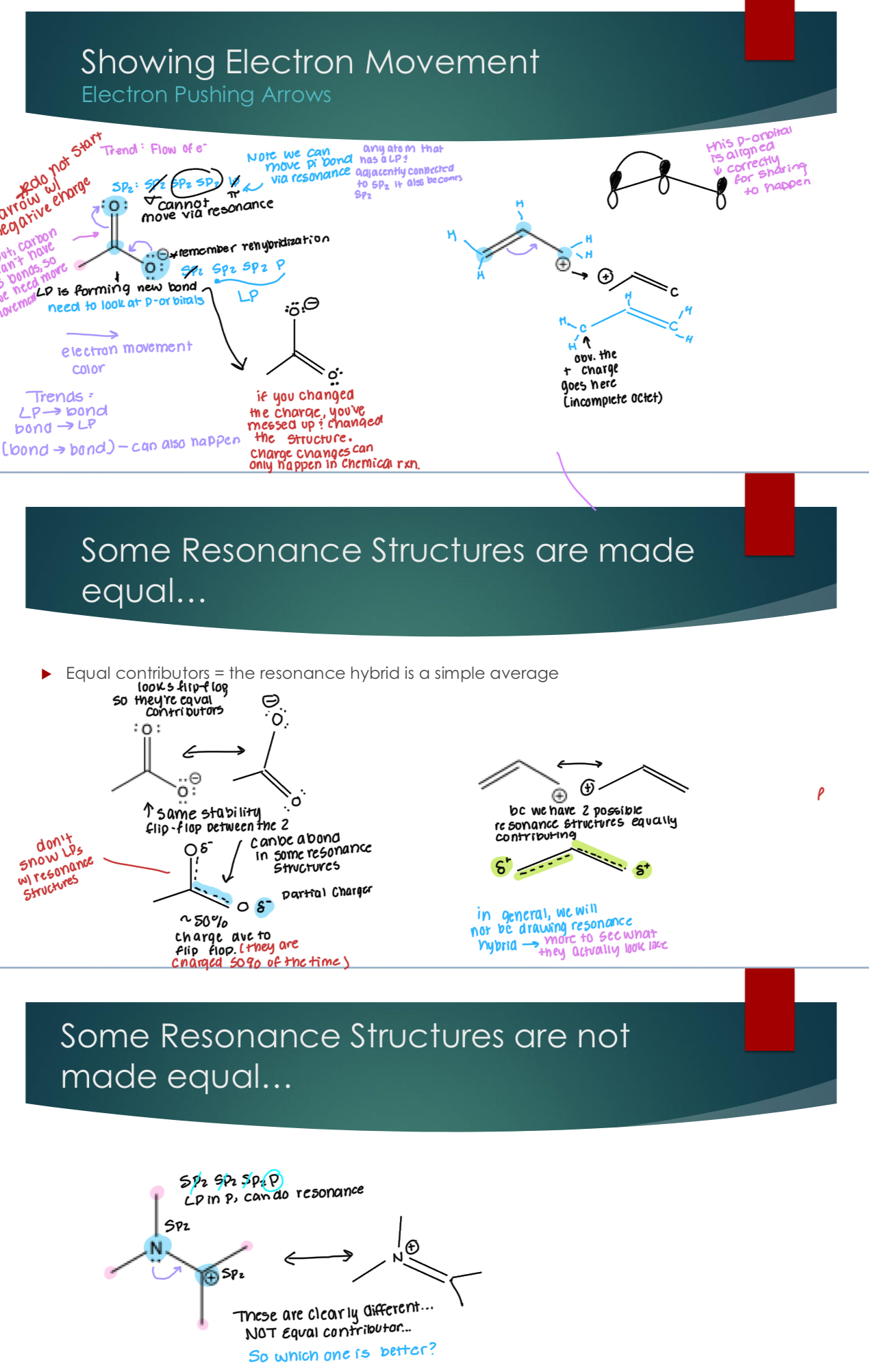
Showing Electron Movement - Electron pushing arrows.
Electron pushing arrows = mechanism arrows : shows how the electrons move to get from one resonance structure to another
They must be:
Curved, always
have to start arrows where the electrons are: aka, must start at a p-orbital.
Some Resonance Structures are made equal so that there are equal contributors = the resonance hybrid is a simple average. And some are made unequal where one is clearly better.
Stabilizing Factors: What leads to better resonance structures?
1. Full electron shells are very stable: Complete octets in the most amount of atoms
Helpful Hint / Strategy: Atoms that do not have charges or lone pairs will typically have complete octets
2. Charges are unstable: Fewer atoms have formal charges
*however, overall charges do NOT change, but FCs do change.
3. Electrons with stronger electrostatic attraction and/ or electrons with more space in their electron cloud are more stable
Negative charge on more electronegative atoms is best
Positive charge on less electronegative atoms is best
Charges on larger, more polarizable atoms is best
- atoms w/ small electron clouds don’t want to be there bc they like to move around
Evaluating Stability of Different Molecules Focus on the charges
Octets are still most important!!
Charges are still bad!
Localized charge is most unstable
Charges are stabilized by Electronic Effects (electrostatic attraction)
- on more EN atoms and + on less
Charges are stabilized by being spread out (delocalization and induction)
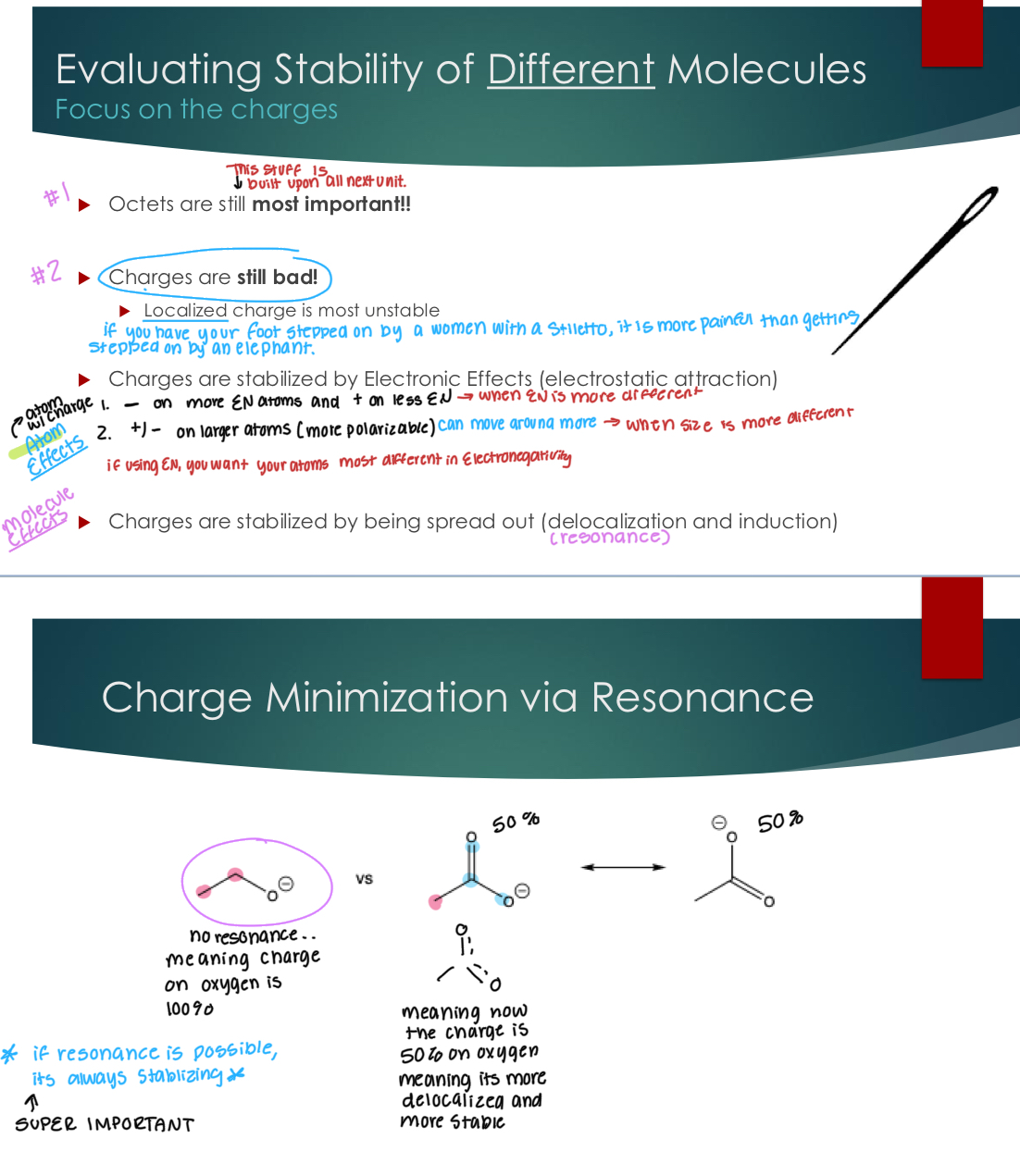
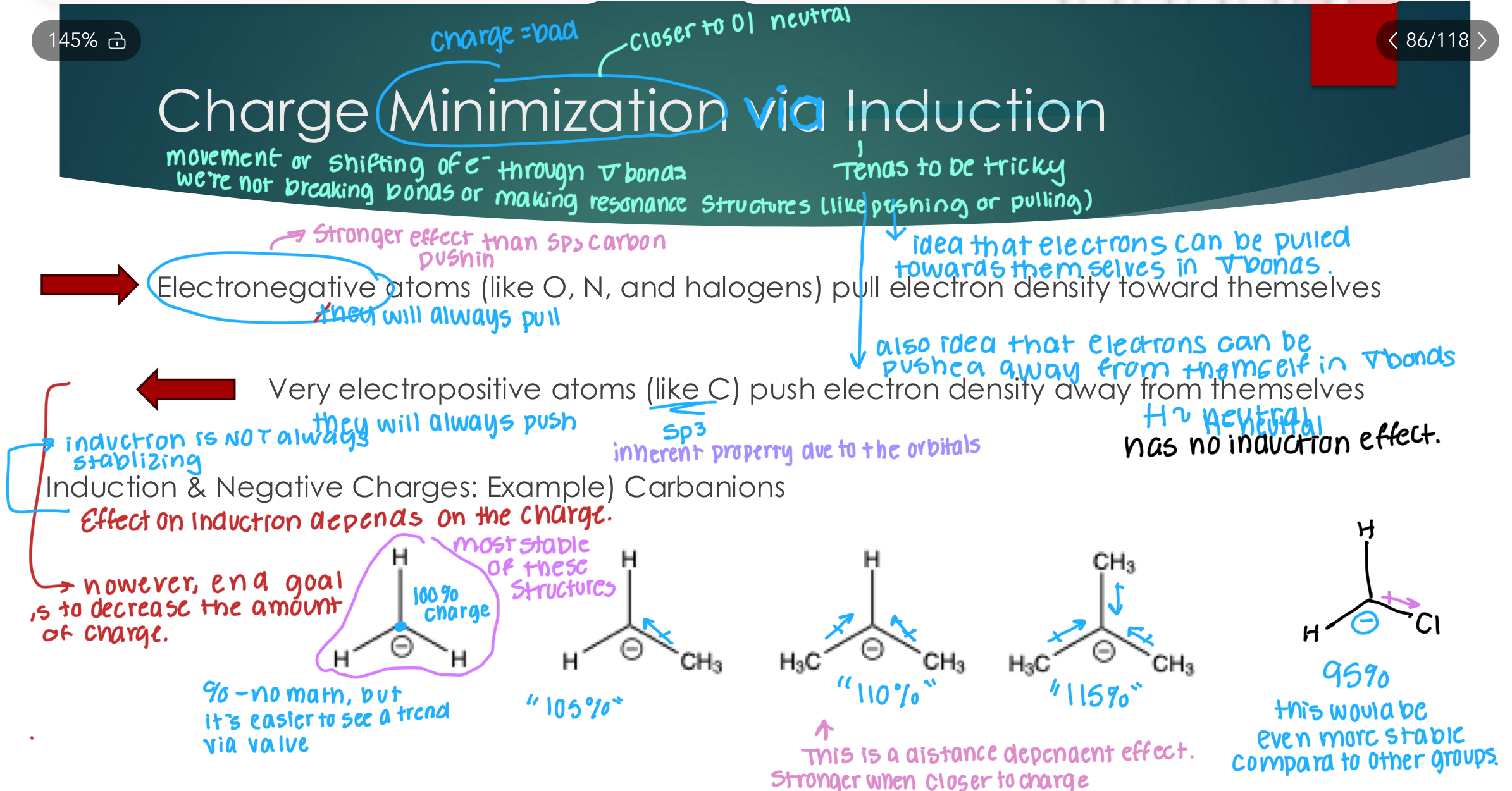
Charge Minimization via Induction
Movement of shifting e- through sigma bonds, we’re not breaking bonds of making resonance structures like pushing or pulling.
Electronegative atoms (like O, N, and halogens) pull electron density toward themselves (in sigma bonds)
Very electropositive atoms (like C) push electron density away from themselves (in sigma bonds)
Induction is not always stabilizing and the effect of it depends on the charge
Induction & Negative Charges: Example) Carbanions
If the charge is positive, you want arrows pointing towards it. If the charge is negative, you want the arrow pointing away from it.
Carbocation Orbitals : Hyperconjugation
Carbocations are also additionally stabilized by adjacent
bonds, most usually C—C or C—H, via hyperconjugation
Hyperconjugation: when electrons in adjacent sigma
bonds spread into the empty p-orbital, adding some
electron density to help stabilize the electron-deficient C
atom, this occurs via orbital overlap between sigma bond
and empty p-orbital

Steps to evaluating the most vs least stable.
In general: atom effects are more important than molecular effects for charge stabilization (but this trend has exceptions)
Step 1: Evaluate the atoms that are charged in each molecule and compare them using electronegativity and size
Step 2: Determine if resonance is possible: think about the number of and the stability of the additional resonance structures
More (stable) resonance structures = more stable
Step 3: Determine if induction is possible: think about the type of induction and the type of charge present: induction is not always stabilizing
good induction = stable
bad induction = unstable
Stable molecules are less reactive because they are already happy with their current structure, and are lower in energy
Less stable molecules are more reactive because they want to get to a more stable state, and are higher in energy
Bond Polarity: Dipoles & Partial Charges
Polar covalent bonds exist between: nonmetal atoms w/ different electronegativity values
These bonds have unevenly shared electrons which creates a bond dipole and partial charges on the atoms participating in the bond
More polar bonds have larger partial charges:

Molecular Polarity: Determining where (and what) the partial charges are
1. Think about (and draw) the bond dipoles
2. Add the bond dipoles (vector math)
3. Assess the charge distribution in the molecule
You can have polar bonds, but they may cancel out and not have a charge.
Do we have a polar bond?
Do we have a symmetrical molecule?
Key points:
Using bond dipoles and molecular shape to determine where partial charges exist in a molecule
Partial charges govern how molecules interact with each other in non-covalent ways (Today)
Partial charges can also help us identify how molecules will react (Week 4)
Intermolecular Interactions
Key Principle: Its all about the electrostatics! (+ wantes to be next to the -)
Key Principle: Attraction between larger charges is stronger
Intermolecular forces are non-bonding forces of attraction that occur between two separate
molecules
Can be two molecules of the same compound
Can be a molecule of one compound interacting with a molecule of another compound
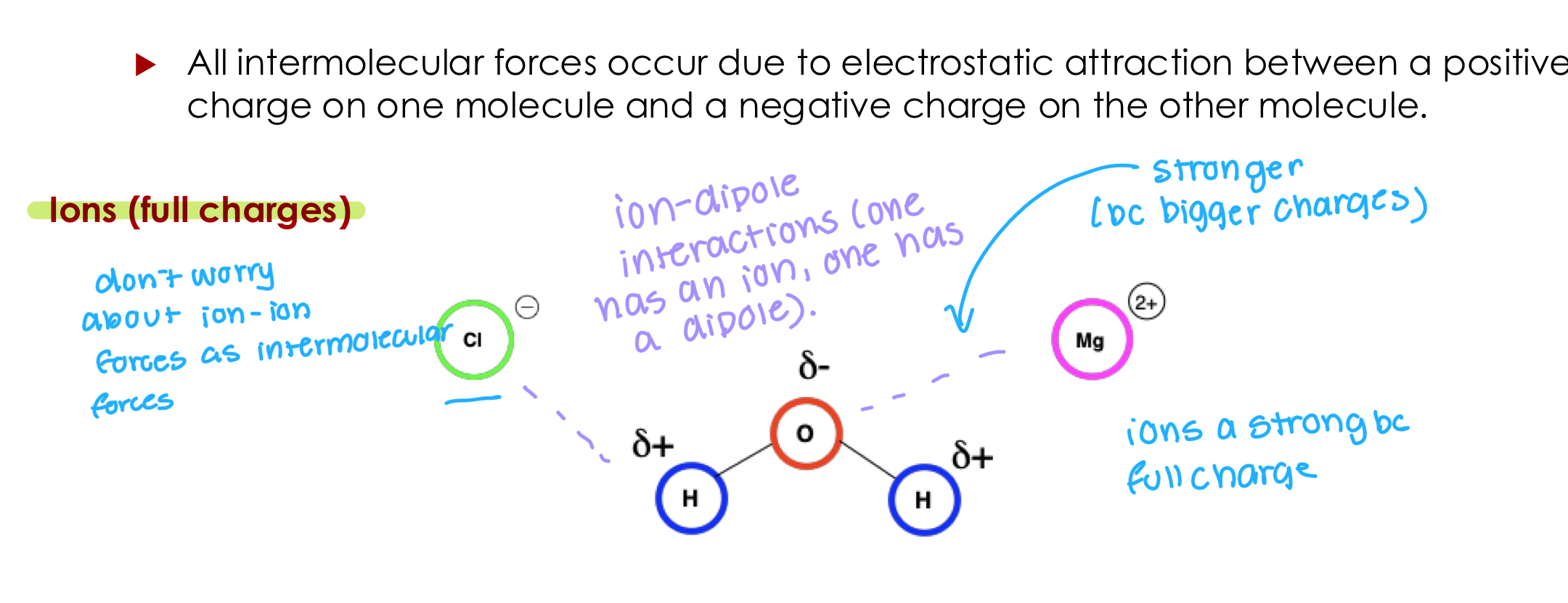
Ion-Dipole
All intermolecular forces occur due to electrostatic attraction between a positive charge on one molecule and a negative charge on the other molecule.
Ion-dipole interactions (one has an ion, one has a dipole).
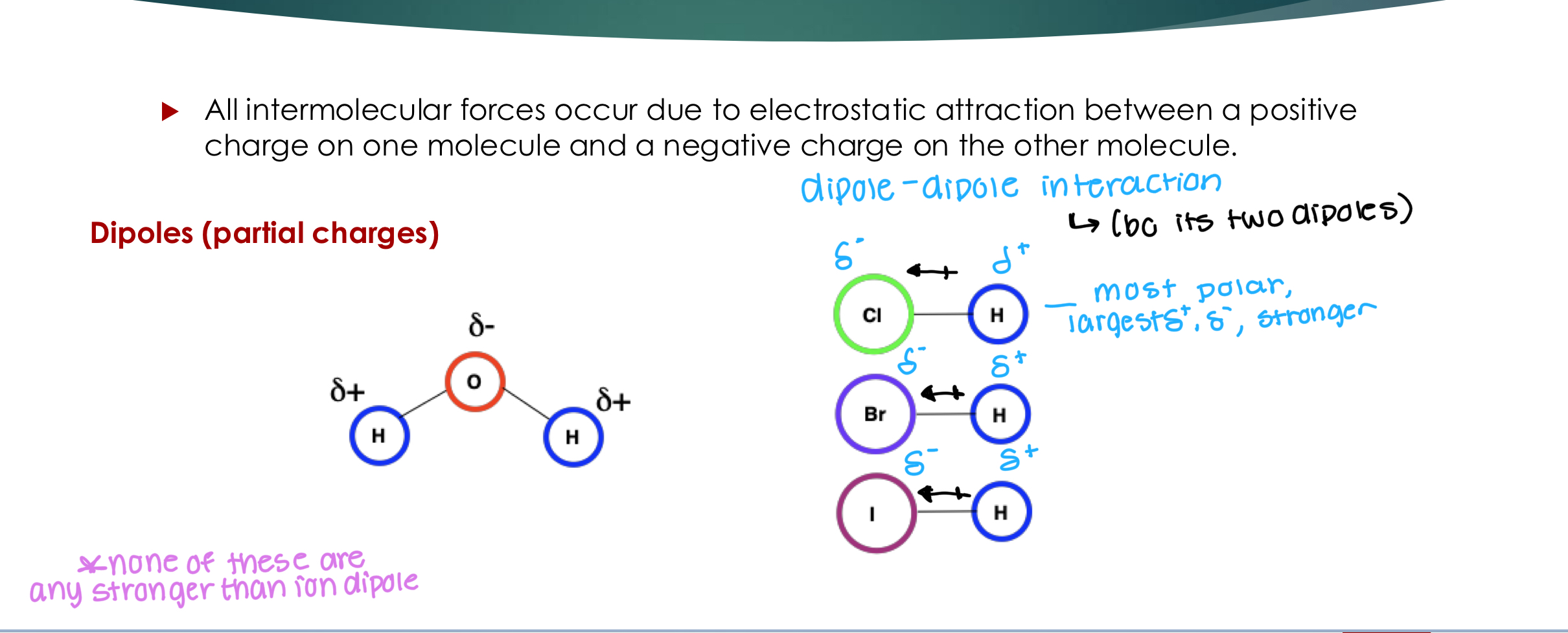
Dipole-Dipole
All intermolecular forces occur due to electrostatic attraction between a positive charge on one molecule and a negative charge on the other molecule.
