M1L2 DNA damage and repair
1/32
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
33 Terms
What causes DNA SSBs?
Radiation and oxidative stress
What causes DNA mismatches?
Replication errors, homologous recombination, cytosine deamination to uracil, 5meC deamination to thymine, adenine deamination to hypoxyanthine (codes like G)
What causes damage to nitrogenous bases?
radiation, oxidative species, alkylation (particularly methylation), deamination
What causes abasic sites?
Occurs spontaneously, particularly for purines, especially if the base is alkylated, also an intermediate in the action of DNA glycosylases
What gives rise to altered bases?
Pyrimidine dimers, alkylated DNA, oxidised bases
What causes DNA DSBs?
Ionising radiation
What produces intra-strand crosslinks?
UV light and cancer drugs
What produces inter-strand crosslinks?
Cancer drugs and metabolites (eg aldehydes)
What are the main types of DNA mutagenesis?
Deletions, insertions, substitutions
Transitions - pyrimidine substituted for a different pyrimidine or purine substituted for another purine
Transversion - pyrimidine substituted for purine or purine substituted for pyrimidine
What is the role of photolyase in direct repair in bacteria, fungi, plants, and certain animals?
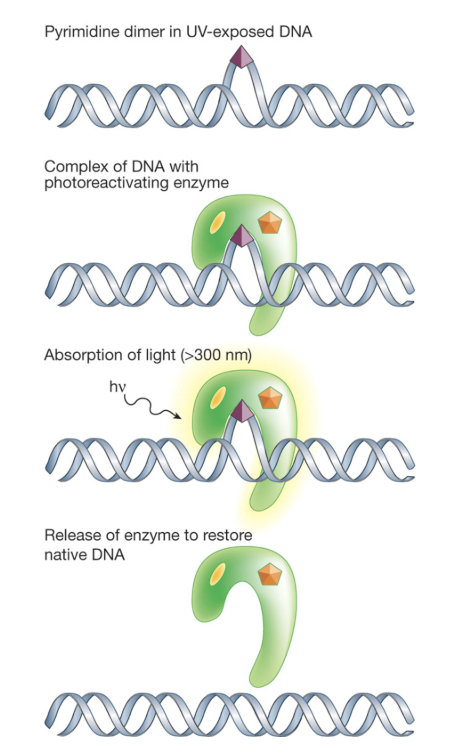
Strong affinity for structure of damaged lesions and can reverse base damage caused by UVB radiation
Pathway has two cryptochromes (FAD and FADH-) which can absorb blue light (300-500nm)
FADH- gets excited, transferring an electron to the UV damaged site
Breaks chemical bonds causing DNA damage and returns pyrimidines to original configuration
How are O6 alkyl groups directly repaired by the suicide protein O6-ATase?
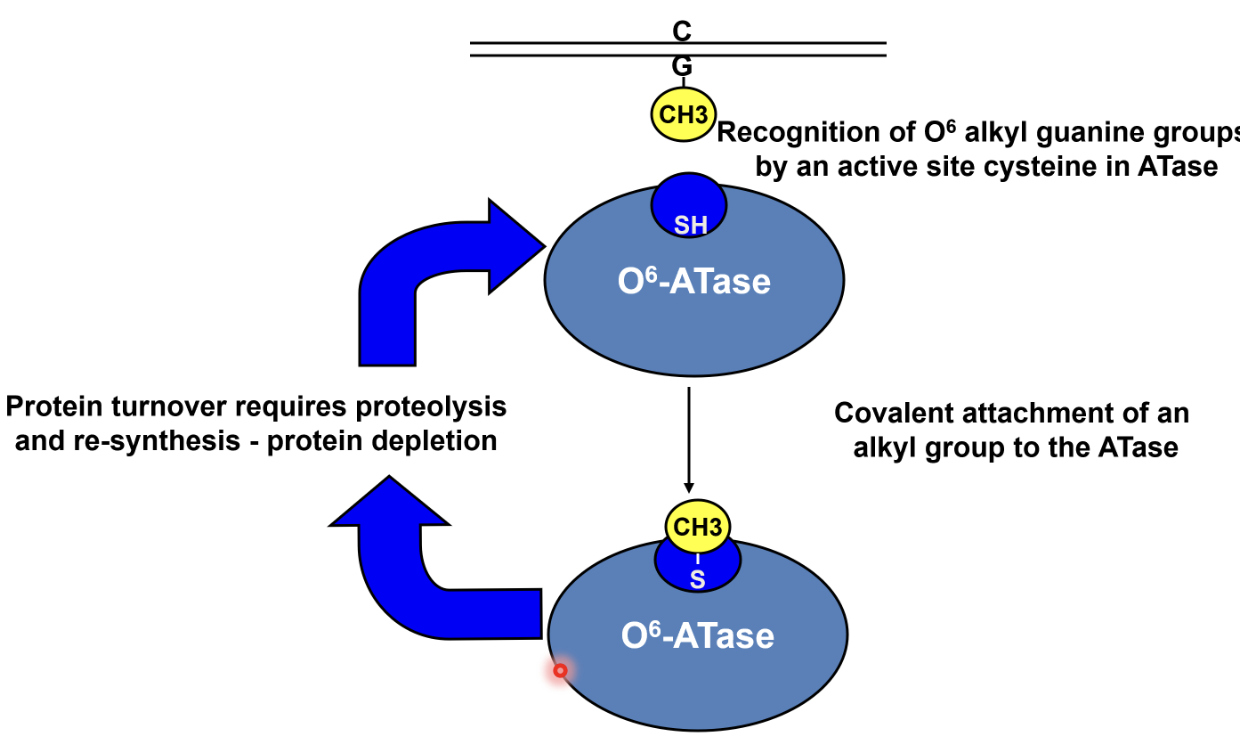
O6 alkyl guanine is recognised by the ATase which transfers the alkyl group to a cysteine residue in its active site, causing covalent attachment of alkyl group to ATase
This corrects the damage but inactivates the ATase, hence protein turnover requires proteolysis and resynthesis
How is O6-ATase implicated in drug resistance?