Not stronger than ion-dipole
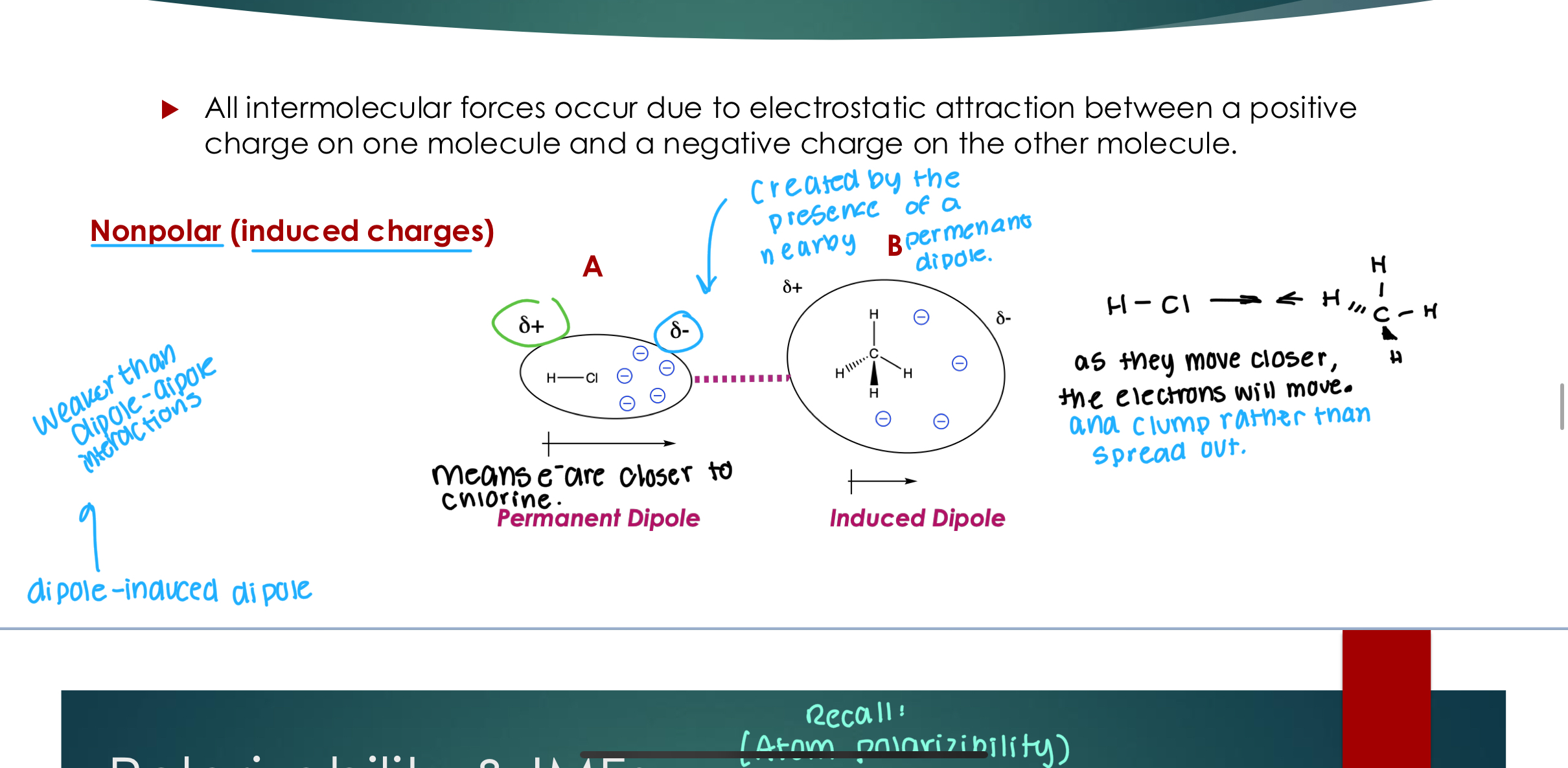
Dipole-Induced Dipole
Nonpolar (induced charges) As the molecules move closer, the electrons will move and clump around the more electronegative atom instead of spread out.
Weaker than Dipole-Dipole interactions
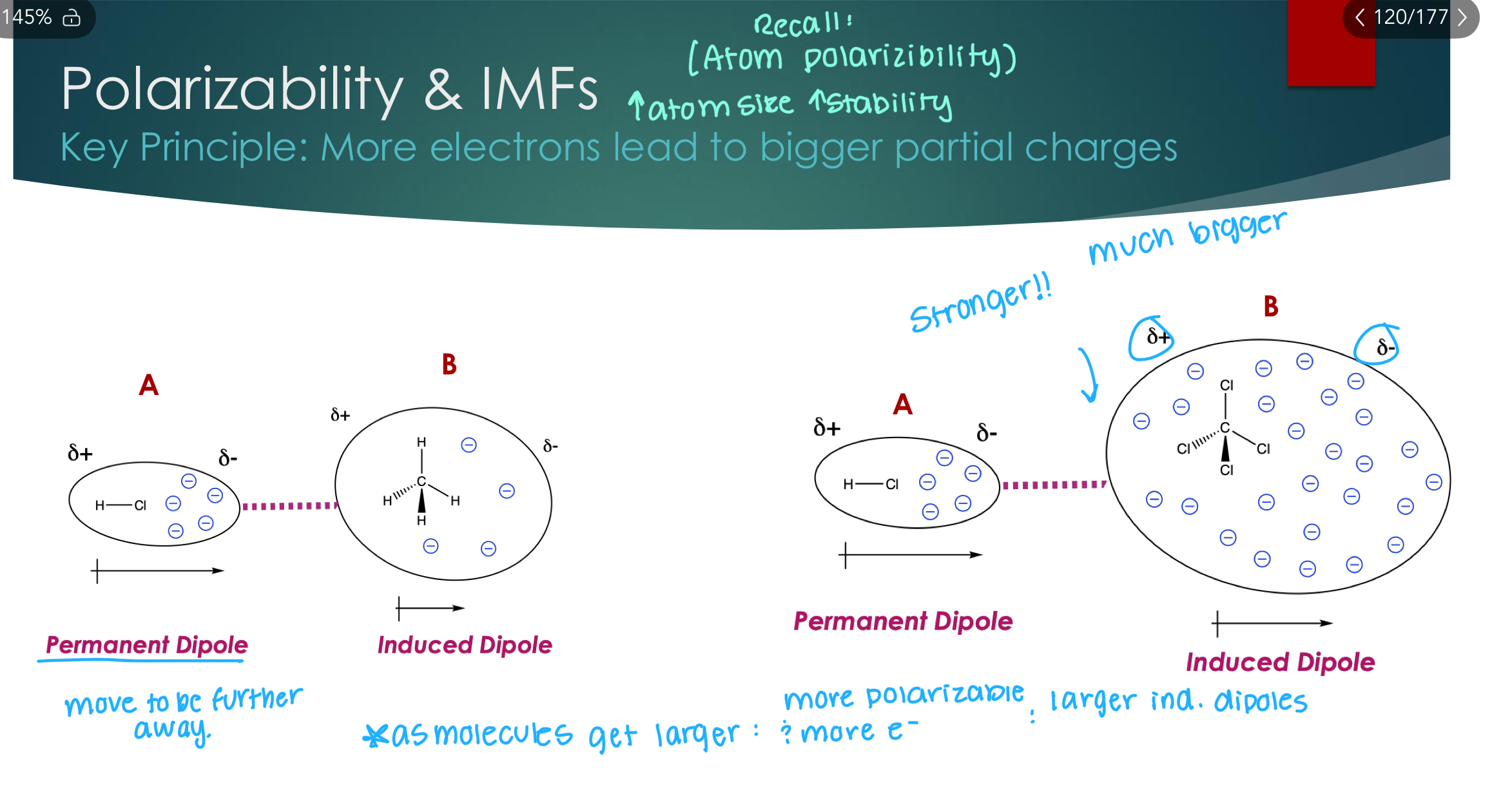
Polarizability & IMFs
Key Principle: More electrons lead to bigger partial charges
Recall: (Atom polarizability → increased atoms size leads to increased stability).
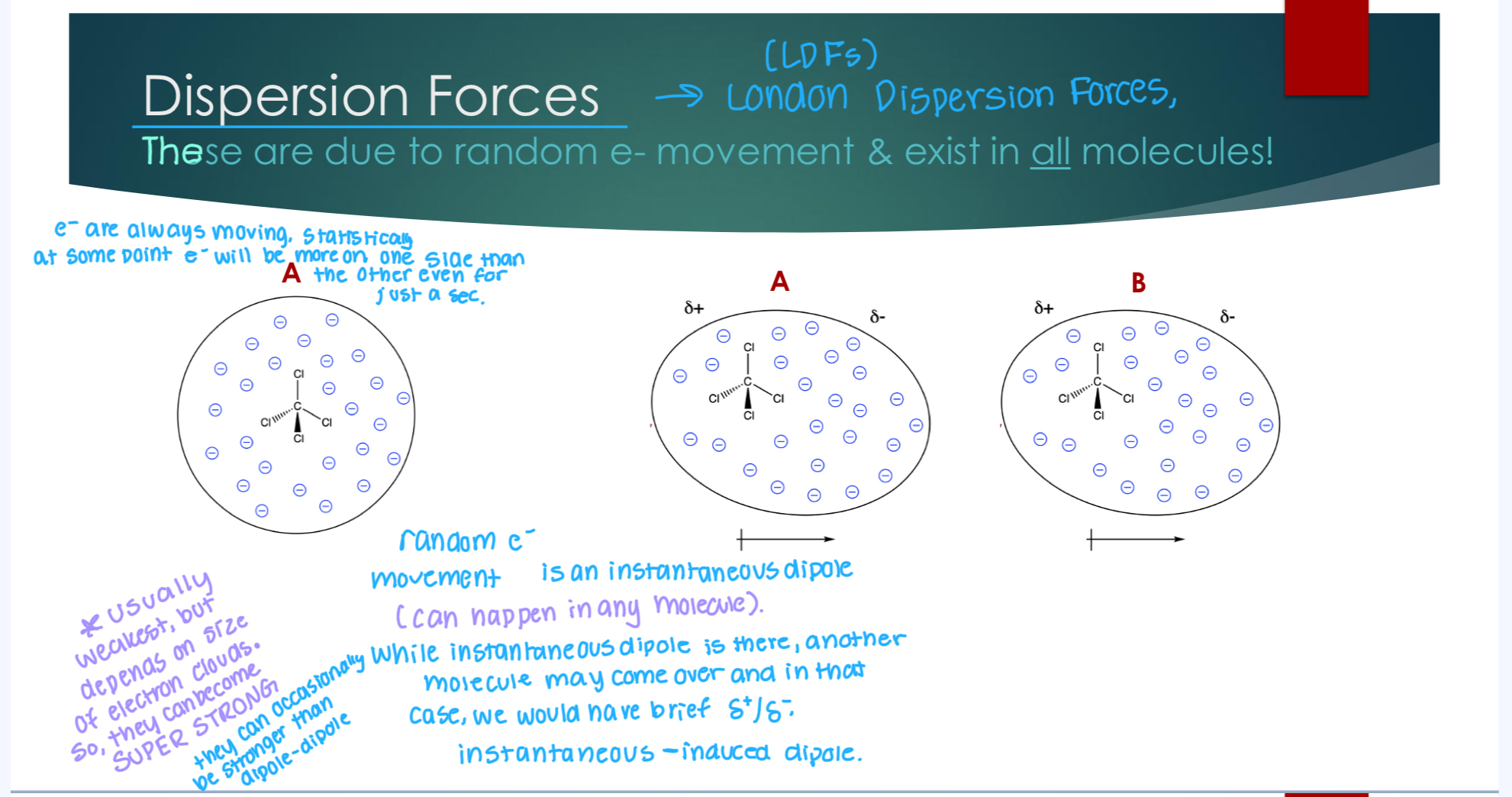
Dispersion Forces
These are due to random e- movement & exist in all molecules! e’ are always moving, and statically speaking, at some point e- will be more of one side than another even just for a second.
Random e- movement is instantaneous dipole (can happen in any molecule) While instantaneous dipole is there, another molecule may come over and in that case we would have a brief partial positive and partial negative.
Usually the weakest, but depends on the size of the electron clouds, so they can become SUPER STRONG Sometimes they can occasionally be stronger than dipole-dipole.
Hydrogen Bonding
A very polar dipole-dipole force
Very large partial charges are created when there is a very large difference in
electronegativity:
A hydrogen bonding (H-bonding) interaction must have a:
H-bond Donor
H-Bond Acceptor

IMFs & Physical Properties of Molecules
Physical State (Phase)
Freezing Point / Boiling point / Vapor Pressure
Gas has the most spread out particles while solids have the most rigid particles. Strong IMFs holds them tight together meaning that ionic compounds are almost always solid.
Stronger intermolecular forces (IMFs) increase a substance's freezing point because more energy is required to overcome these attractions and allow molecules to form a rigid solid structure
Stronger IMFs also indicate a higher boiling point, because it requires more energy to break those bonds.
Stronger IMFs lead to lower vapor pressure because molecules are held together more tightly, making it harder to escape into the gas phase, while weaker IMFs result in higher vapor pressure, as molecules can escape more easily
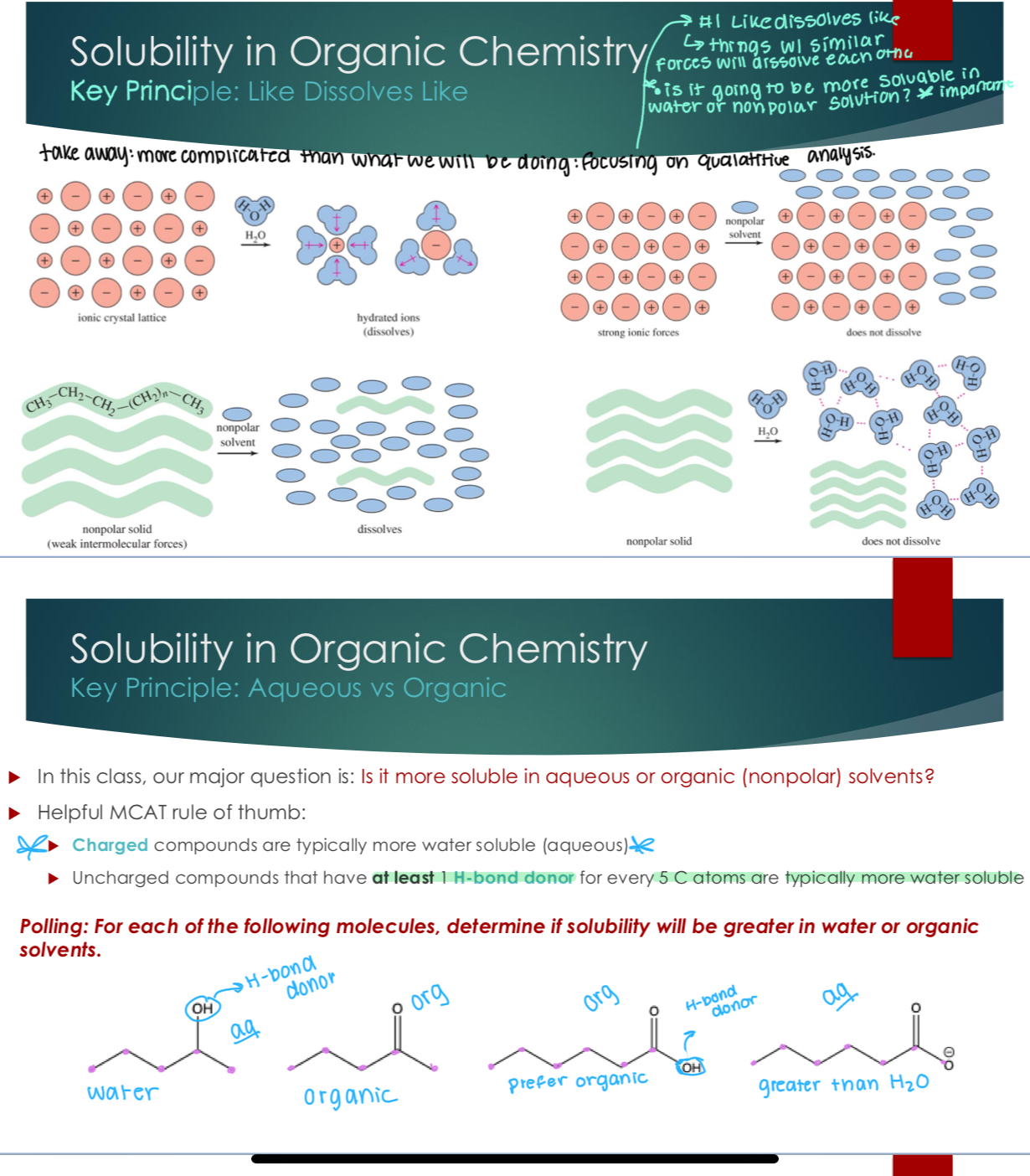
Solubility in Organic Chemistry
Key Principle: Like Dissolves Like
Key Principle: Aqueous vs Organic
In this class, our major question is: Is it more soluble in aqueous or organic (nonpolar) solvents?
Helpful MCAT rule of thumb:
Charged compounds are typically more water soluble (aqueous)
Uncharged compounds that have at least 1 H-bond donor for every 5 C atoms are typically more water soluble
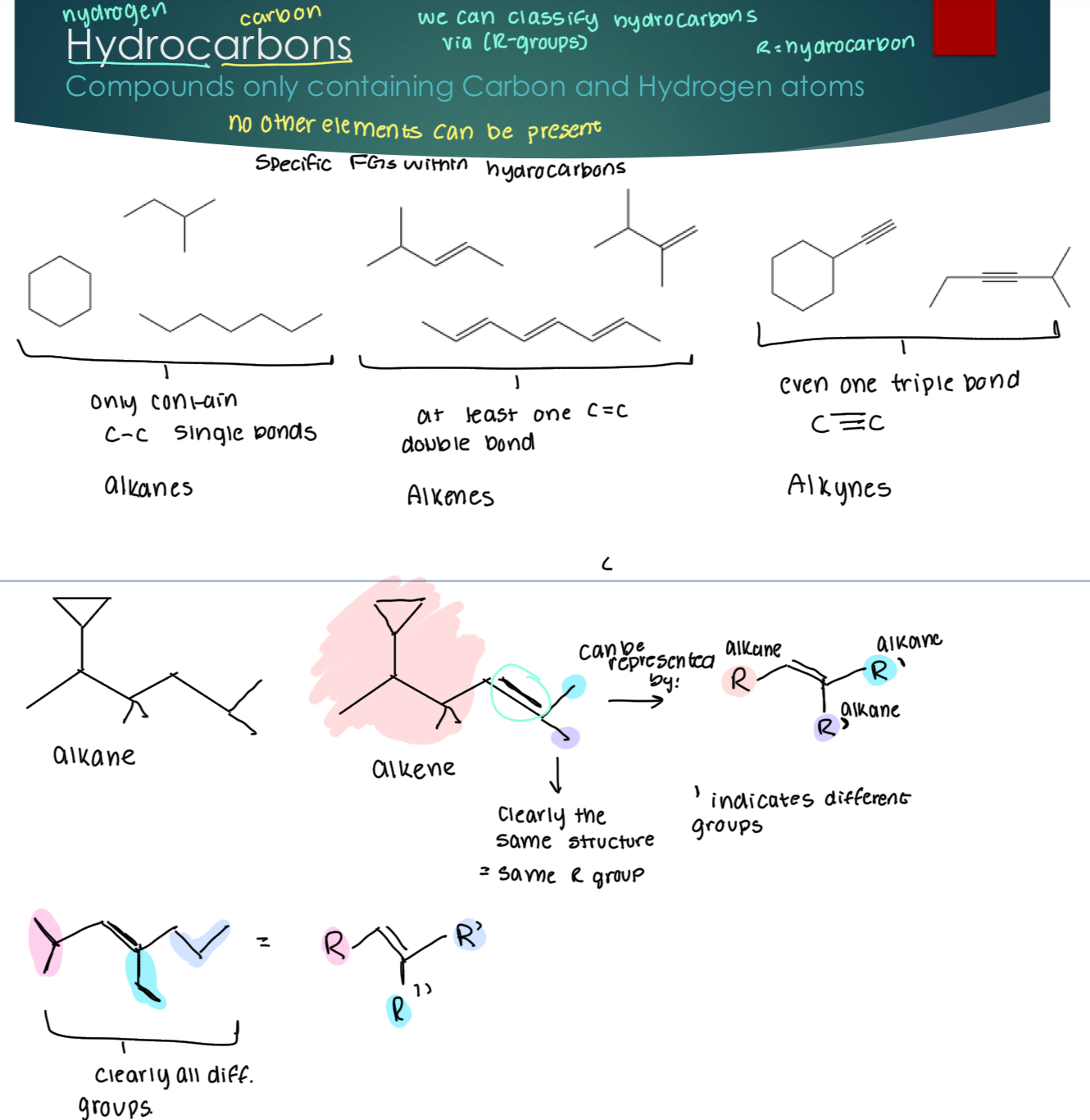
Hydrocarbons
Compounds only containing Carbon and Hydrogen atoms
R= hydrocarbon.
Alkanes only contain single bonds.
Alkenes contain at least one double bond
Alkynes contain at least one triple bond.
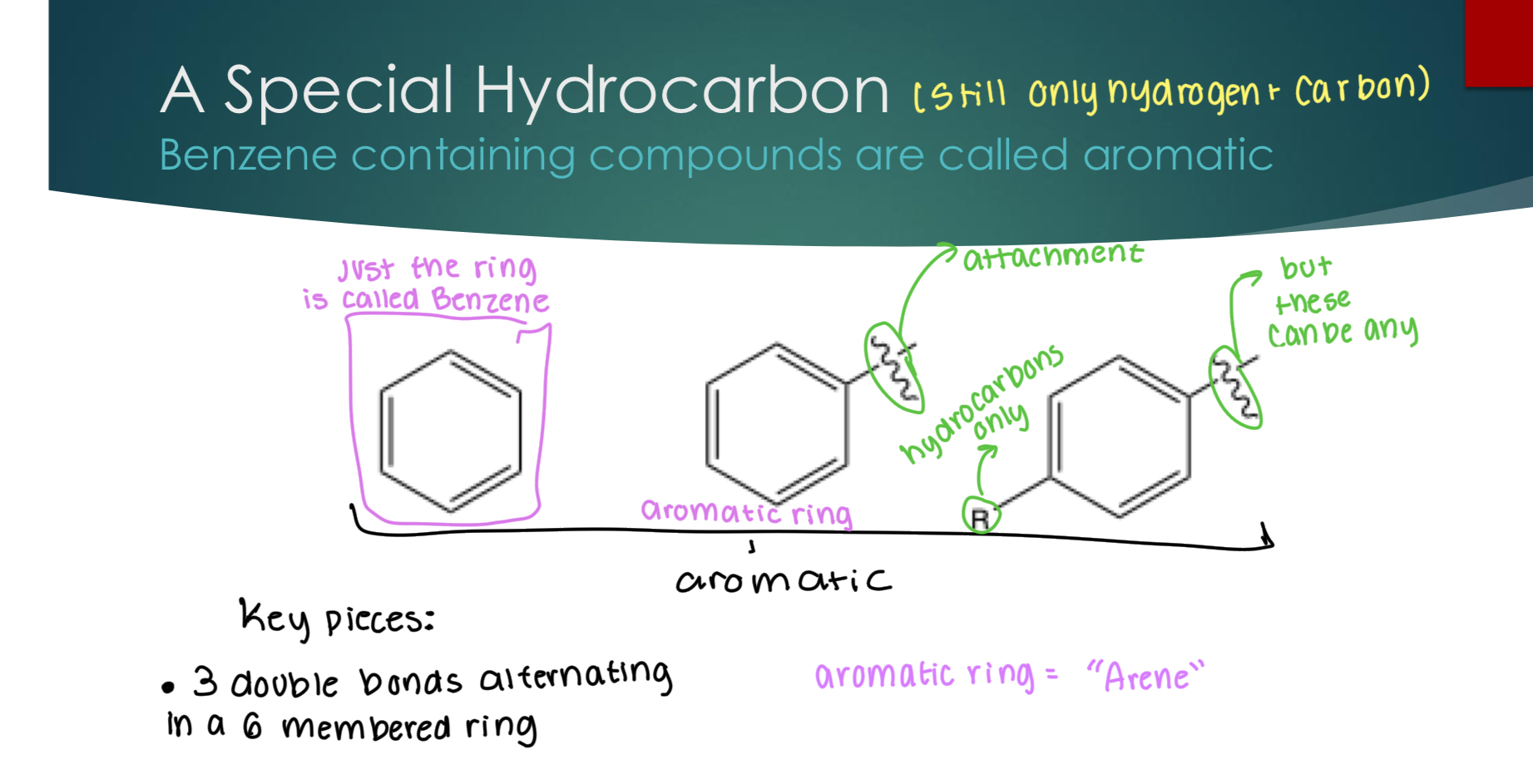
A special hydrocarbon:
Benzene containing compounds are called aromatic.
3 Key pieces:
3 double bonds alternating in a 6-membered ring.
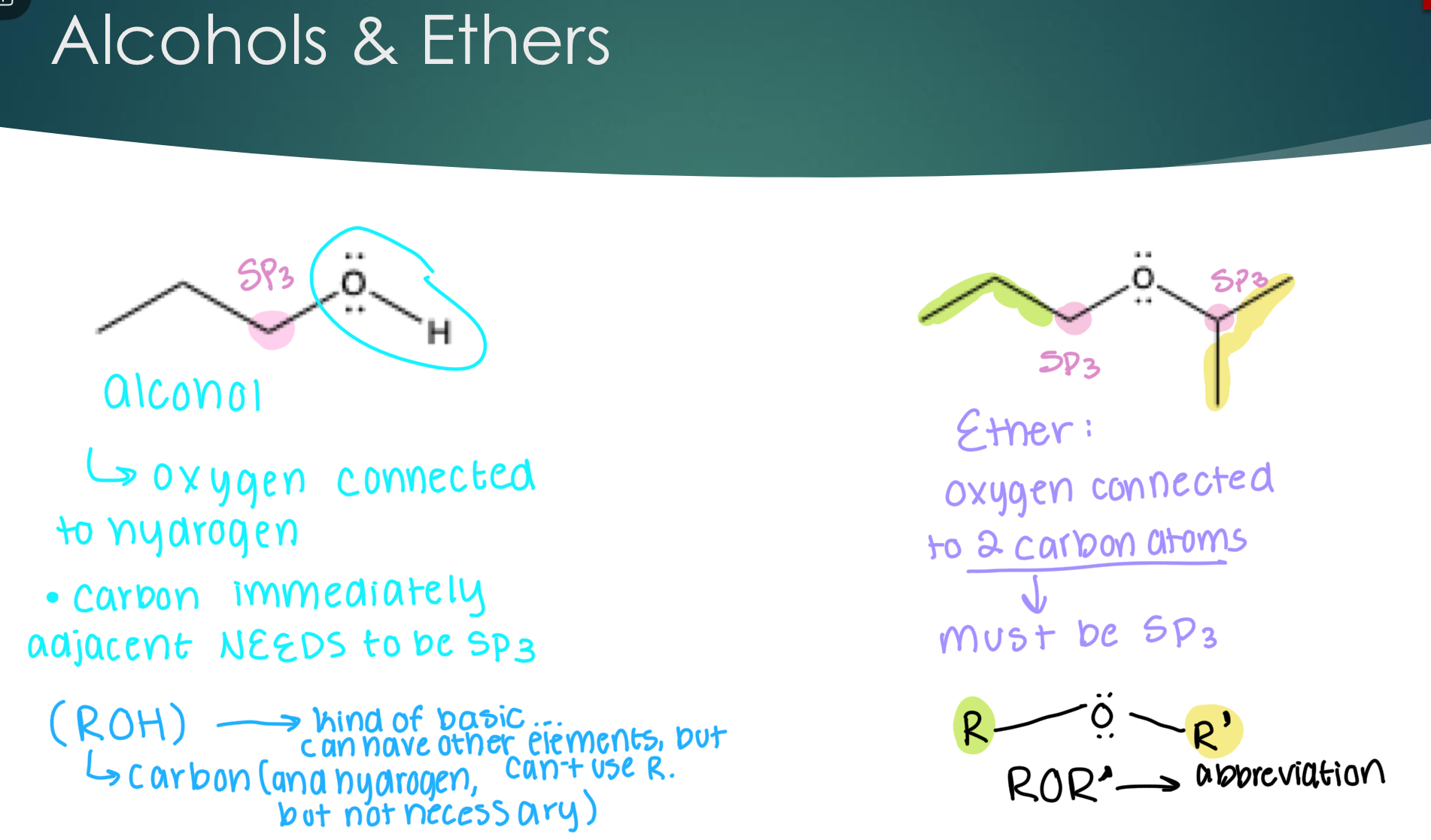
Alcohols & Ethers
Alcohol-oxygen connected to hydrogen. Carbon immediately adjacent NEEDS to be SP3,
(ROH) → kind of basic, can have other elements but carbon (and hydrogen can’t use R.)
Ether- oxygen connected to 2 carbon atoms (must be Sp3)
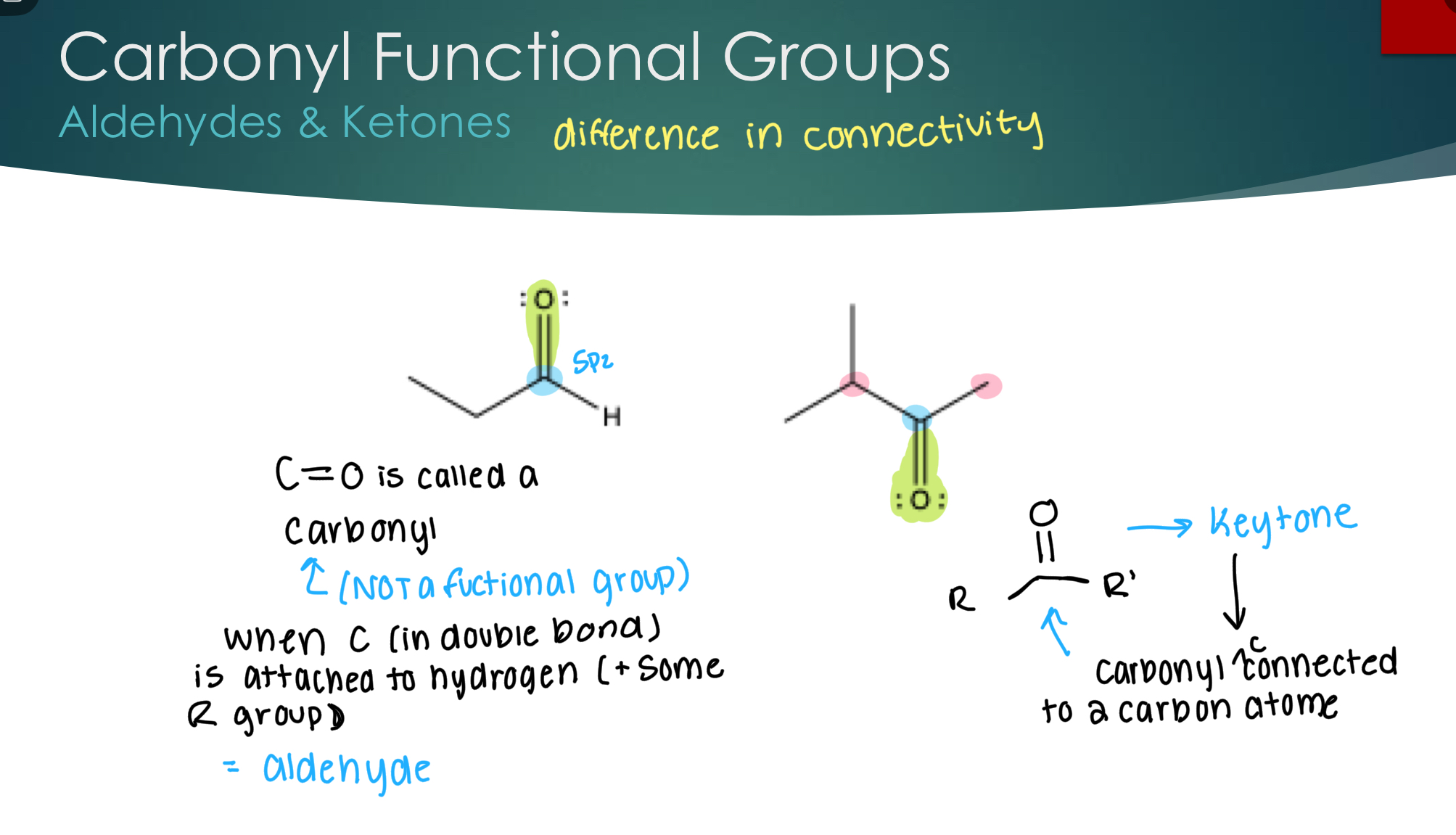
Carbonyl Functional Groups (Aldehydes & Ketones)
Aldehydes & Ketones
C=O is called a carbonyl (not a functional group).
Aldehyde
When C (in double bond) is attached to hydrogen (+ some R group)
Ketone
When the carbonyl group is connected to 2 carbon atoms.
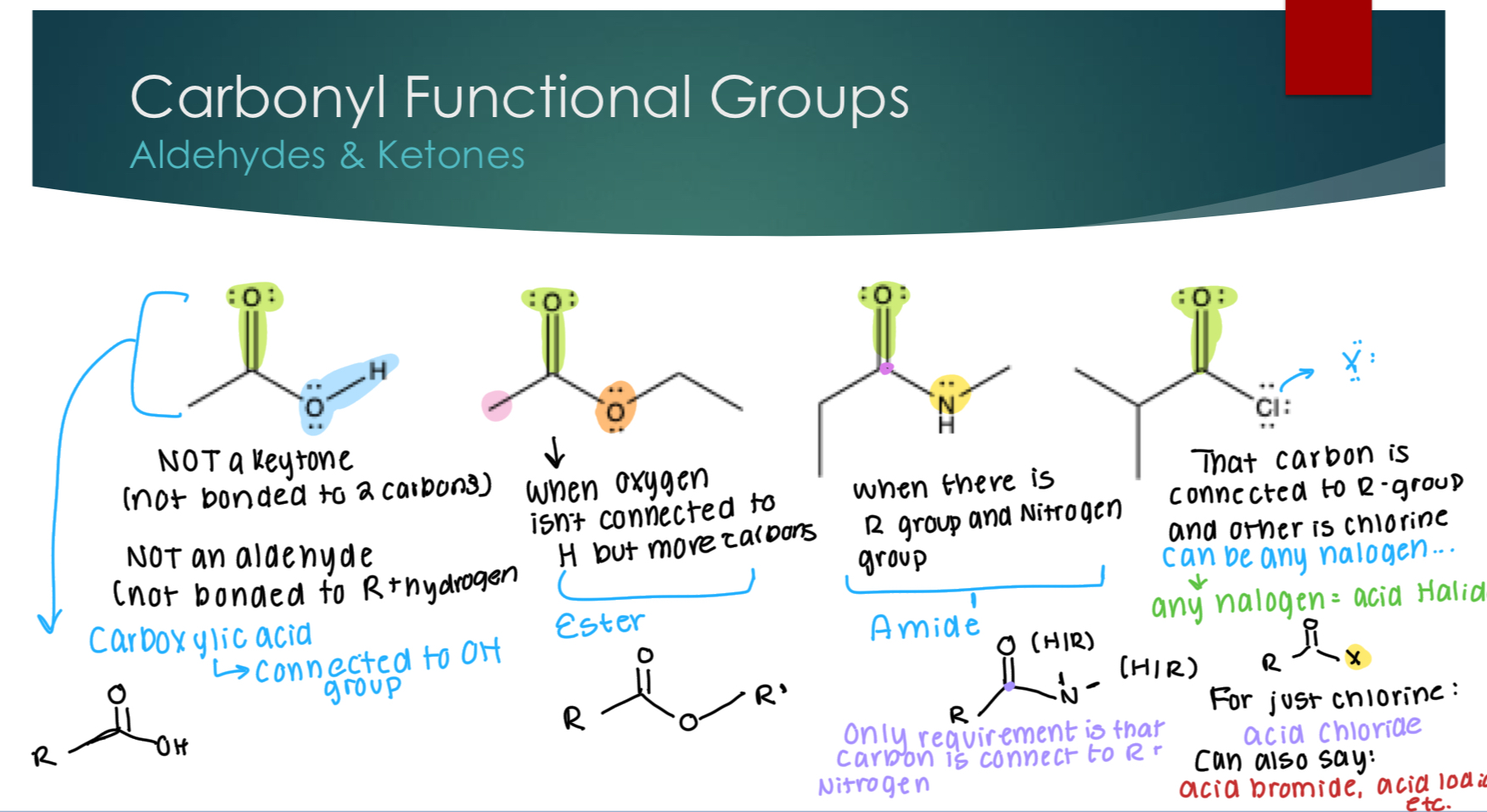
More Carbonyl functional groups.
Carboxylic Acid- when the carbonyl group is attached to an OH group.
Ester- When the carbonyl group is attached to one carbon atom that is attached to another oxygen atom.
Amide- When the carbonyl group is attached to one carbon atom that is attached to a nitrogen
Acid Halide- When the carbonyl group is attached to one carbon atom that is attached to any of the halides.
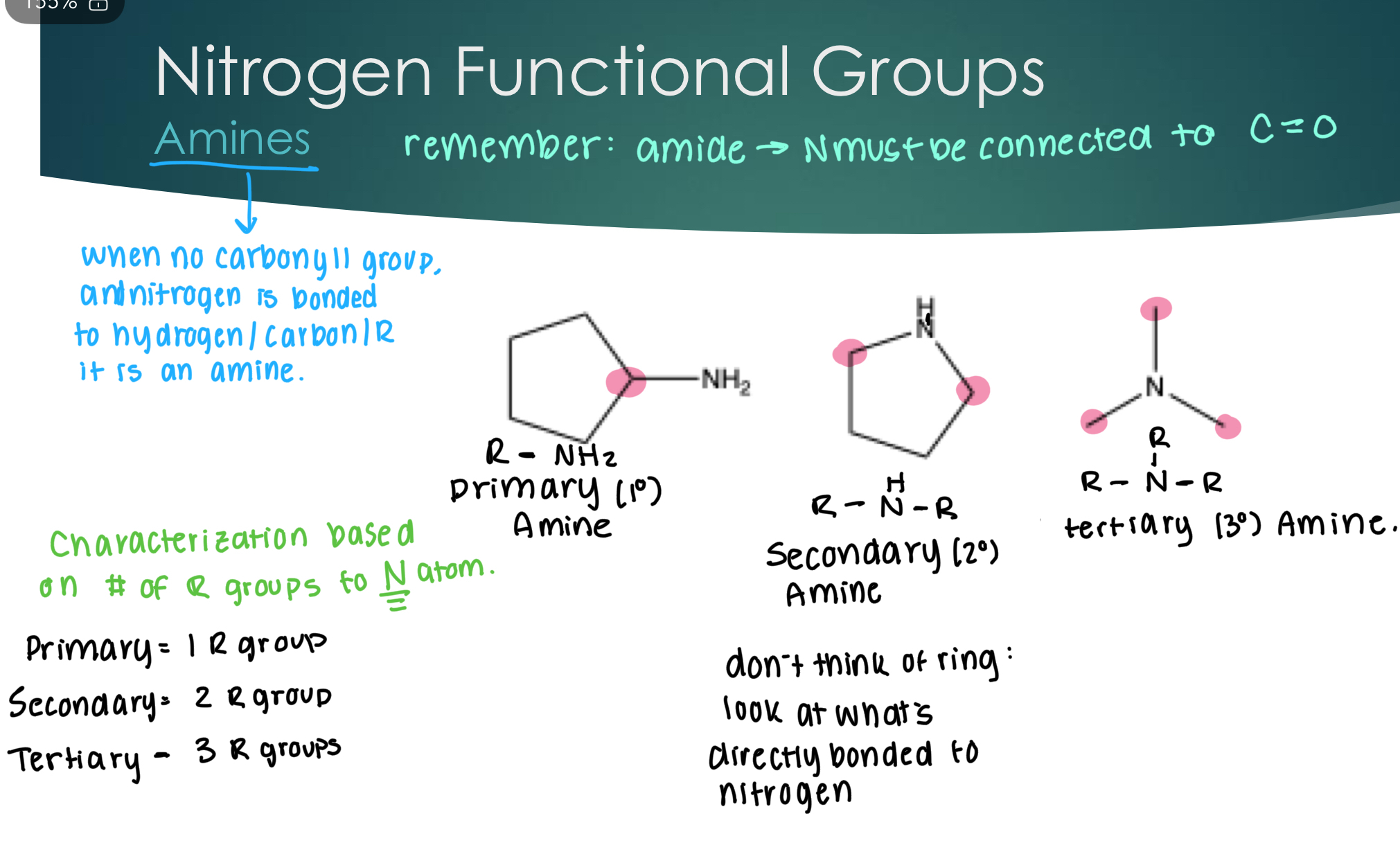
Nitrogen Functional Groups: Amines
Amines: When no carbonyl group and nitrogen is bonded to hydrogen/carbon/R group.
Characterization based in # of R groups to N atom:
Primary = 1 R group
Secondary = 2 R groups
Tertiary = 3 R groups
Nitrogen Functional Groups : imine, nitrile, and pyridine
Imine: when there is a double bond to a carbon
Nitrile N ≡ C
pyridine 6 membered rings w/ 3 double bonds.
Functional Group Priorities: Naming & Reactivity
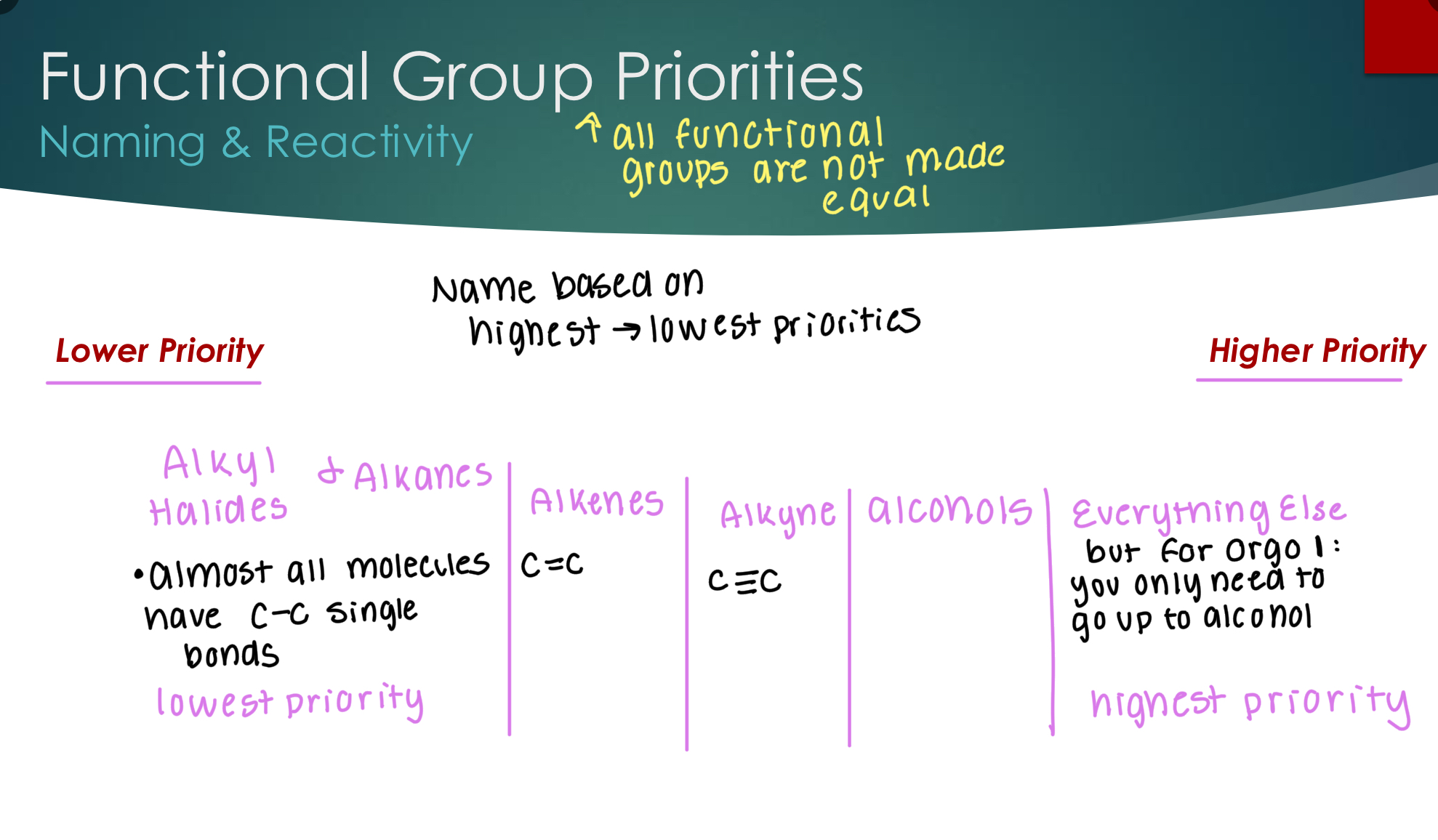
Intro to Nomenclature
Organic molecules are named based on the highest priority functional group present
That functional group is used to determine the “Base Name” :
Carbon number prefix for longest carbon chain containing the highest priority functional group
+
Suffix related to highest priority functional group
name, and usually a carbon number for where
the highest priority group is attached to the
longest carbon chain
Any other lower priority groups or smaller carbon branches attached to the longest carbon chain are called substituents
Substituents are named based on their functional group or total carbon count (for carbon branches) are added to the front of the base name with the format:
C# on the longest carbon chain where the substituent is attached
—
Substituent name with suffix that indicates
substituent instead of highest priority group
Nomenclature: Alkanes! w/ no branching
When you have an alkane with no substituents & no branching...