Temozolomide induces DNA methylation in the treatment of brain cancers, however cancer cells tend to upregulate this ATase as a resistance mechanism
Drug development investigating inhibitors of these ATases to prevent this, but there are issues with bypassing the blood brain barrier and tumour evolution
What are two pathways that can be used to repair O6-alkyl guanine?
Direct repair and base excision repair
What are the three broad steps of BER?
Lesion recognition
Removal of damage nitrogenous base
Short/long patch BER
How are DNA lesions recognised in BER?
The enzymes have a motor protein that slides rapidly up and down the double helix
Damaged base can be recognised by shape, hydrogen bonding potential and electric charge distribution
How are damaged bases removed in BER?
By monofunctional glycosylases which have high specificity for small chemical base modifications (eg deaminated cytosine, oxidised guanine…)
When sliding along the phosphate backbone glycosylases can flip out bases from the interior of the helix
Once flipped into the enzyme binding pocket, damaged bases but not normal bases get cleaved by glycosylase
Normal bases will have a short residency time in the active site but damaged bases have a long residency which permits the enzyme to attack the N-glycosidic bond to cleave the base
Nitrogenous base gets removed by glycosylase, producing an AP site (abasic site)
AP endonuclease cleaves the AP site, exposing a 3’-OH on one side of the cleavage site and deoxyribose phosphate (dRP) on the other side
Describe short patch BER
DNA pol β (has dRP lyase activity) removes the phosphorylated ribose to restore normal DNA biochemistry by exposing 5’-phosphate but leaves a 1-nt gap
Pol β extension fills the gap and DNA ligase III/XRCC1 seals the nick
Describe long patch BER
DNA pol β/δ/ε extends from the 3’-OH for 3-12 nt which generates a displaced flap
Flap endonuclease cleaves the flap and the dRP is thus removed
Nick is sealed by DNA ligase I/PCNA
What is Xeroderma pigmentosum?
a condition associated with defects in NER causing severe photosensitivity, skin and non-skin cancers, and neurological abnormalities
How is DNA damage recognised in NER?