These molecules are referred to as “straight chain alkanes” and can be designated with “n” which stands for normal
Carbon number prefix for carbon chain + ane (suffix for alkane)
Example: 8 carbons = octane
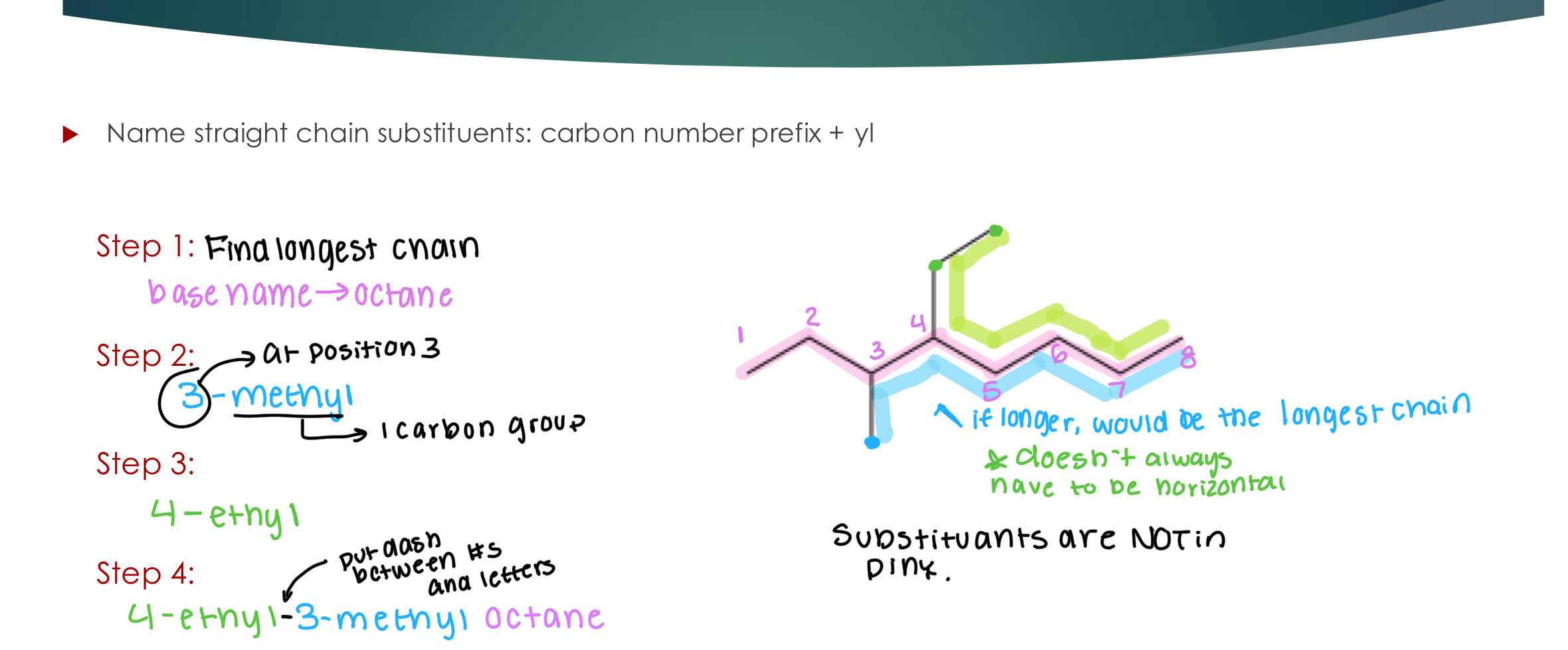
Nomenclature: Alkanes! w/ branching
Step 1: identify and name the “base chain” :
Carbon number prefix for longest carbon chain + ane
Step 2: identify and name the alkyl substituents (the branching groups off the main chain)
Step 3: number your “main chain” carbons (best practice: so that the most substituents are
attached to lower carbon numbers)
Step 4: add the substituents to the front of your base chain name (best practice: list them in alphabetical order)
C# where the substituent is attached — Substituent name with suffix that indicates substituent (-yl) + Base name
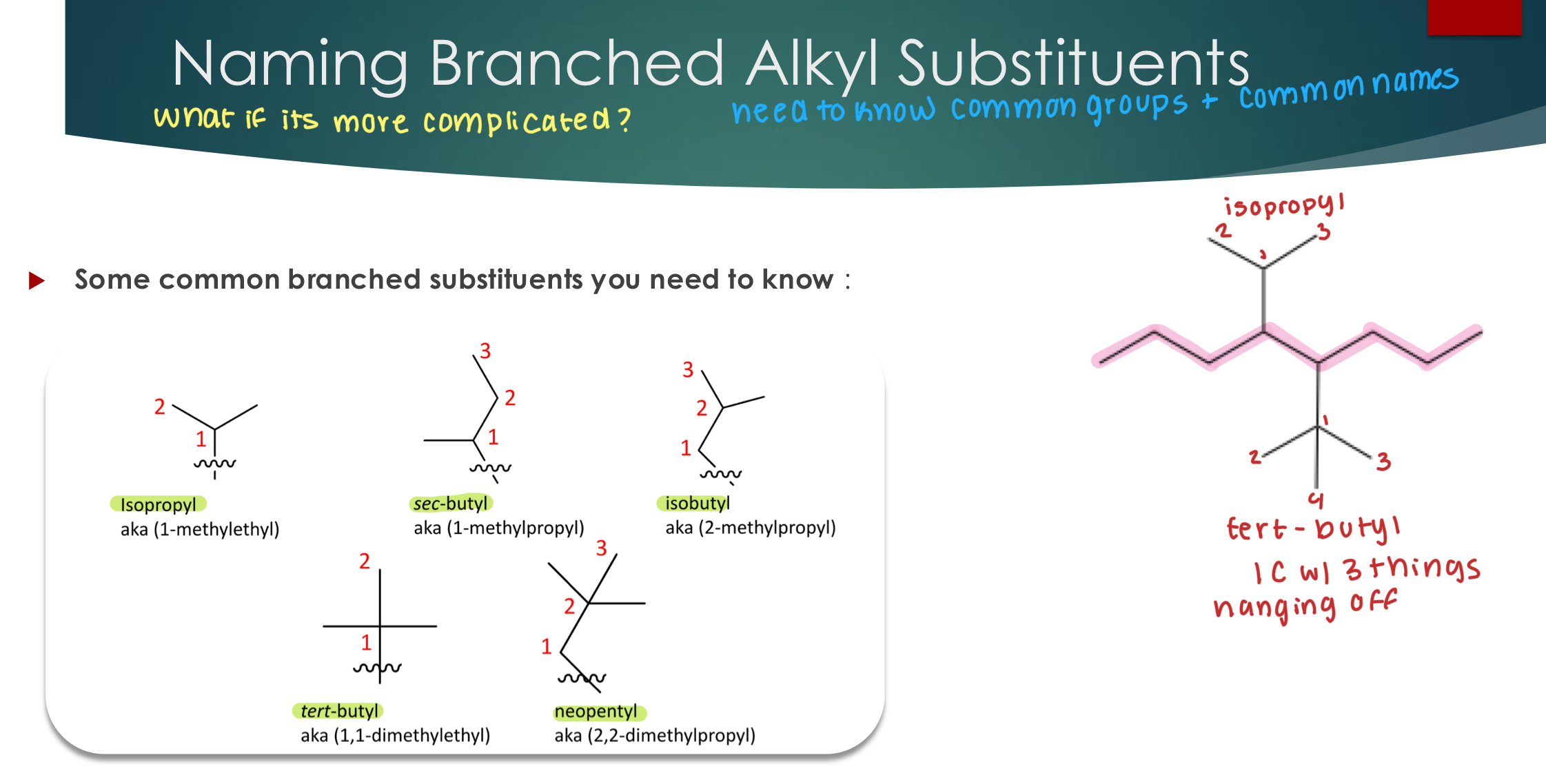
Naming Branched Alkyl Substituents
When you have more than 1 of the same substituent, you must add a quantity prefix to the substituent name and include the carbon numbers for each substituent together
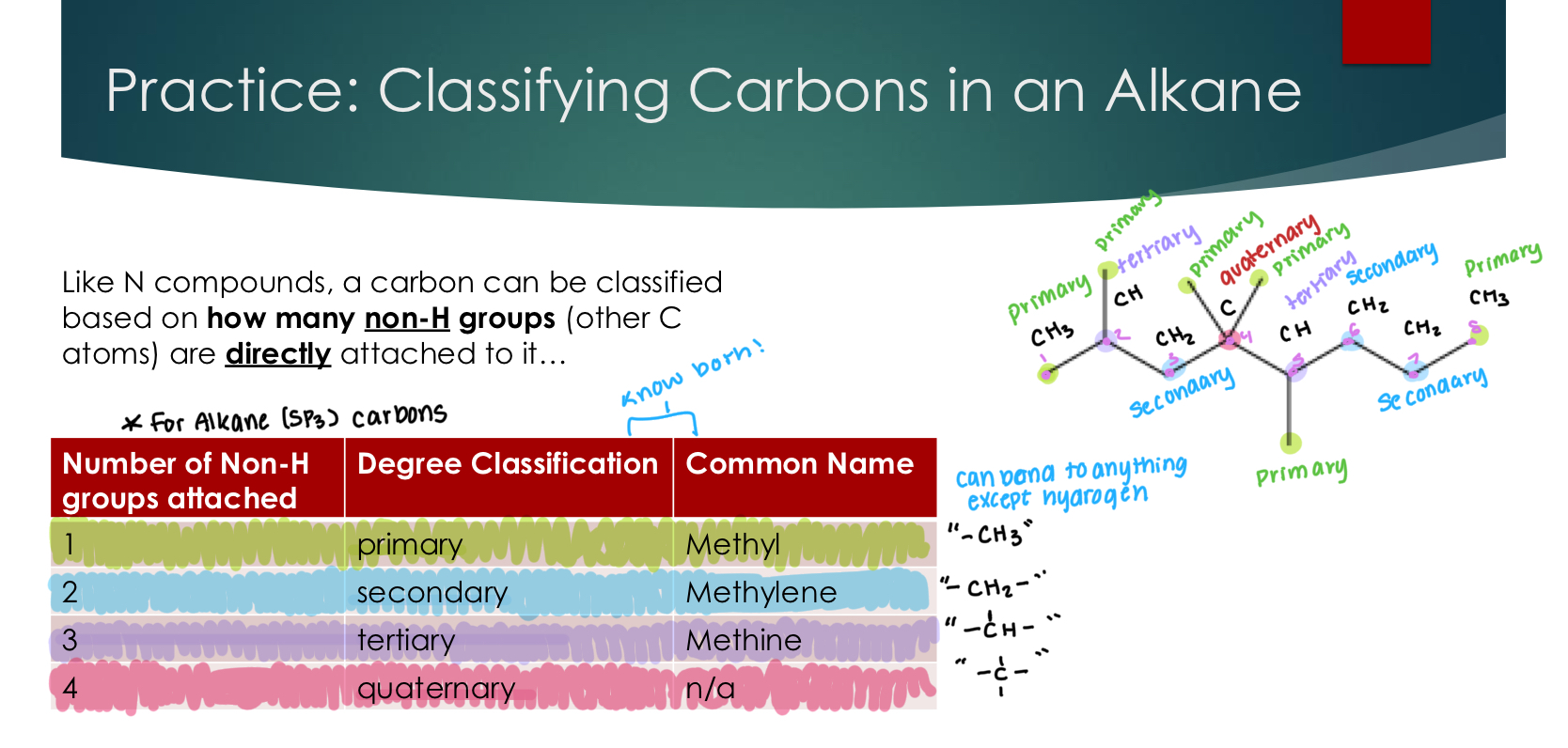
Classifying Carbons in an Alkane
Like N compounds, a carbon can be classified based on how many R groups (other C atoms) are directly attached to it…
Halogen Substituents: Alkyl Halides
Halogens are even a lower priority than alkanes, so IUPAC naming rules consider the halogen as a substituent
As a substituent, halogens are named with the ending “o” :
Fluorine Chlorine Bromine Iodine
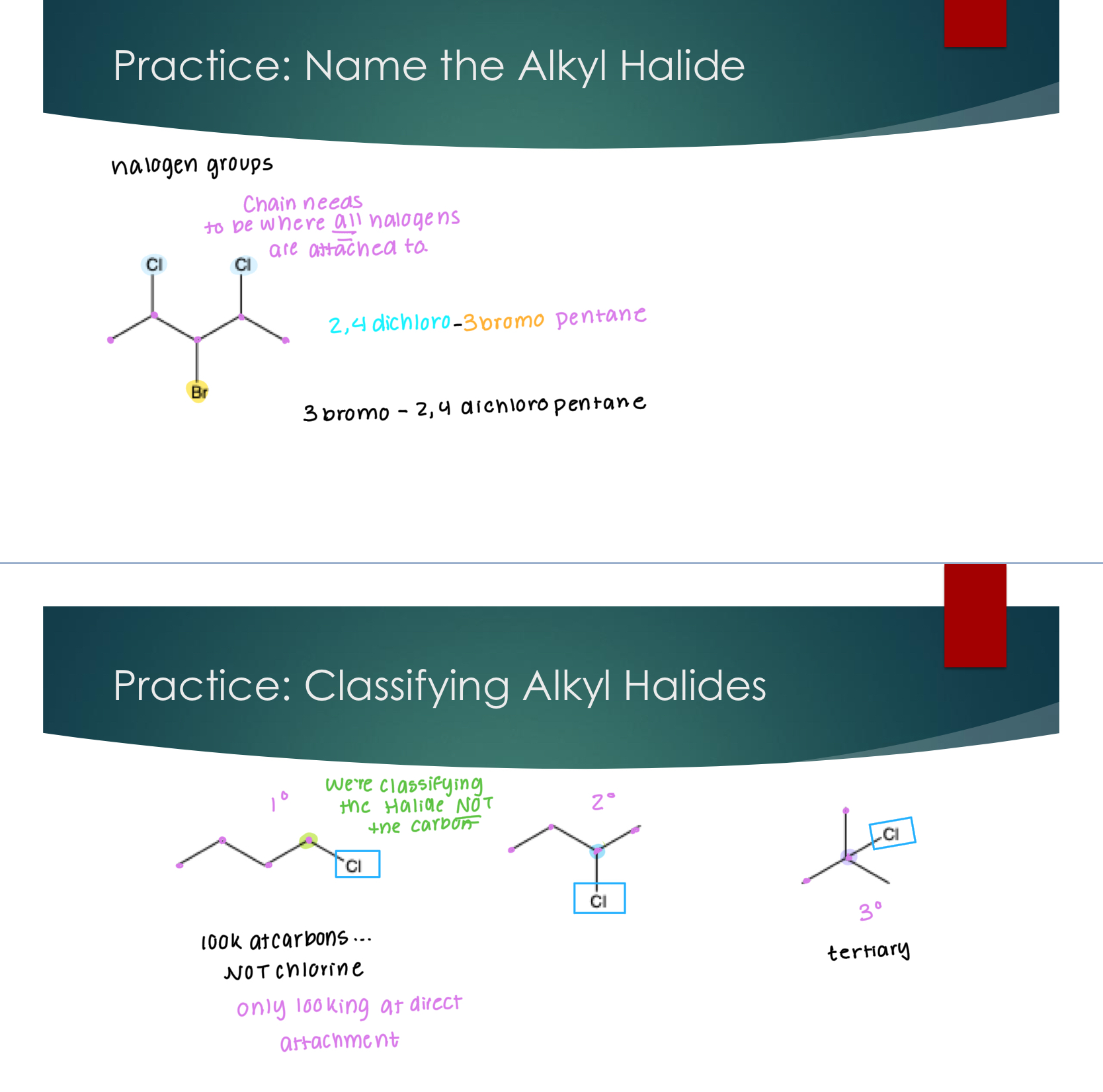
Isomers
Very broadly, isomers are defined as chemical compounds that share the same chemical formula
But this definition can be broken down to further classify these compounds into types of isomers, and this process is centered on answering the question “how do these isomers differ in structure”
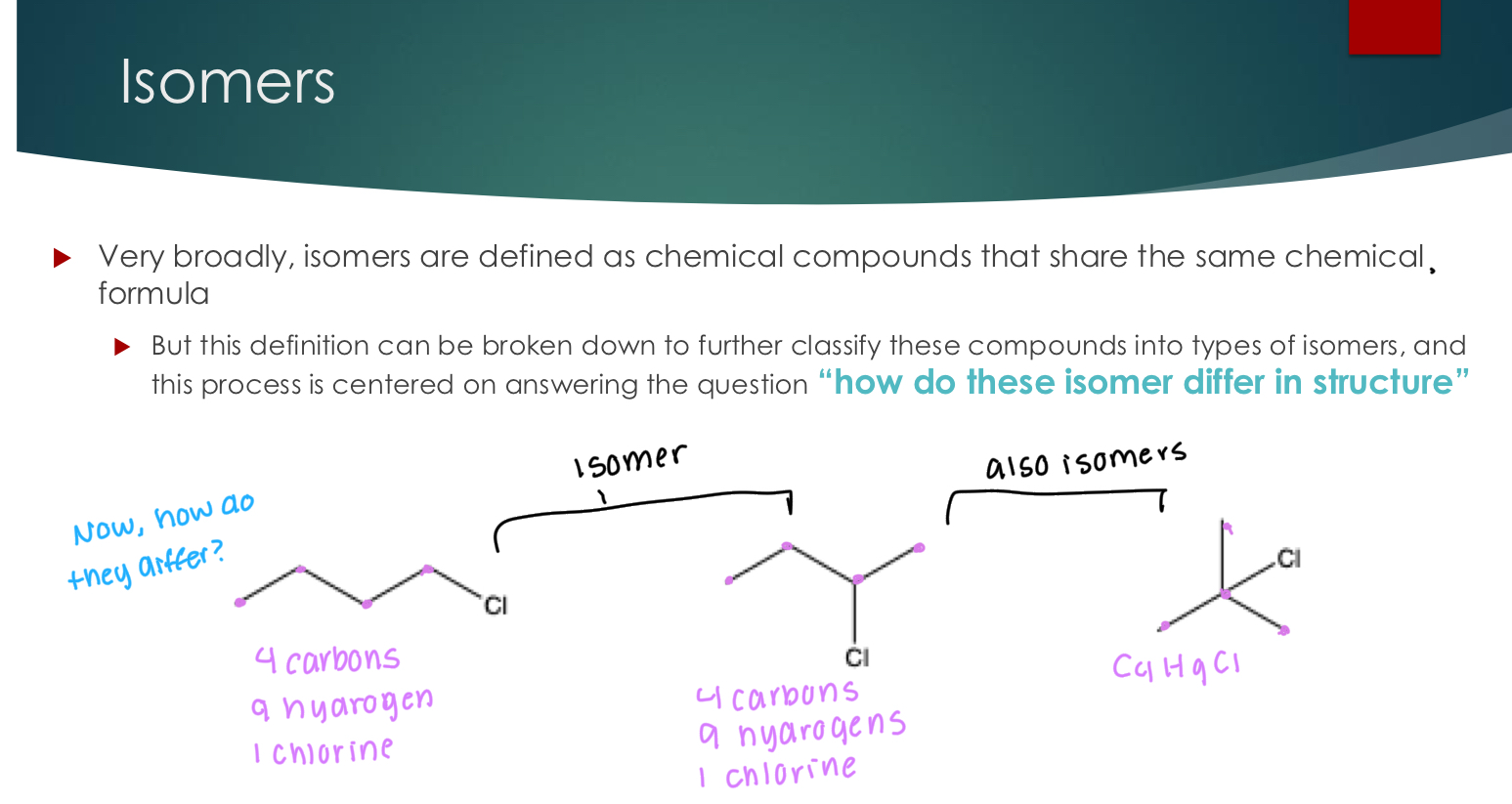
Constitutional Isomers
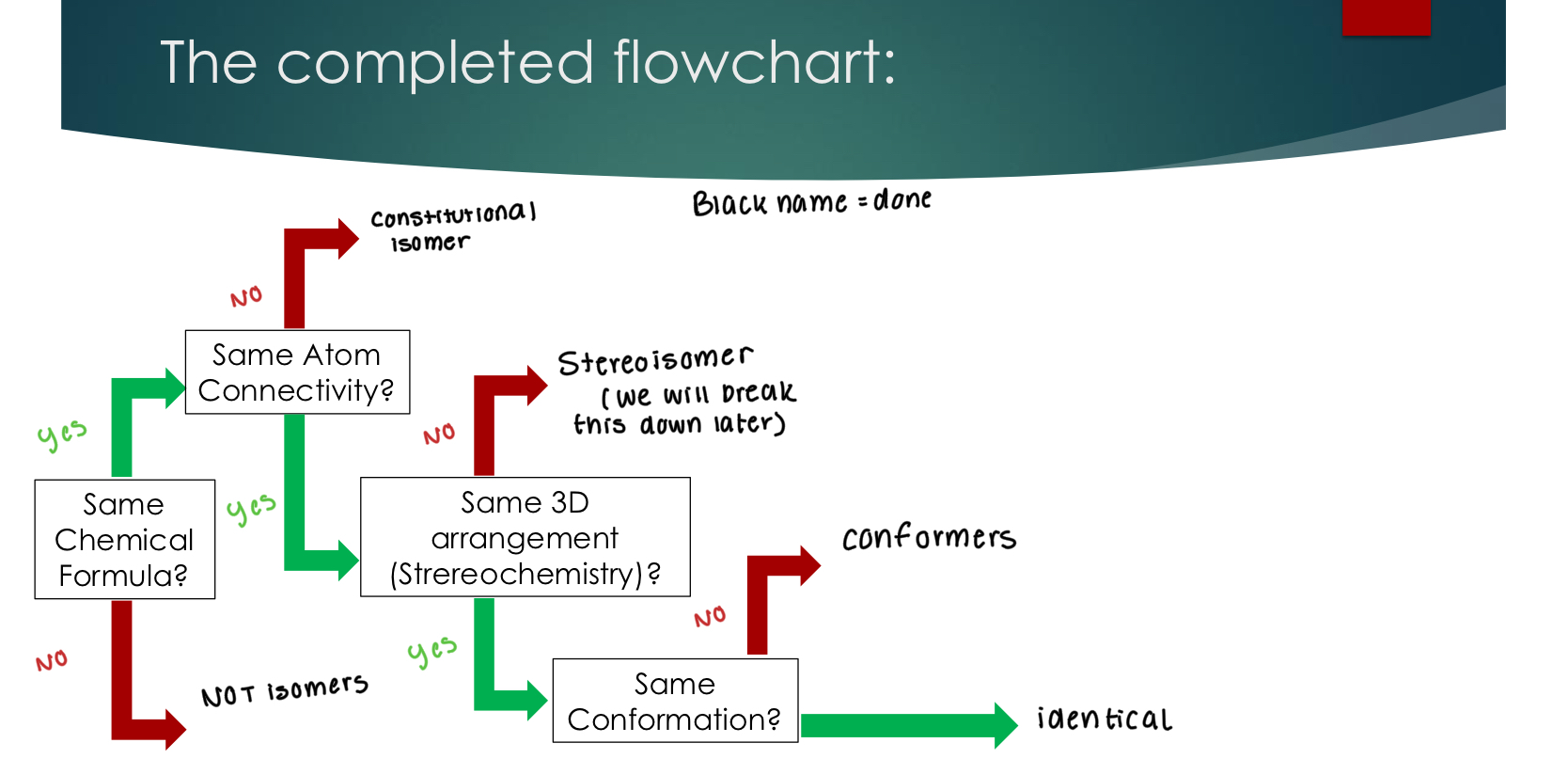
Step 1: Does atom connectivity change: Constitutional Isomers
Used when atom connectivity is different.. in other words… are the bonded the same way?
priority #1
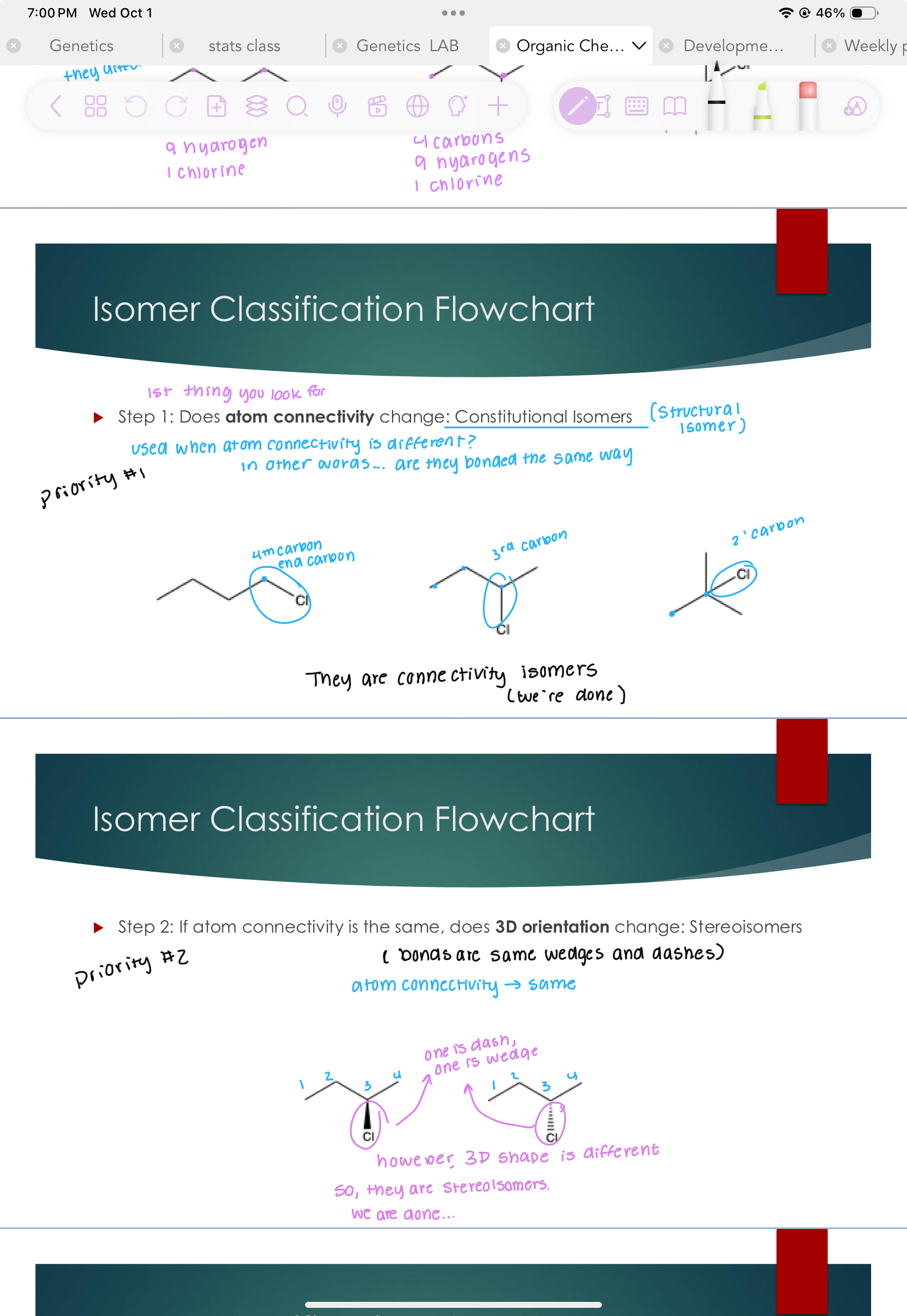
Stereoisomers
Step 2: If atom connectivity is the same, does 3D orientation change: (bonds are same wedges and dashes) priority #2
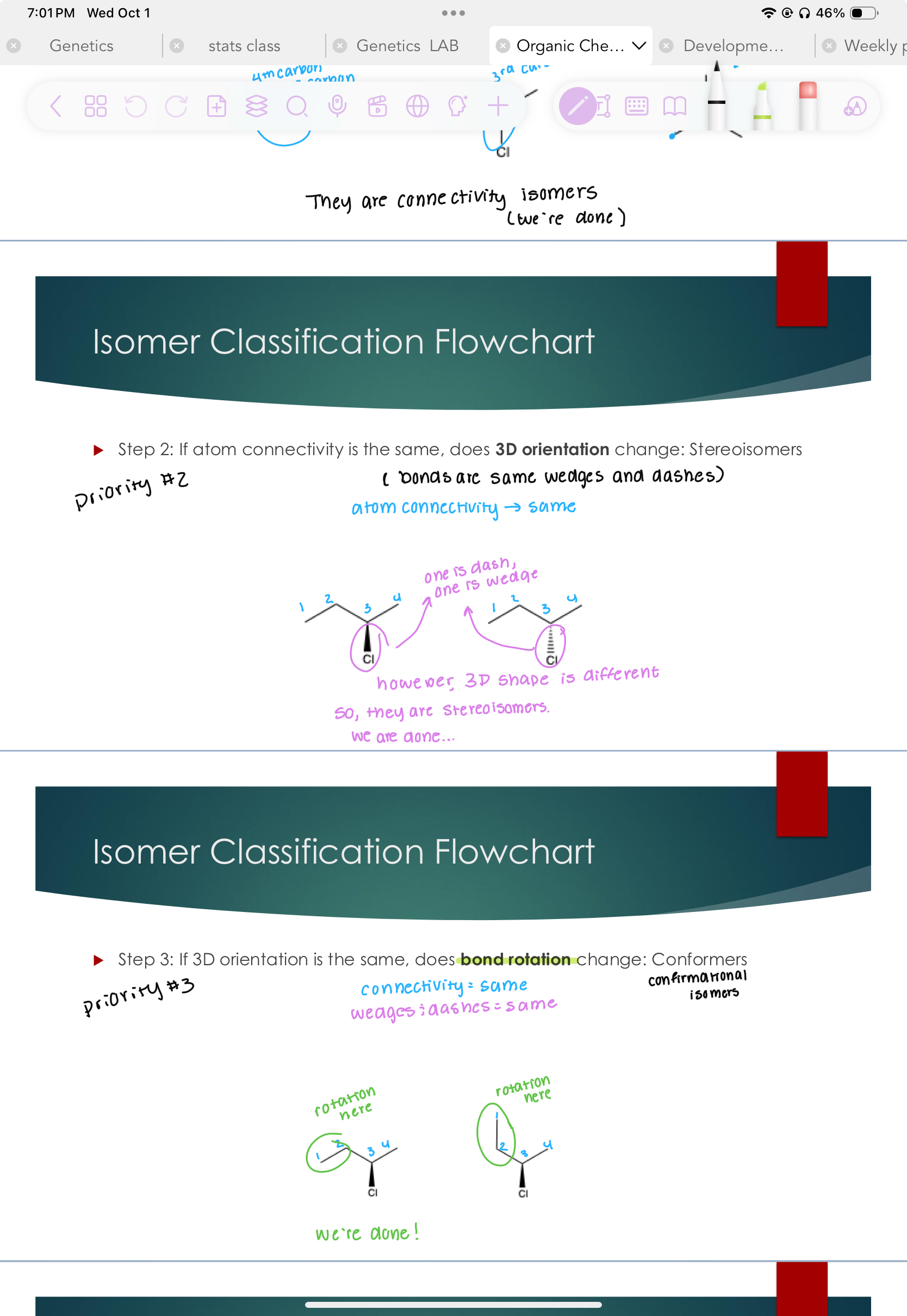
Conformers
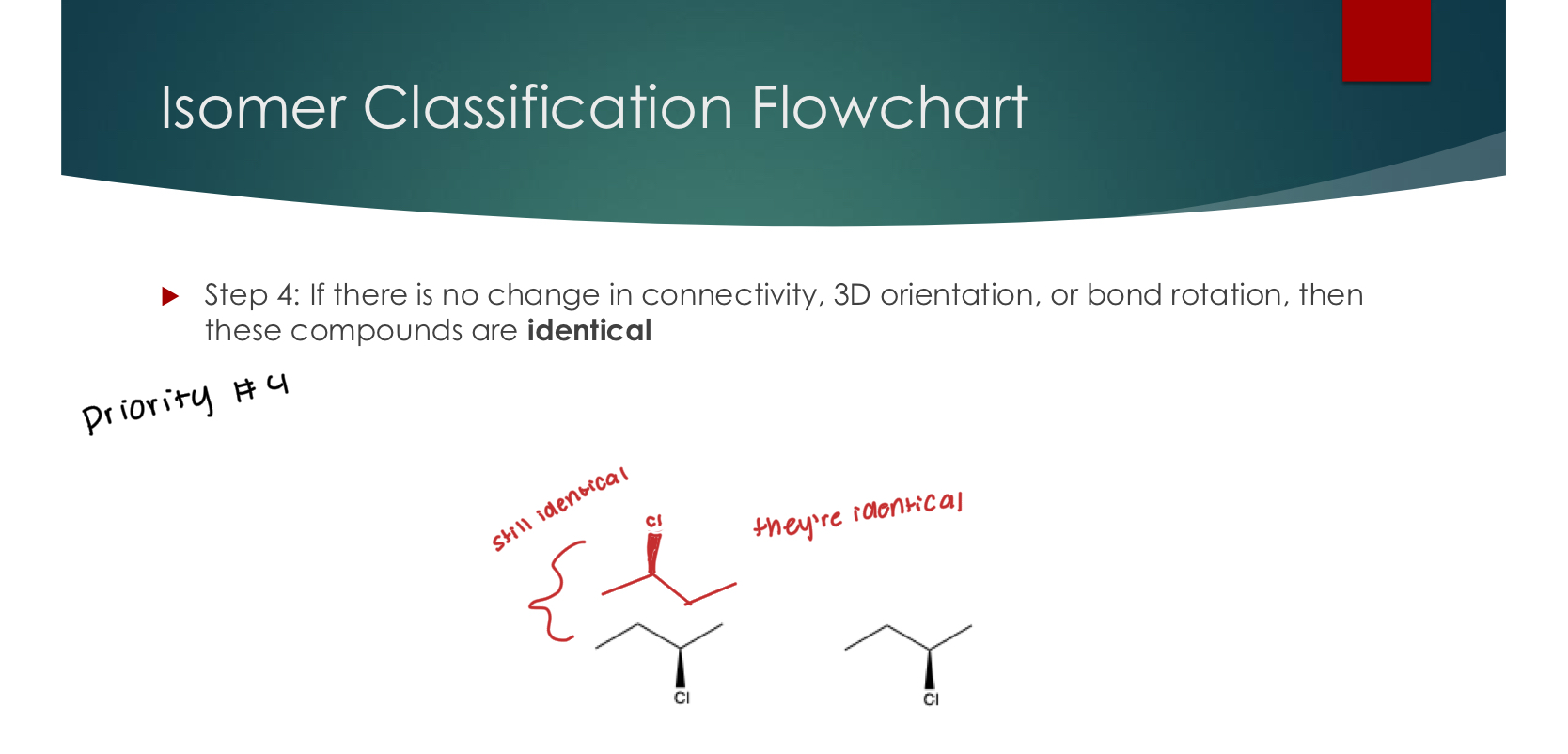
Step 3: If 3D orientation is the same, does bond rotation change:
#3 priority

Identical
Step 4: If there is no change in connectivity, 3D orientation, or bond rotation, then these compounds are identical
Isomer Flowchart
Isomer Relationship affects the relationship between chemical and physical properties
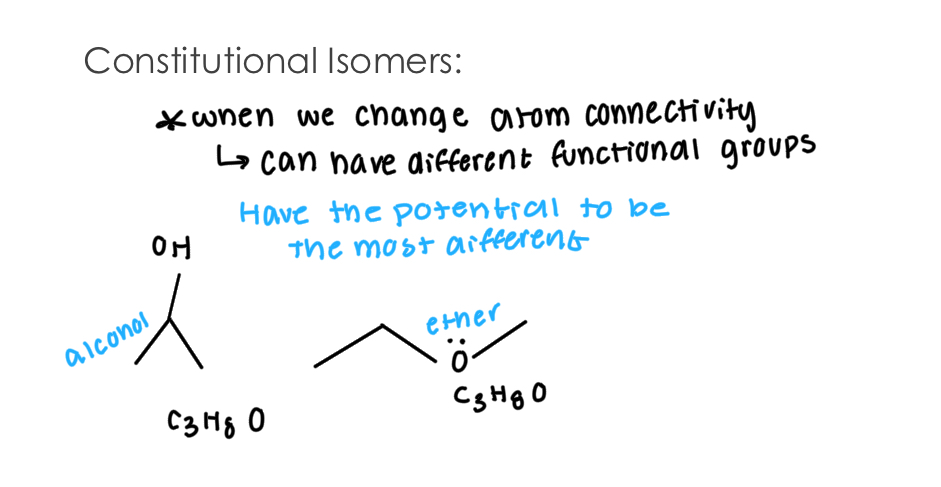
Constitutional Isomers:
When we change atom connectivity, we can attain different functional groups
Atom connectivity include atoms that have the potential to be the most different.
Bond Rotation & Conformation
What bonds can rotate?
Rotation occurs when rotating the bond does not change the amount of orbital overlap
For example, sigma bonds that are head-to-head can rotate.
Pi bonds which rely on side-to-side overlap cannot rotate.
Molecules that differ by only allowed rotation around a bond are called conformers

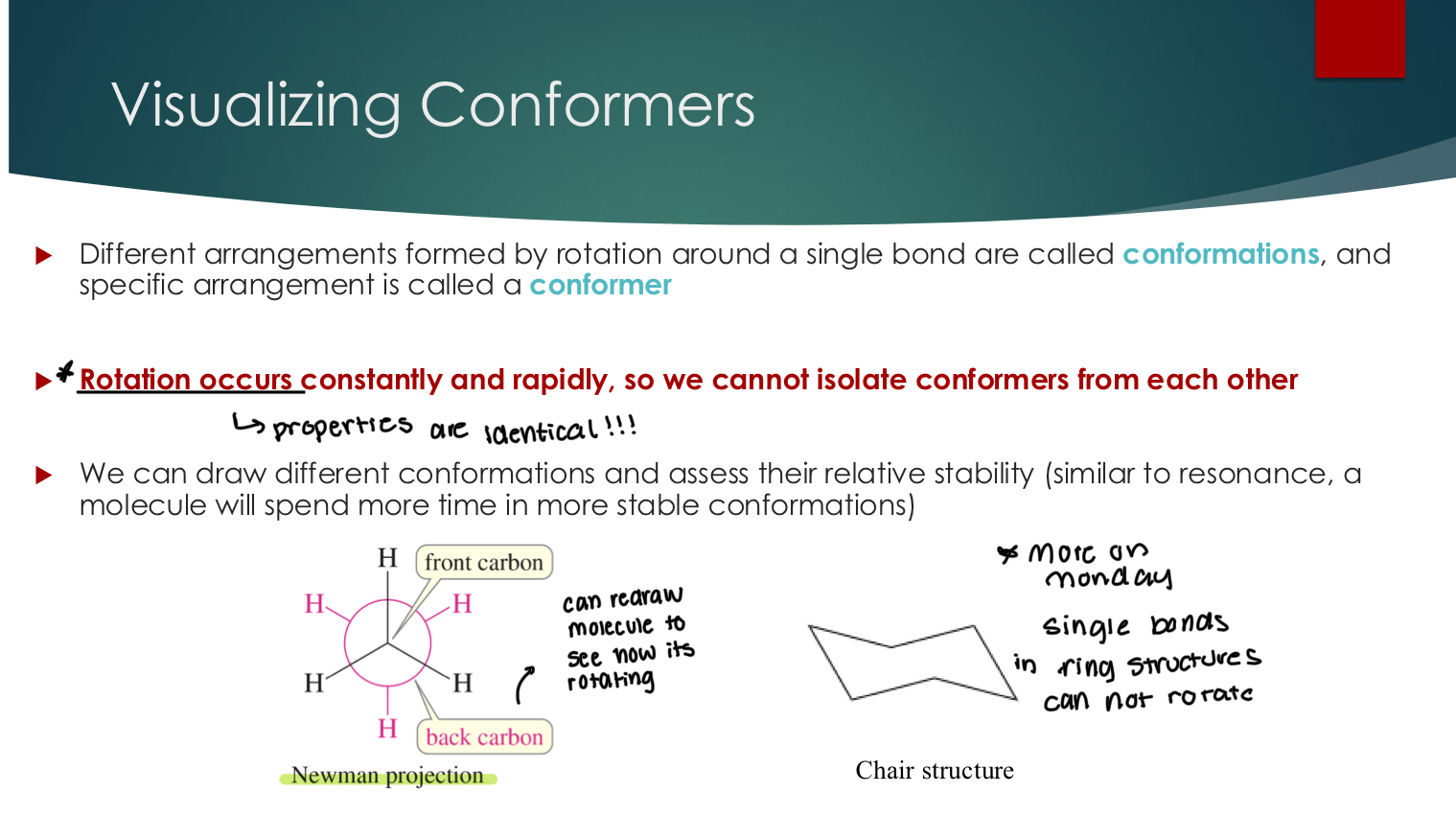
Visualizing Conformers
Different arrangements formed by rotation around a single bond are called conformations, and a specific arrangement is called a conformer
Rotation occurs constantly and rapidly, so we cannot isolate conformers from each other Properties are identical!! for this reason.
We can draw different conformations and assess their relative stability (similar to resonance, a molecule will spend more time in more stable conformations)
Assessing Stability: Electronics vs Sterics
When considering the stability of a molecule we have two main parameters to consider... electronics & steric.
The key principle to both: everybody wants their own space!