Detects bulky, large chemical damage (eg. crosslinked adjacent pyrimidines due to UVB)
Damage is detected by structural distortion and defamation of the DNA structure as bulky chemical damage alters base pairing and DNA geometry, causing local unwinding/denaturation and lack of base pairing at the damaged site
XPC recognises the lack of base pairing
XPB (moving 3’ to 5’) and XPD (5’ to 3’) helicases unwind DNA around the lesion
XPD particularly acts as a damage verification helicase and moves towards the damage and its unwinding activity is arrested by DNA damage, generating a static bubble which can be cut by the endonucleases
If there is no damage XPD would continue to translocate along the DNA and continue unwinding which collapses the NER apparatus, avoiding incision
What are the steps of NER?
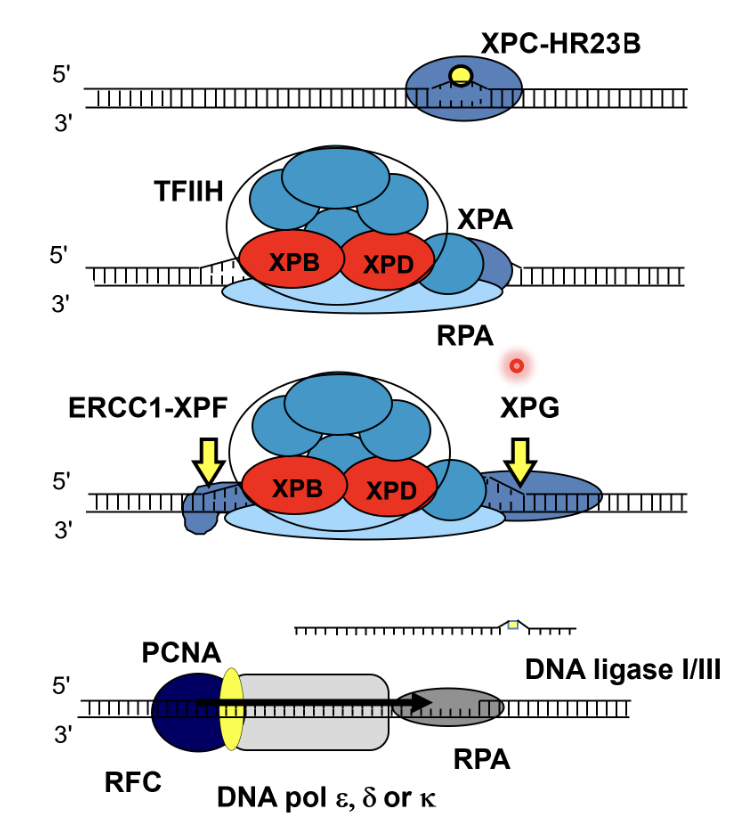
DNA damage is recognised by XPC-HR23B and the DNA duplex is opened up
TFIIH (related to another form of TFIIH involved in initial RNA pol II transcription), XPA (structural role), XPB and XPD helicases (create a bubble structure of DNA) and RPA (single stand binding protein) are recruited to the damaged strand
Bubble structure creates landmarks whereby endonucleases ERCC1-XPF (cuts 5’ to the damage) and XPG (cuts 3’ to the damage) can excise the strand
ERCC1-XPF cuts first, followed by DNA synthesis, and then XPG cuts the flap (rather than a flap endonuclease)
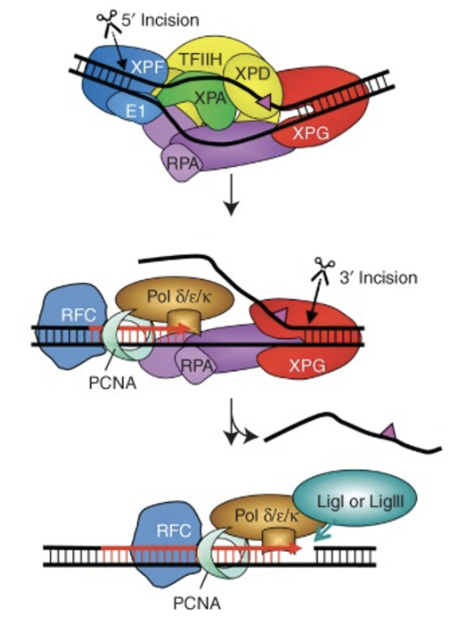
20-30nt stretch of DNA strand containing the damage is removed, leaving a gap which gets filled by DNA pol ε/δ/κ and PCNA (which is supported by RFC acting as a clamp loader) using the undamaged strand as a template
The nick is sealed by DNA ligase I/III
How can NER be transcription coupled?