Electronics: How stable & delocalized (spread out) are the electrons
➢ Octet analysis
➢ Atom electrostatics (EN)
➢ Resonance
➢ Induction
Sterics: How spread out are the atoms
➢ 3D conformational analysis
➢ Bond angle / ring strain analysis

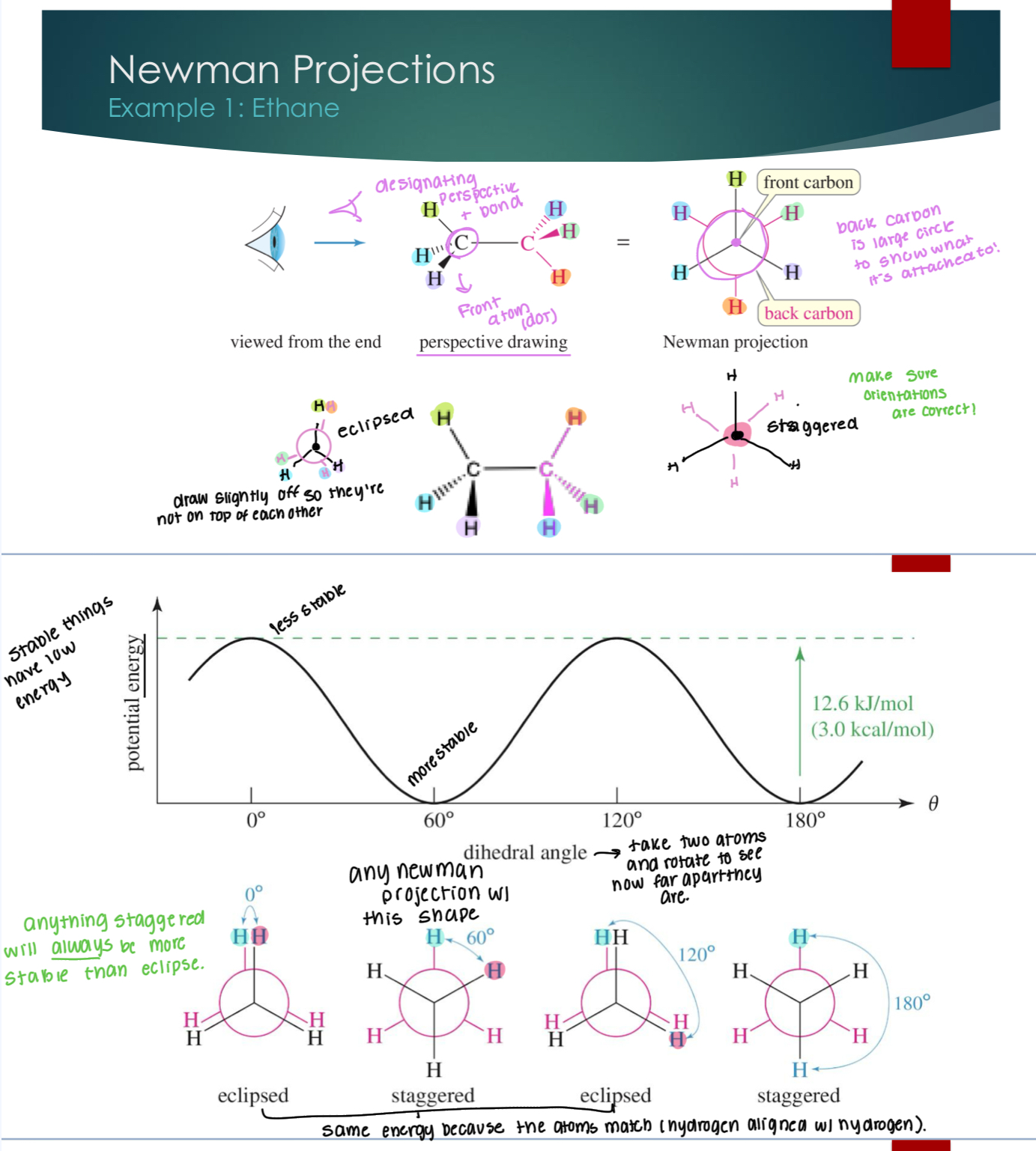
Newman Projections with Ethane
First, we need to designate a perspective and a bond. Back carbon is represented by a large circle to show what it is attached to.
Eclipse structures are on top of each other (but, we draw them slightly off to show them) while staggered are 60 degrees from each other.
Staggered are always more stable than eclipses.
Dihedral means that you’re taking the atoms and seeing how far apart they are.
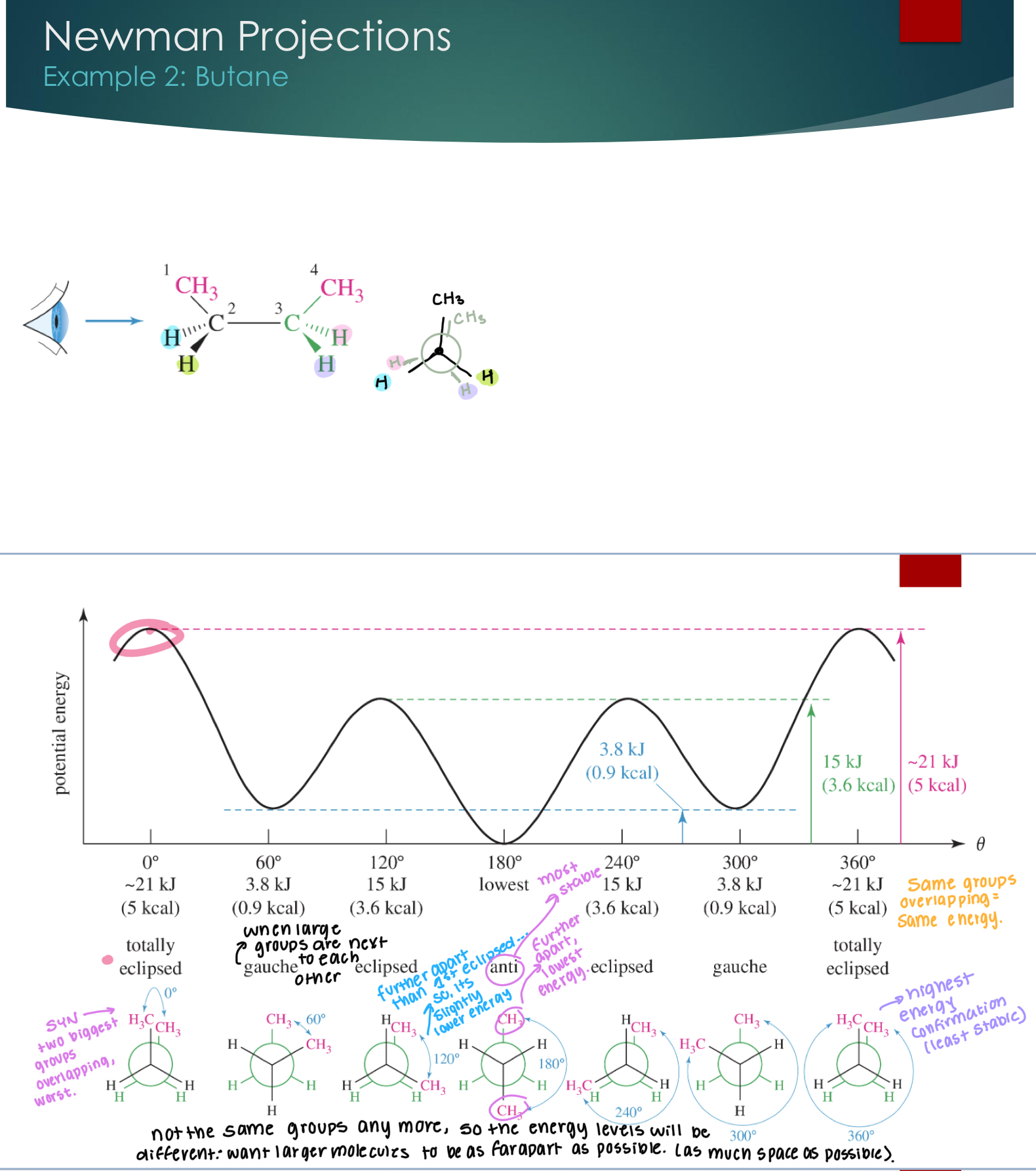
Different types of orientation names and stability using butane.
SYN eclipsed → two biggest groups are overlapping, which is the worst.
Gauche → when large groups are next to each other
Eclipsed → Where the biggest groups are further apart than the 1st eclipsed
Anti → most stable. Furthest from each other and the lowest energy.
Not the same groups anymore, so the energy levels will be different: want larger molecules to be as far apart as possible (as much space as possible).
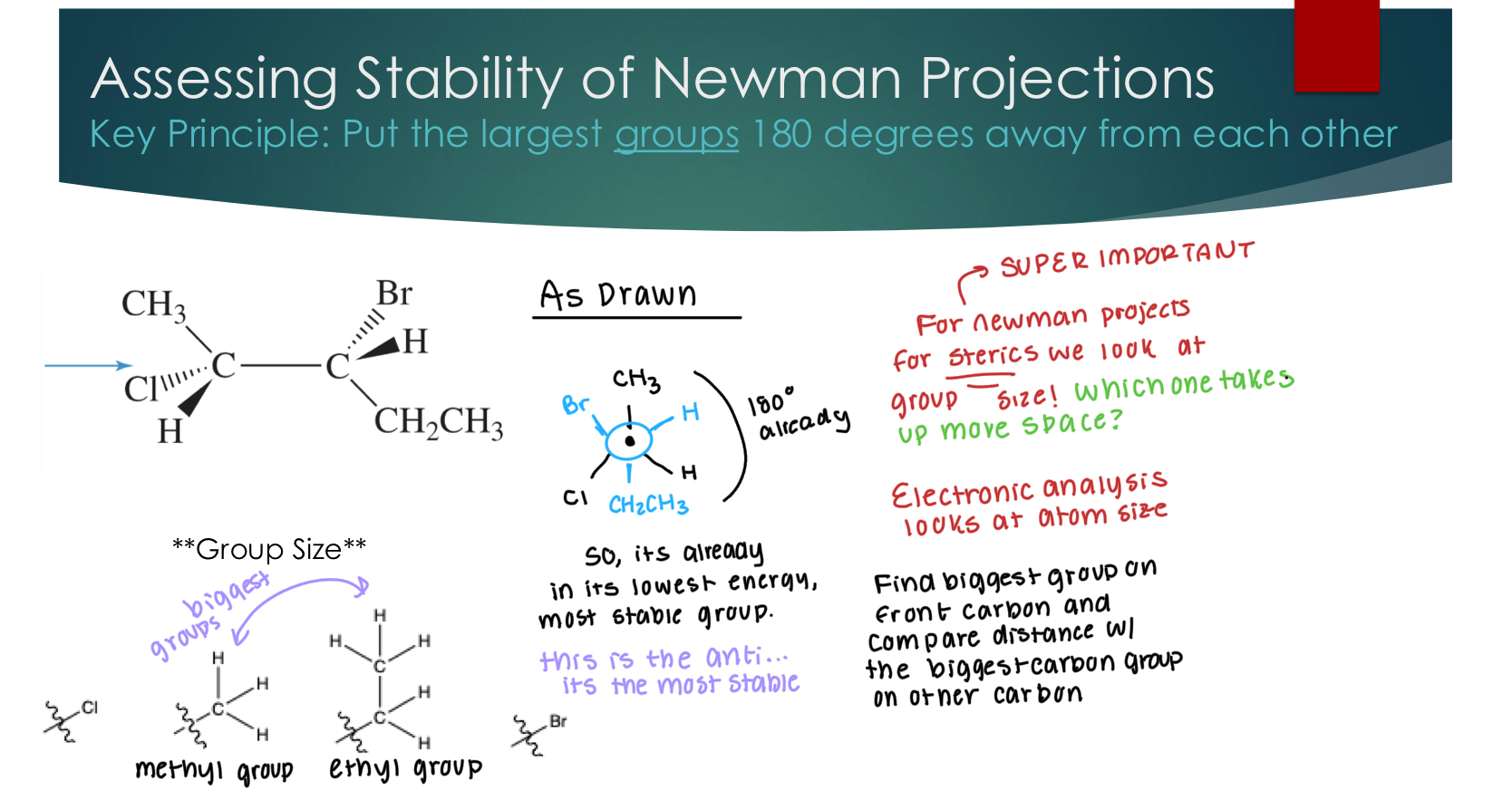
Assessing Stability of Newman Projections
Key Principle: Put the largest groups 180 degrees away from each other
SUPER IMPORTANT
For Newman projects for steric, we look at the group size: Which one takes up the most space in front and back?
Electronic analysis looks at atom size
Find the biggest group on from carbon and compare distance w/ the biggest carbon group on other carbon.
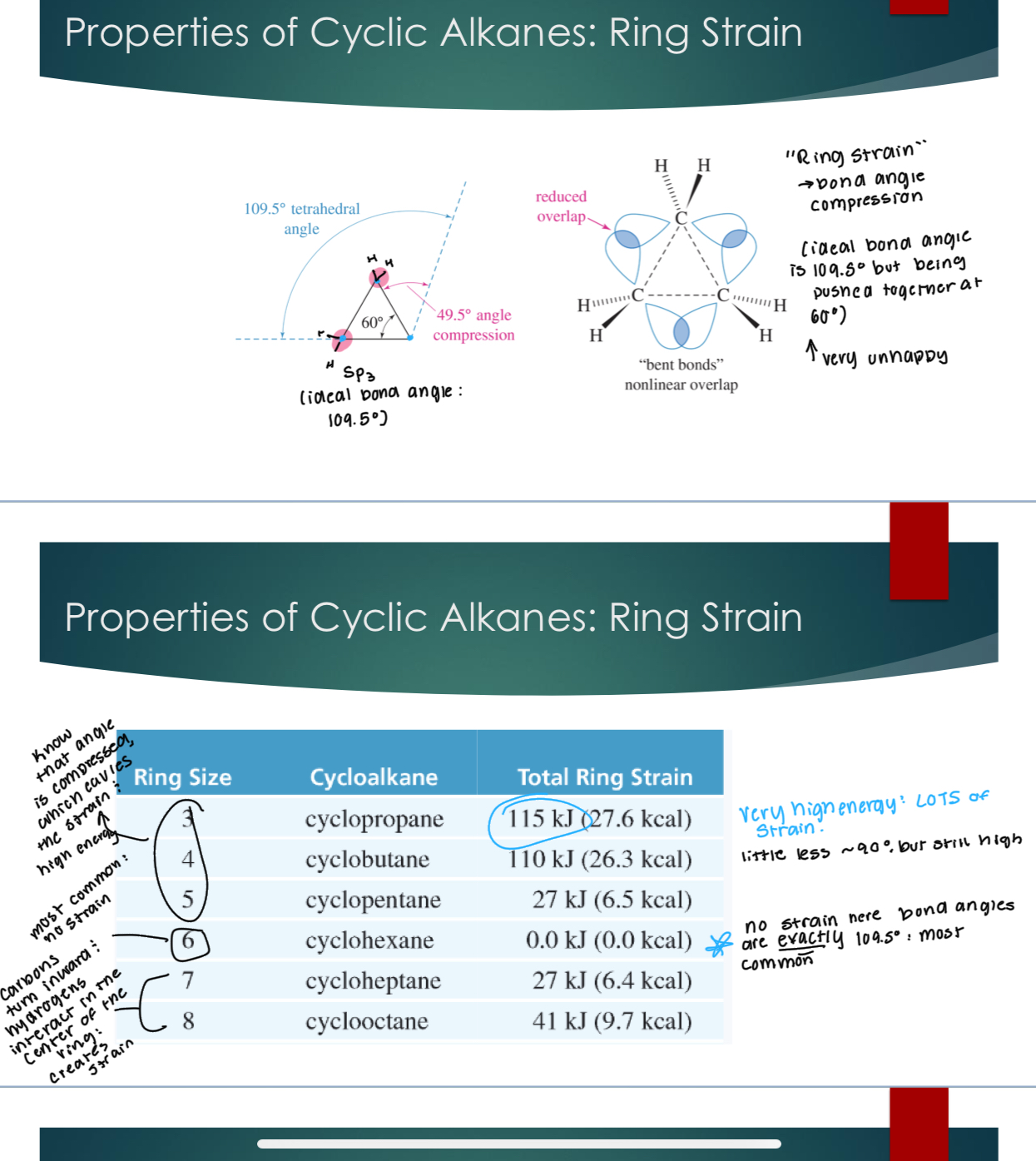
Cyclic Alkanes: naming and structure
Add a “cyclo” prefix to the name
Properties of Cyclic Alkanes: Ring Strain → just means bind angle compression. Example, ideal bond angle is 109.5, but being pushed together at 60° which is very unstable because carbons are very unhappy.
For the chart, know that the angle is compressed for 3,4, and 5, which causes the strain and the high energy. 6 has no strain, and 6,7,8 have carbons that turn inward and hydrogens interact in the center of the ring, which created the strain.
Conformation of Cyclic Molecules
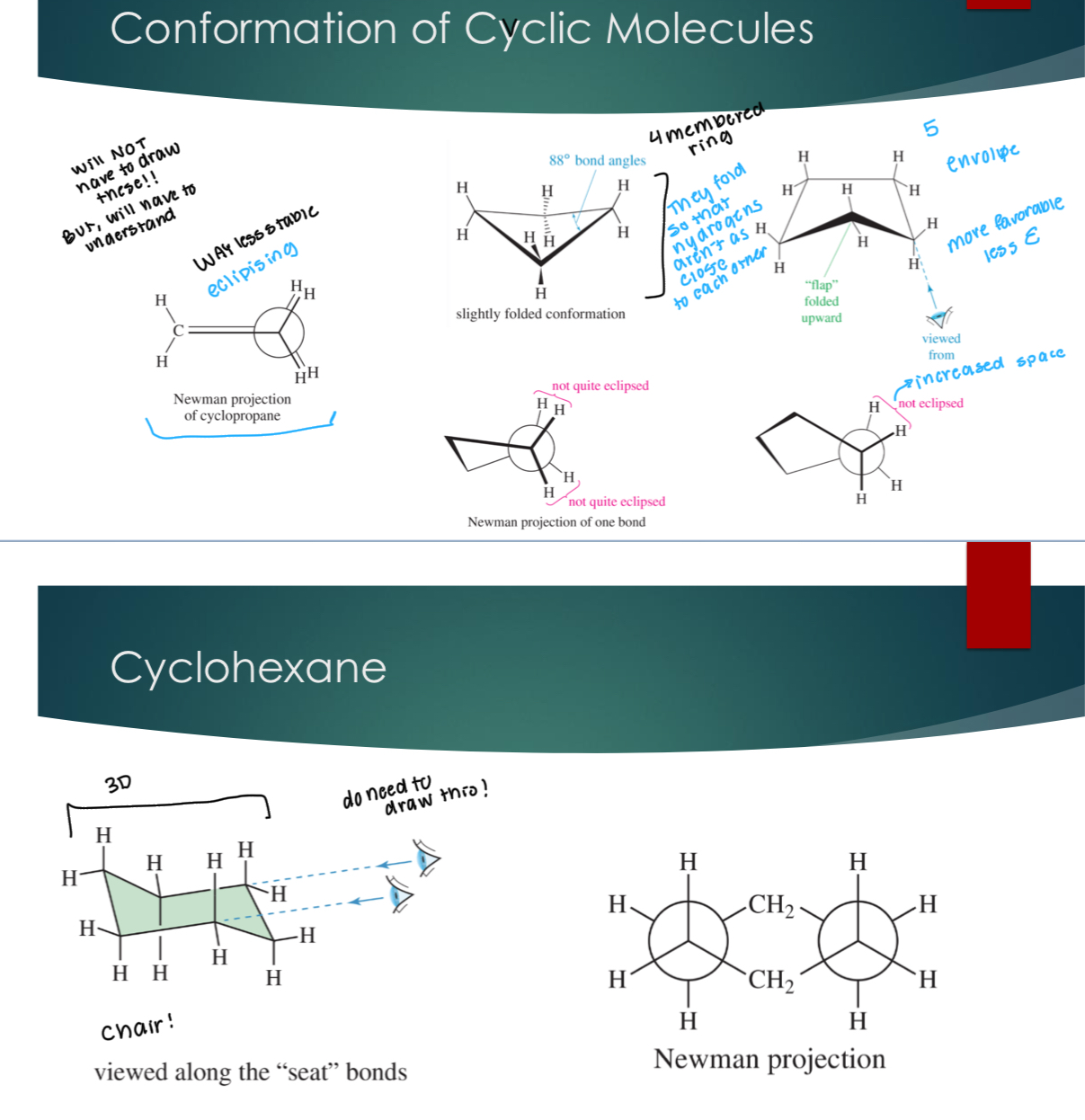
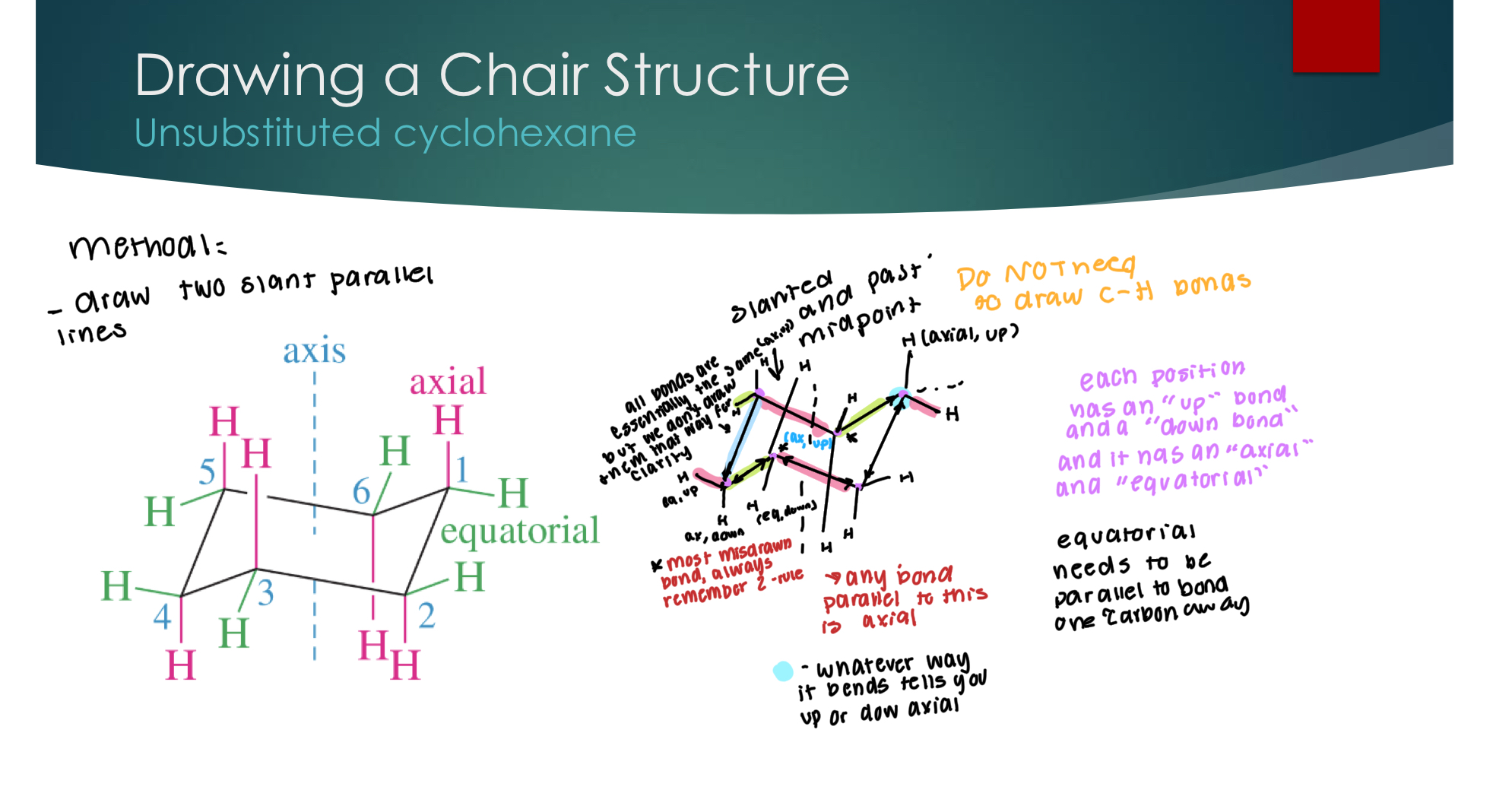
Drawing a Chair Structure
Unsubstituted a cyclohexane
each position has an “up” and “down” bond, and it has an “axial” and “equatorial”.