A subpathway of NER operates independently of XPC-HR23B as it is triggered by RNA pol II arrest at a lesion
Cockayne syndrome A and Cockayne syndrome B proteins (CS factors) are needed to couple the arrest of RNA pol II to the NER factory
CSA is a motor protein which hydrolyses ATP and can move things around on DNA, could possibly push RNA pol II back away from the lesion to allow space for the NER apparatus to work
They may be involved in recruiting and positioning other NER proteins
NER on UV damage induced regions are is more efficient in rapidly transcribed regions of the genome due to RNA pol II
Cells degrade RNA pol II as a last resort if transcription coupled repair fails which may enable global genome repair function
What happens in Cockayne syndrome?
Cockayne syndrome - characterised by growth failure, impaired development, photosensitivity, premature aging, hearing loss/eye abnormalities, sometimes associated with Xeroderma pigmentosum
Due to defective transcription coupled repair (80% due to defective CSA, some due to defect in core NER factors)
Tend to be protected from skin tumours and reach maturity before phenotypic consequences
One theory is that these individuals do not deal with RNA pol II stalling well and its breach of DNA damage causes permanent stalling and a gradual breakdown of the gene expression profile
Explain how translesion synthesis (TLS) polymerases enable DNA damage tolerance
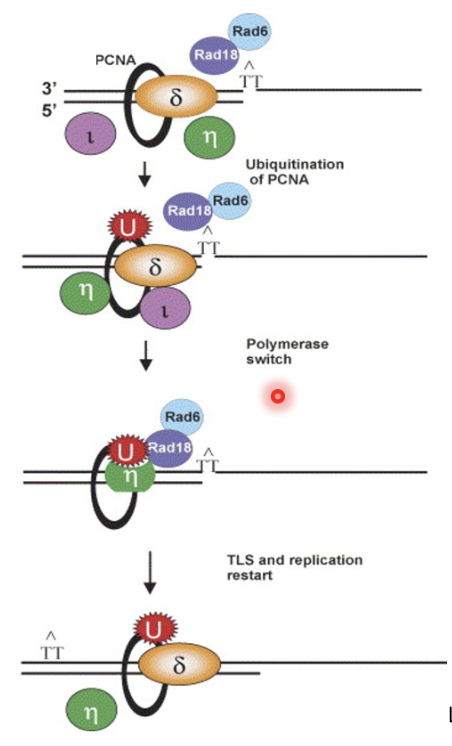
Upon fork stalling, replicative pol is removed/remodelled at PCNA, causing monoubiquitination of K164 on PCNA by Rad18 and Rad6
Causes recruitment of TLS pols (Pol ι and η) which continues extension and later switches back with replicative pol
Deubiquitination of PCNA is time consuming if it at all occurs, so not strictly necessary for re-exchange of TLS pol for replicative pol
Pol η has evolved to insert AA opposite TT adduct so does not necessarily cause mutation
What may be the result of TLS defects?
When TLS by Pol η goes wrong it causes Xeroderma pigmentosum variant form, characterised by defect in conversion of newly synthesised DNA from low to high MW after UV irradiation
Describe what is Fanconi anaemia
Recessive chromosomal instability syndrome
Bone marrow failure and childhood AML
Cancer predisposition up to x10,000
Developmental abnormalities
Genetically heterogeneous - 22 genes identified (FANCA and FANCC mutations being most common)
Exclusive hypersensitivity to ICL-inducing agents
What causes Fanconi anaemia?
Defects in ICL repair leads to Fanconi anaemia
Sources - endogenous aldehydes, oxidised lipids… etc
What are the clinical implications of mutations in FANCD1 (BRCA2) or Rad51C?