Wedge means up, dash means down.
Confirmation Change in Cyclohexane (The Chair Flip)
Chairs flipping does NOT change the bond type… everything must be up or down. Only axial/equatorial can change. Axial will become equatorial and equatorial will become axial.

Assessing Stability of Chair Structures
Key principle: Put the substitutants equatorial
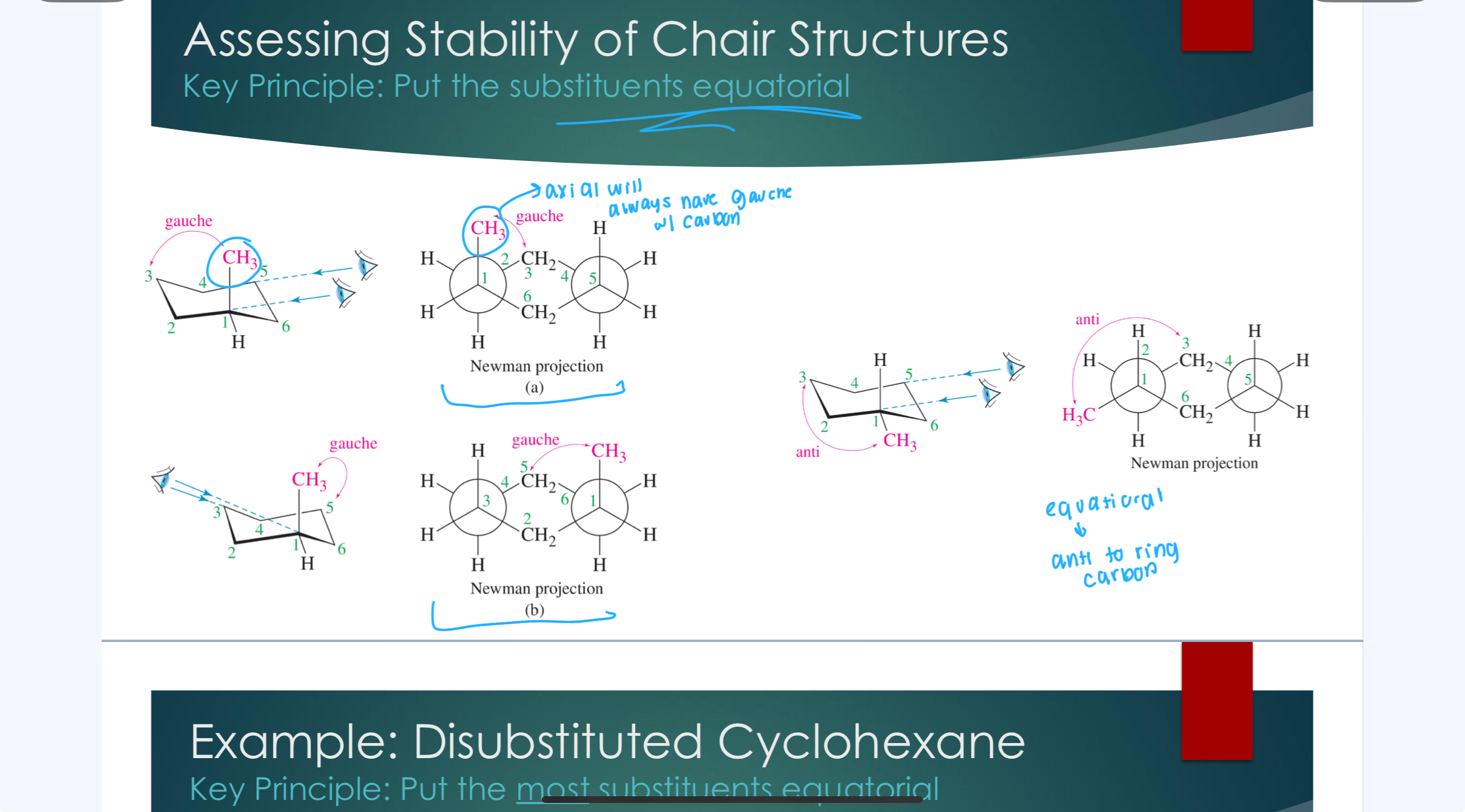
Disubstituted Cyclohexane
Key Principle: Prioritize putting the larges substituents equatorial
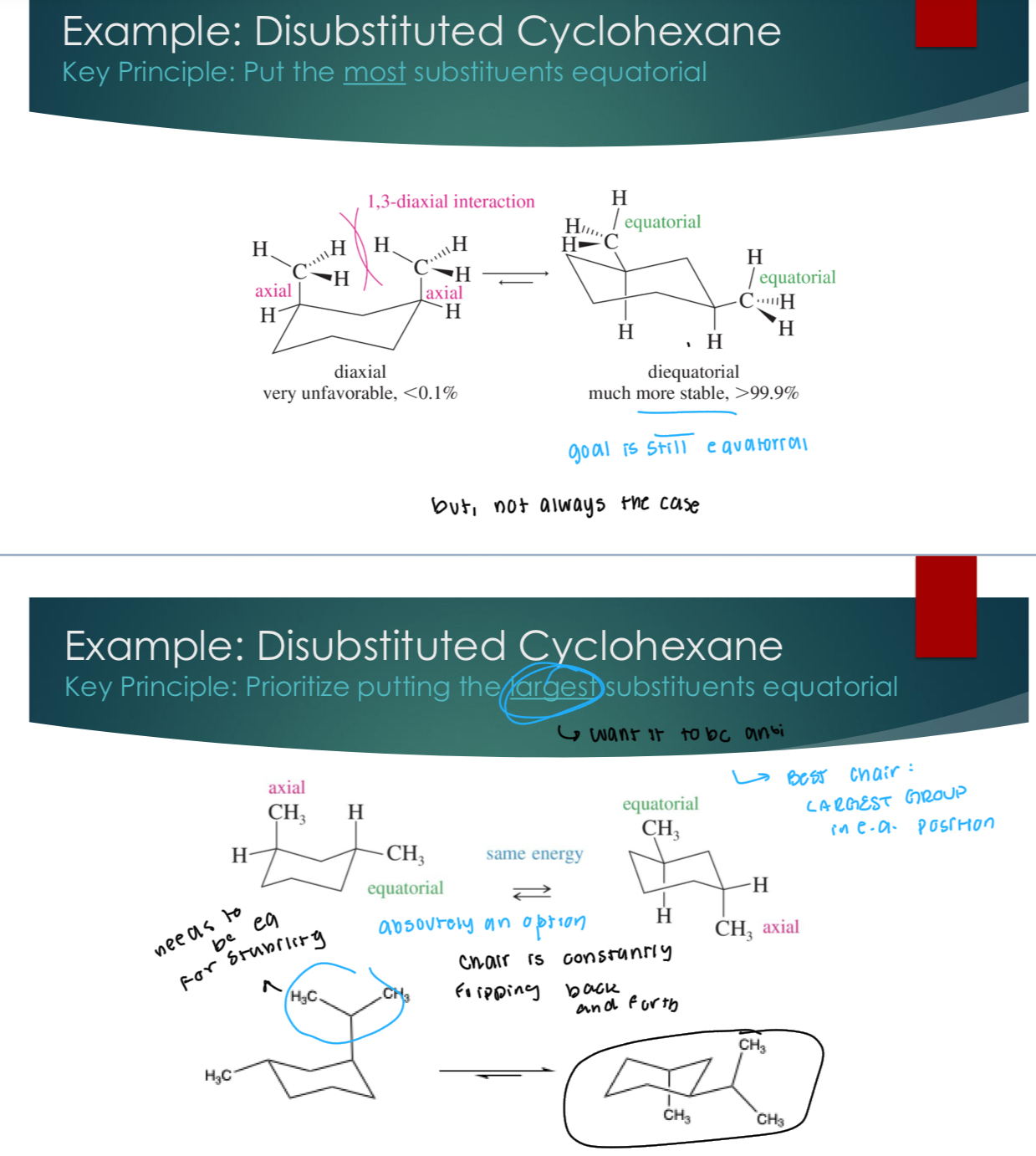
Some more aspects of chair stability — 1,3 Diaxial strain
1:3 diaxial relationship is terrible because they’re still really close to each other in space. If you are going to be axial, it’s better to be a hydrogen.

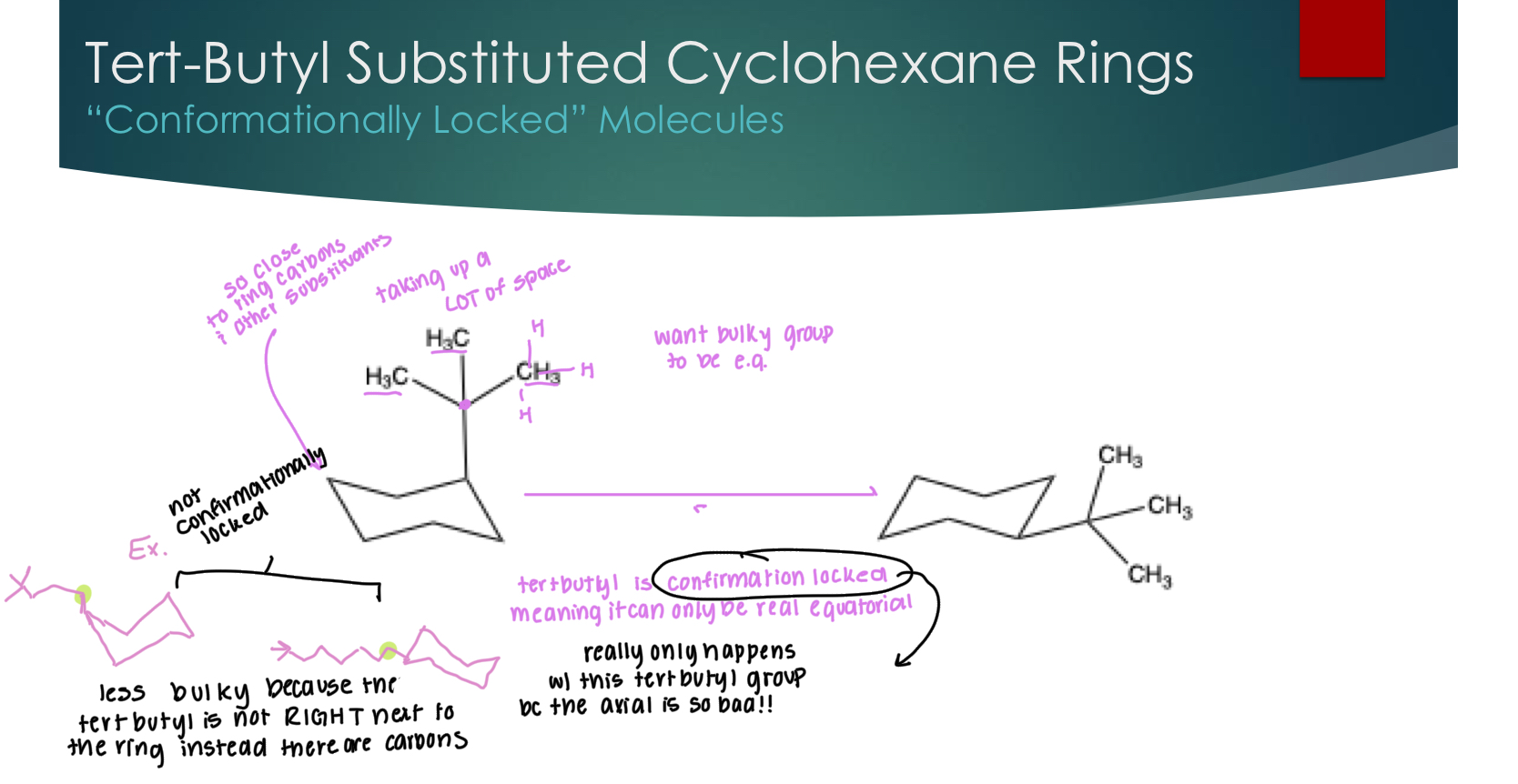
Tert-Butyl Substituted Cyclohexane Rings
“Conformationally Locked” Molecules
tertbutyl is confirmation locked, meaning it can only be equatorial, this really only happens with trtbutyl because the axial conformation is so incredibly unstable.
If there are more carbons attached to the ring and then the tertbutyl, it is not confirmationally locked
Chirality
Something is chiral if it has a non-super-imposable mirror image and no plane of symmetry
Chiral interactions a different depending on if it is interacting with something chiral and in what order. For example, you can put either sock on either foot because it’s not chiral, but you’re going to have an uncomfortable day is you put your shoes on the wrong foot.
Why do we care?
Think of the drug that was chiral and the drug caused birth defects for the babies, as opposed for just anti nausea medicine for the mothers.

Other Properties of Chiral Molecules: Optical Activity
Chiral molecules are optically active, meaning they can rotate plane polarized light
If the plane polarized light is rotated in a clockwise direction, the molecule is referred to as dextrorotatory and notated with: (+)
If the plane polarized light is rotated in a counterclockwise direction, the molecules is referred to as levorotatory and notated with: (-)
*****Neither the amount or direction of optical rotation can be predicted by looking at a chemical structure, it must be experimentally determined*****
Measuring Optical Activity
To determine optical activity, or the degree of optical rotation, we use a polarimeter
Optical rotation of a chiral molecule is reported in the format:
(+) 35°C (rotated light by 35°C)
(-) 70°C (rotated light counterclockwise by 70°C)
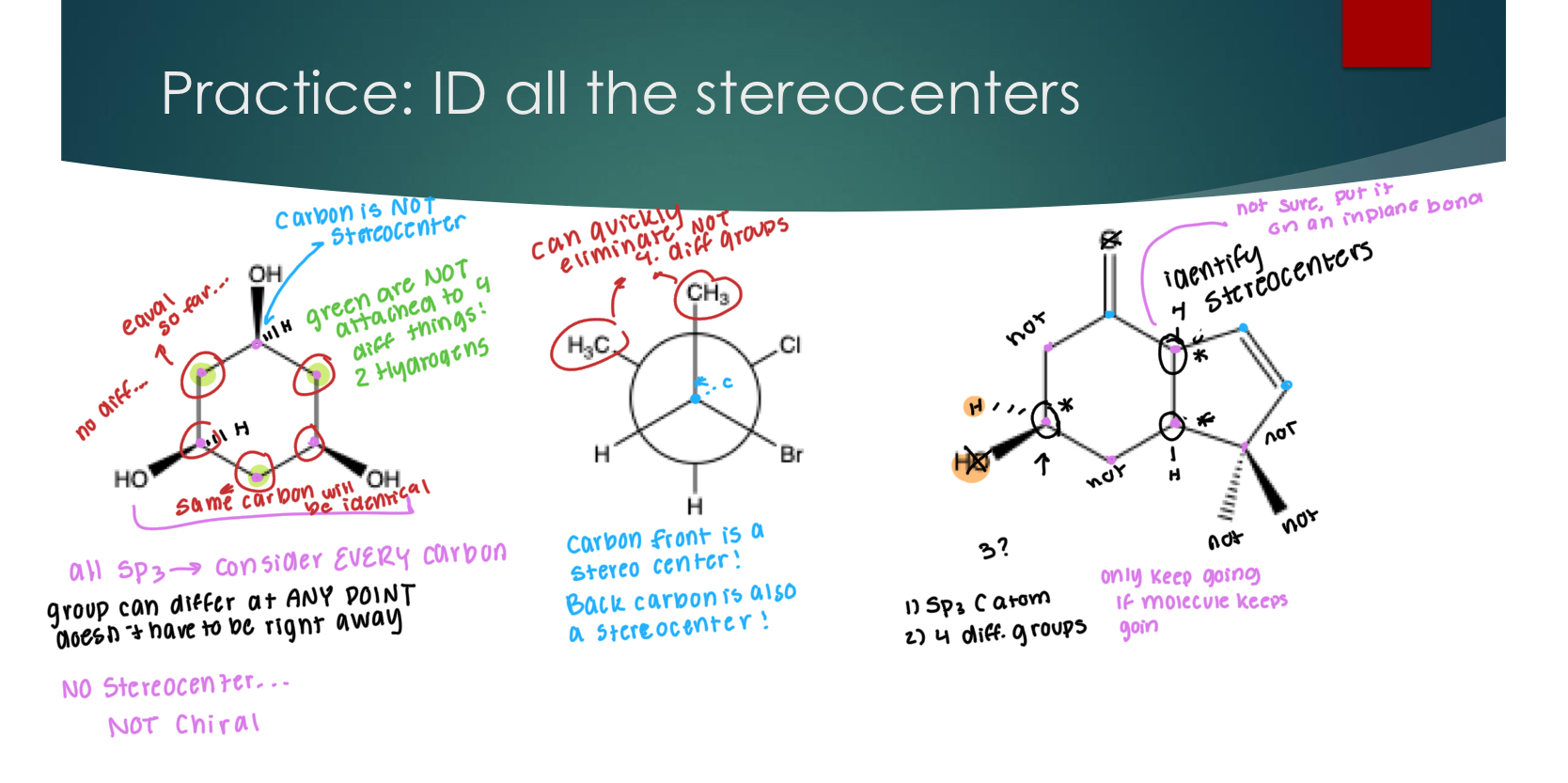
Identifying Chiral Compounds
Identifying Chiral Compounds
A more practical definition of chirality: a molecule that contains at least one stereocenter, and no plane of symmetry
A Stereocenter = chiral carbon = asymmetric carbon:
Looking for carbon with 4 different groups (bc then it can’t be superimposable)
It must be Sp3 and
Other atoms besides carbon can be chiral, but we will not be discussing them in this class
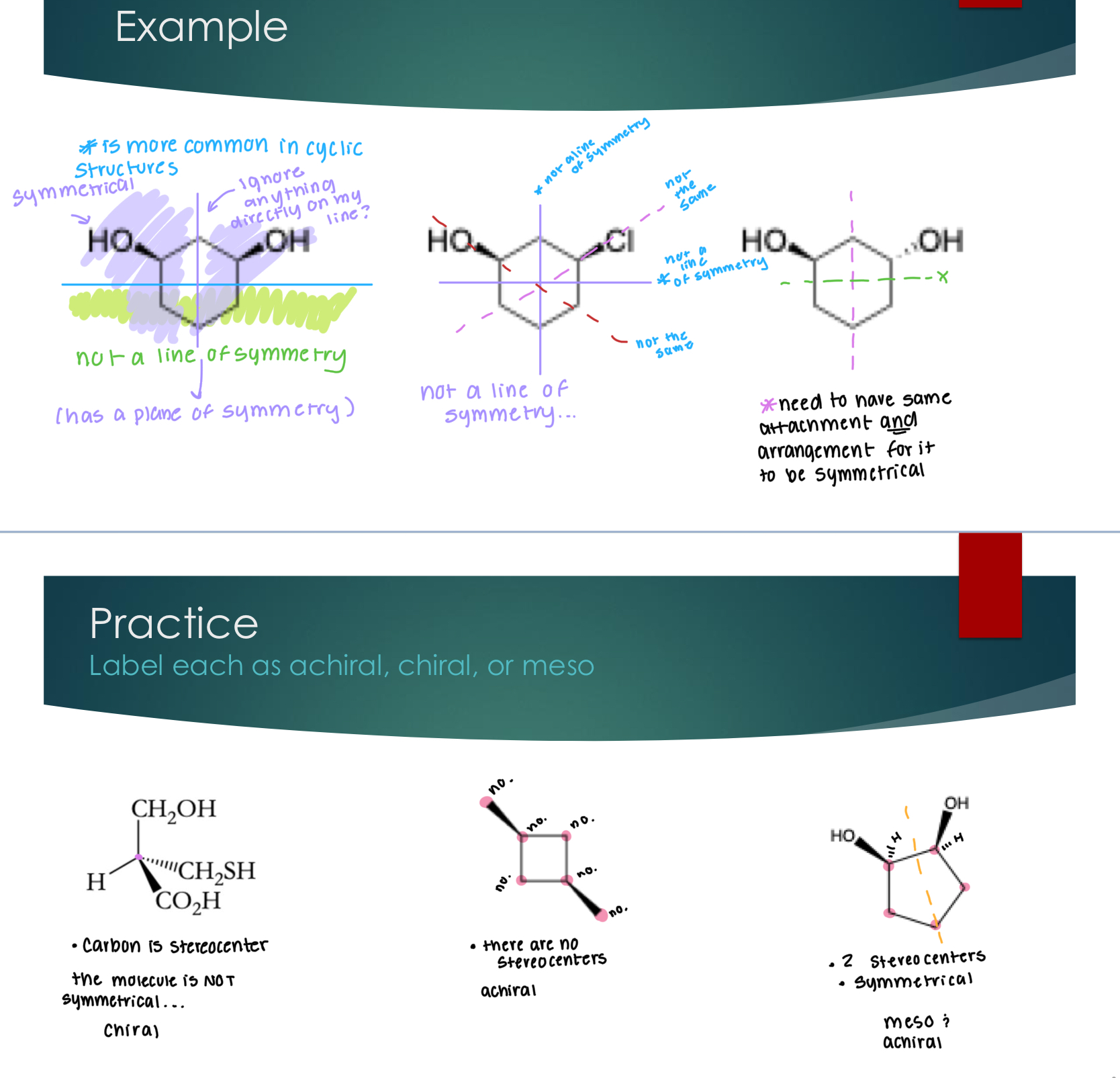
Meso Compounds
Molecules are chiral if they have at least one stereocenter AND no plane of
symmetry
If a molecule has at least one chiral center, but does have an internal plane of symmetry, it is called a meso compound
Achiral means:
A molecule that has no stereocenters is achiral
Meso compounds are achiral (meaning it has no optical activity (symmetry))
Classifying Compounds with Chiral Centers
Optical Rotation: all chiral molecules will have some optical rotation: denoted by (+) or (-)
* Must do an experiment to figure out
Absolute Configuration: R or S per stereocenter: we can determine this on structure alone.
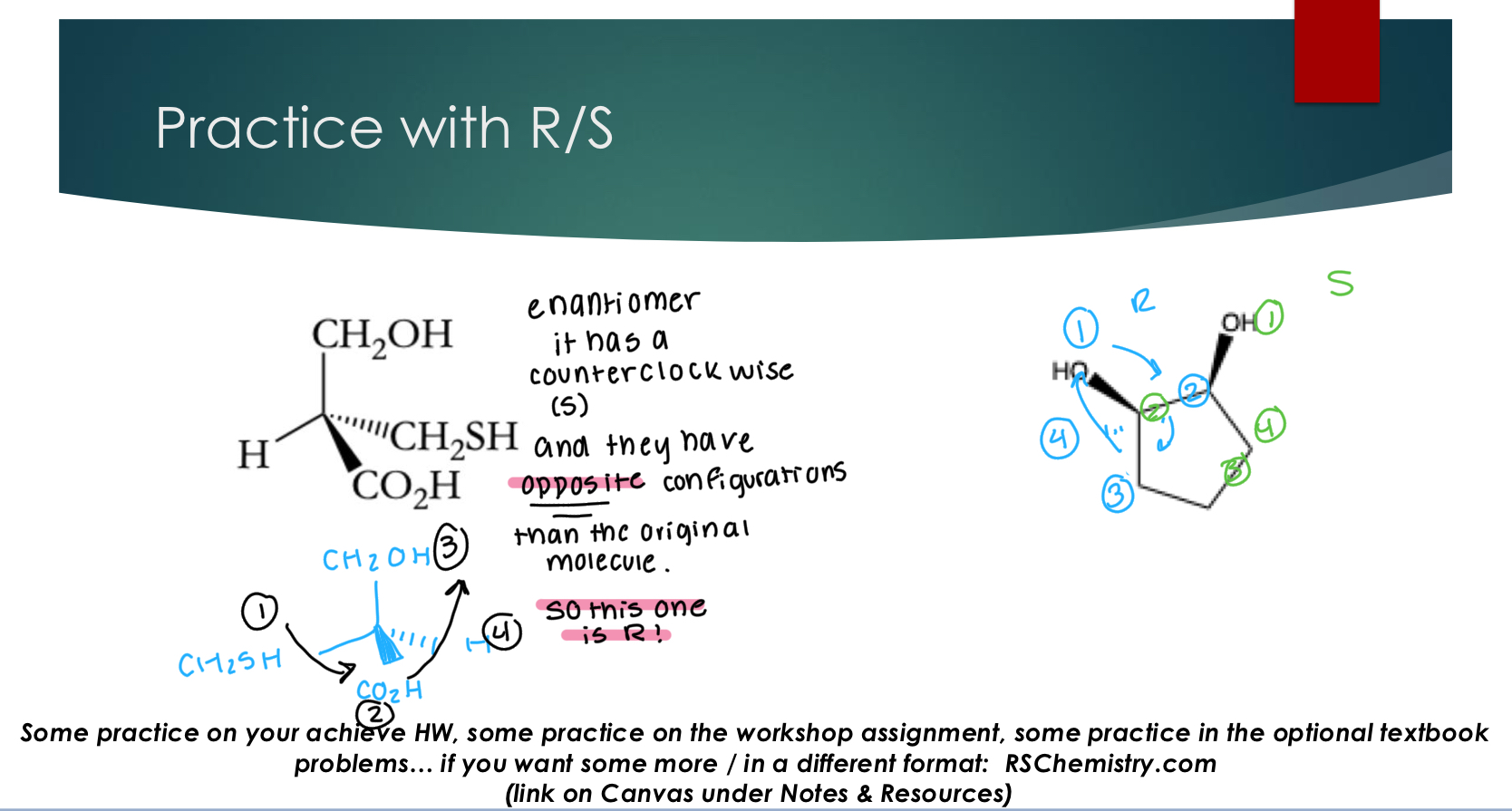
Determining Absolute Configuration
Step 1: Identify the stereocenter assign priority (1→4) of each substituent
1 = highest priority and 4 = lowest priority
Start with the atom directly bonded to the chiral center for this analysis
Prioritization:
1. Heavier atoms have priority (higher atomic number or heavier isotope) noting lower priority level than hydrogen: keep moving until there is a difference
2. If the atom directly bonded to the chiral center is the same, then go to the next atom and find the
first point of difference. (do not look at the whole group) parentheses method
3. Multiply bonded atoms count as an equivalent number of singly bonded atoms
Suggested trick / method: prioritized parentheses of attached atoms
Step 2: Orient the lowest priority group (#4) “away” i.e. on the dash position
Reorienting your molecule:
❖ If the #4 group is on the dash: you can skip to step 3 with no adjustments
❖ If the #4 group is on the wedge: you can skip to step 3 and follow the appropriate adjustment
❖ If the #4 group is on an in plane bond: redraw the molecule, switching the location of #4 group and the group currently on the dash. Do not change any other orientations or positions. → when you switch exactly two things around a stereocenter, you are drawing the enantiomer.
Then follow the appropriate adjustment in step 3
Step 3: Draw an arched arrow from group 1 → 2 → 3 on your correctly oriented molecule
If this arrow is going in a clockwise direction then the stereocenter is ‘R’
If this arrow is going in a clockwise direction then the stereocenter is ‘S’
Don’t forget any adjustments!!!
❖ #4 group on the wedge adjustment: pick the opposite configuration as the analysis gives you
❖ #4 group is on an in plane bond adjustment: the original molecule will have the opposite configuration letter as the redrawn molecule you analyzed
(Finally) Finishing Our Isomer Flowchart
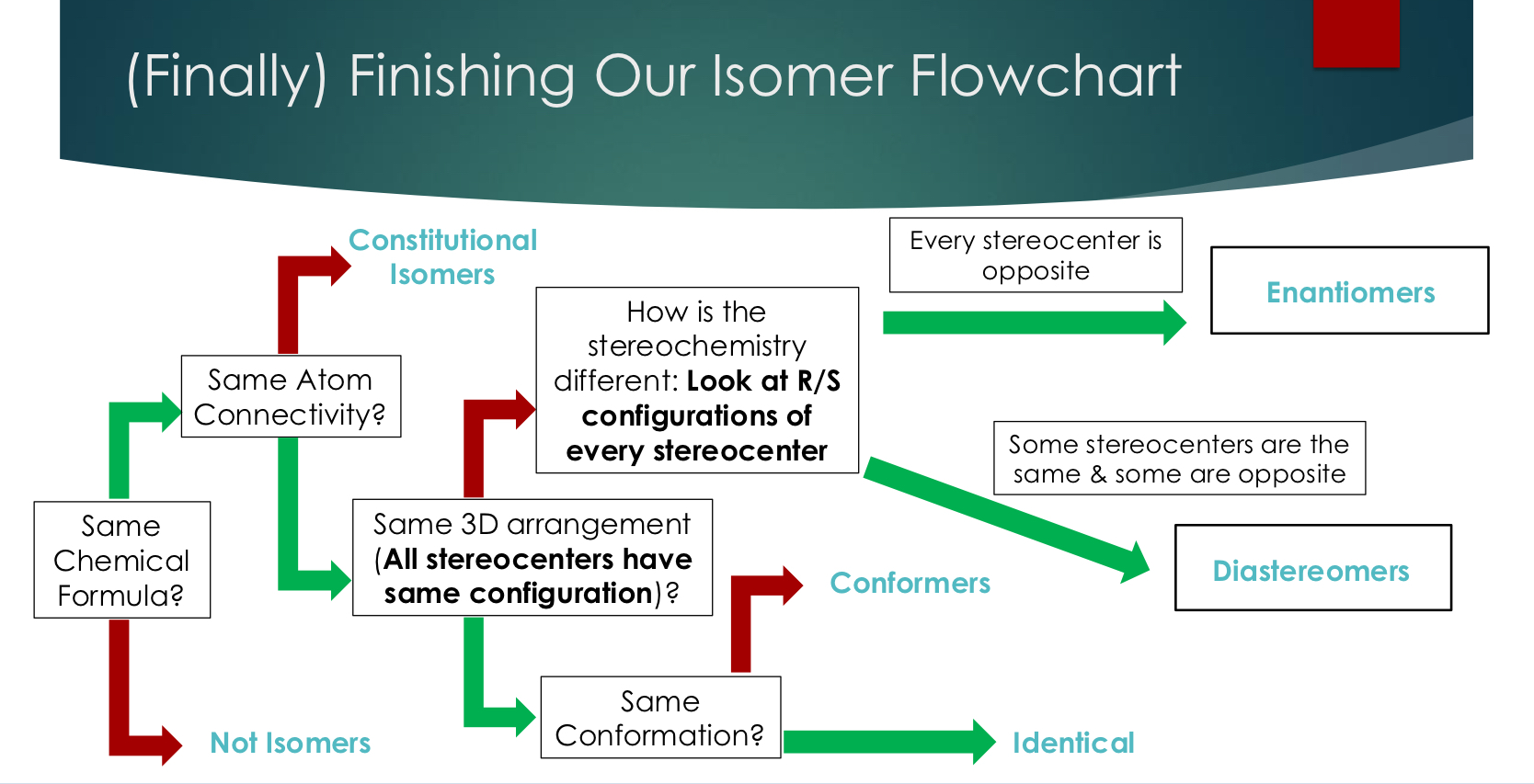
Enantiomers
Stereoisomers that are non-superimposable mirror images of each other (perfectly opposite)
Every chiral will have exactly 1 enantiomer
All chiral centers have the opposite absolute configuration (R/S)
Special property of enantiomers: equal magnitude (same number) and opposite sign optical rotation (+) or (-) this is an exception
All other chemical properties of enantiomers are identical → most challenging isomers to separate (we can actually separate) → resolution.
Note: we can’t separate conformers
Diastereomers
Stereoisomers that are not non-superimposable mirror images of each other (not perfectly opposite)
Some but not chiral centers have the opposite absolute configuration (R/S) (SSR maybe diastereomer is SSS)
Diastereomers have unrelated optical rotations!
All other chemical properties of enantiomers are similar but different → can separate but typically tricky
a typical difference would be 80°C vs 82°C
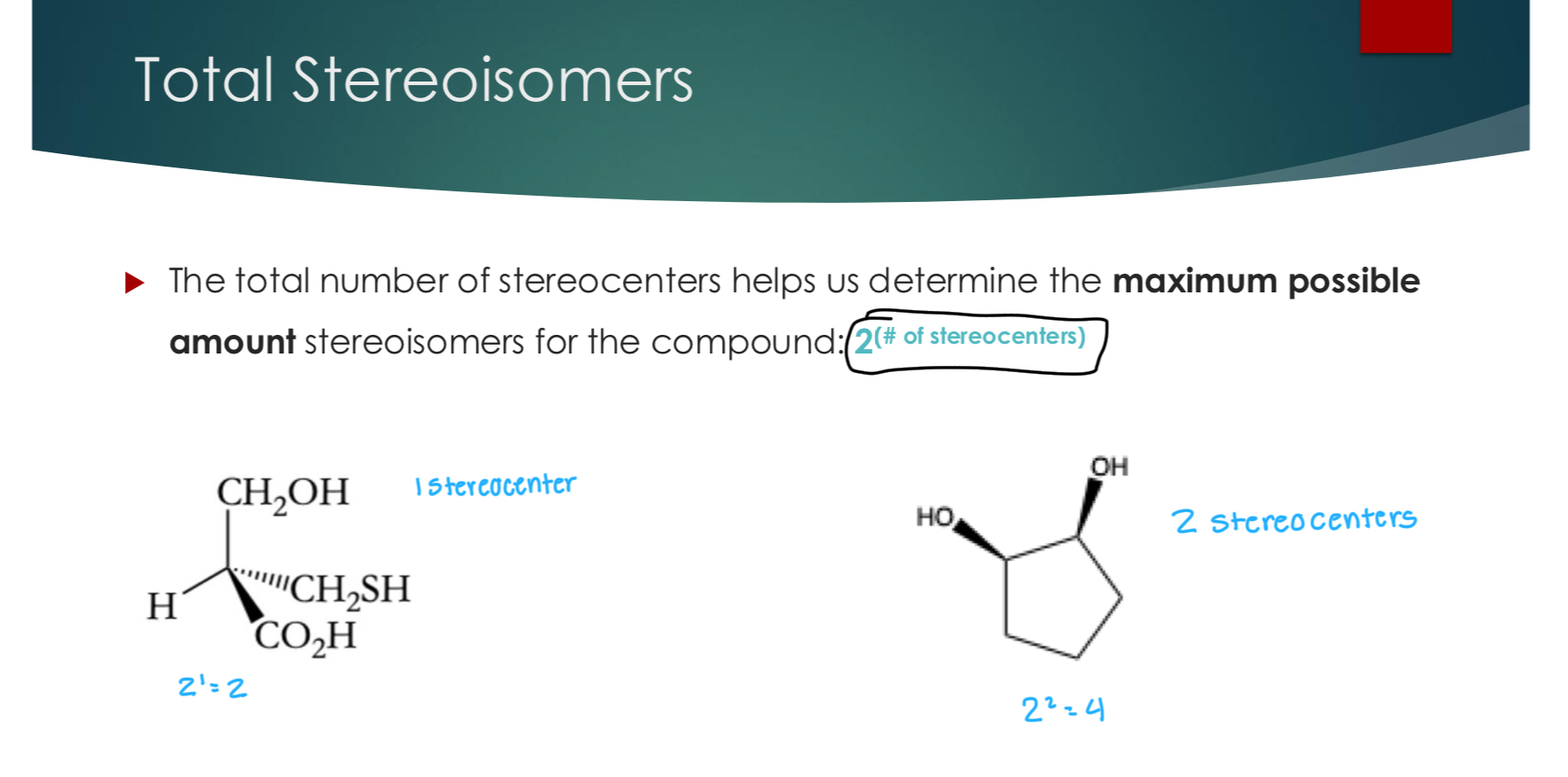
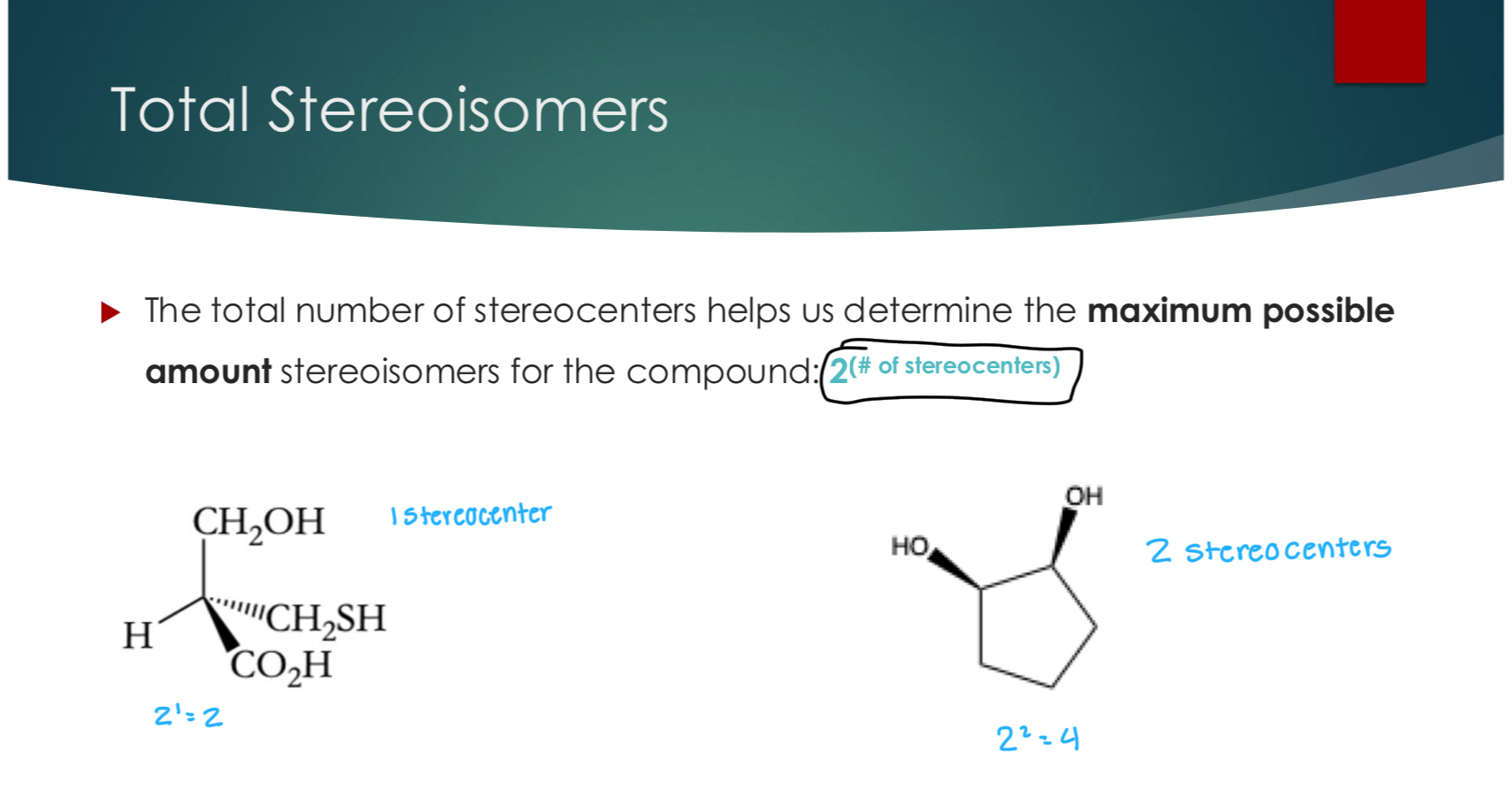
Total Stereoisomers
The total number of stereocenters helps us determine the maximum possible amount stereoisomers for the compound: 2(# of stereocenters)
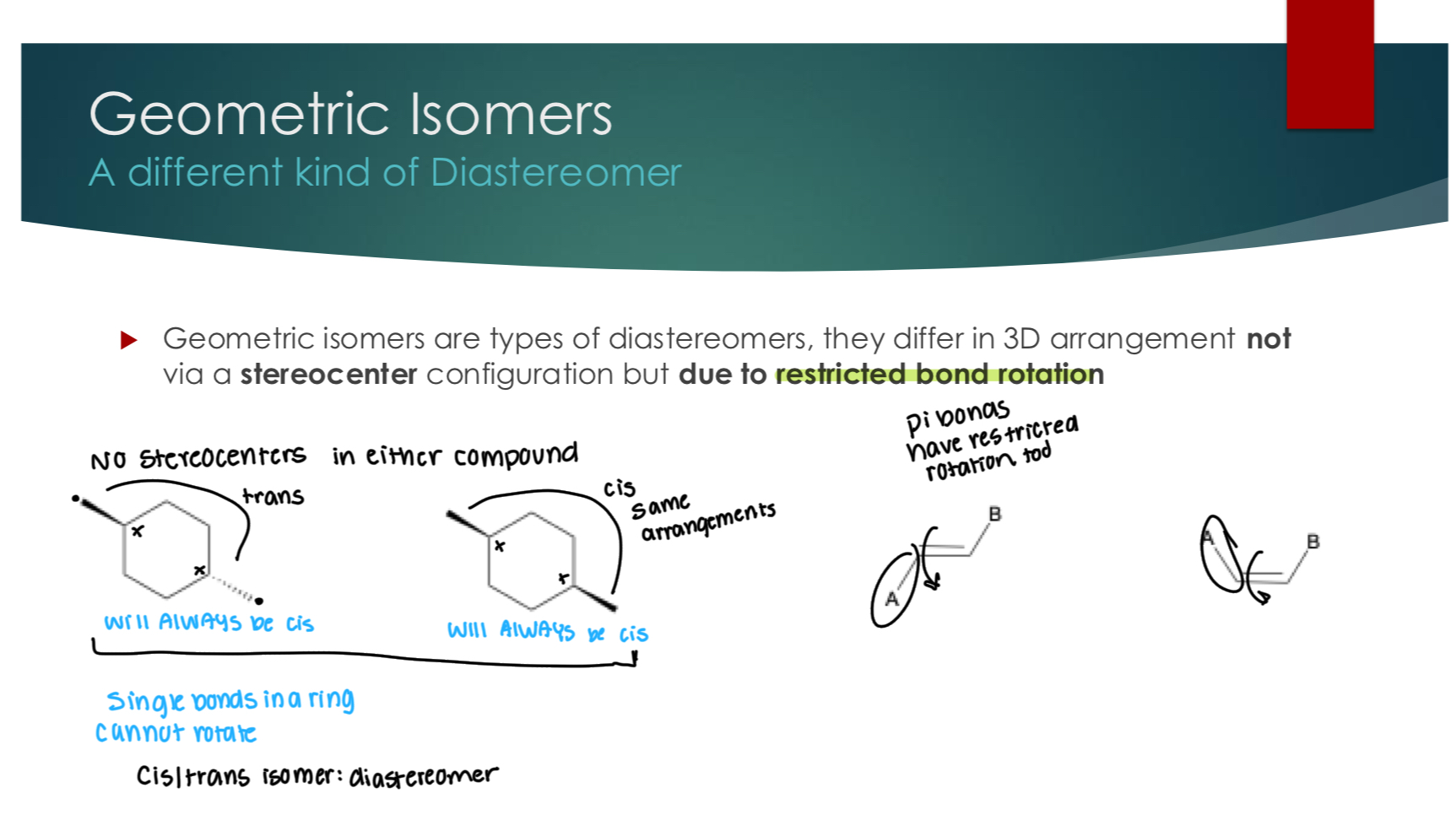
Geometric Isomers A different kind of Diastereomer
Geometric isomers are types of diastereomers, they differ in 3D arrangement not via a stereocenter configuration but due to restricted bond rotation
pi bonds have restricted bond rotation.
Single bonds in a ring cannot move.