High penetrance breast cancer susceptibility alleles
What are the clinical implications of mutations in FANCJ or FANCN?
Low penetrance breast cancer susceptibility alleles
What are the clinical implications of mutations in FANCQ (XPF)?
Multiple inherited cancer prone/developmental disorders
What happens in the FA pathway?
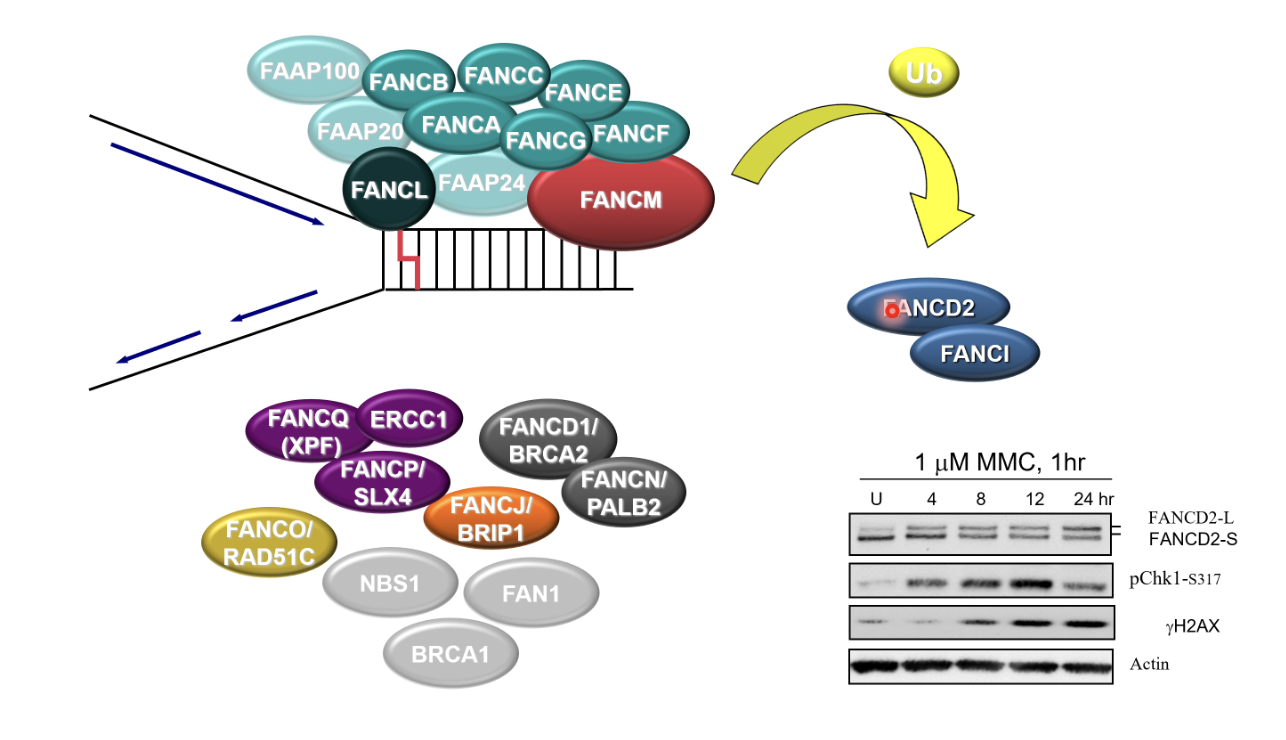
FANKL monoubiquitinates FA ID complex (FANCD2/FANCI)
FANCD2/FANCI bind to dsDNA around the damaged replication fork and bind to it to recruit other proteins
ICL incision, TL synthesis, HR
But only FANCM (translocase), FANCL (E3 ubiqutin ligase) and XPF (nuclease)
What are the steps of the FA pathway?
Convergence of replication forks signals the presence of the lesion and triggers FA pathway
FANCM may function to rewind the DNA behind the stalled fork (fork reversal) which generates a region of dsDNA adjacent to the crosslink
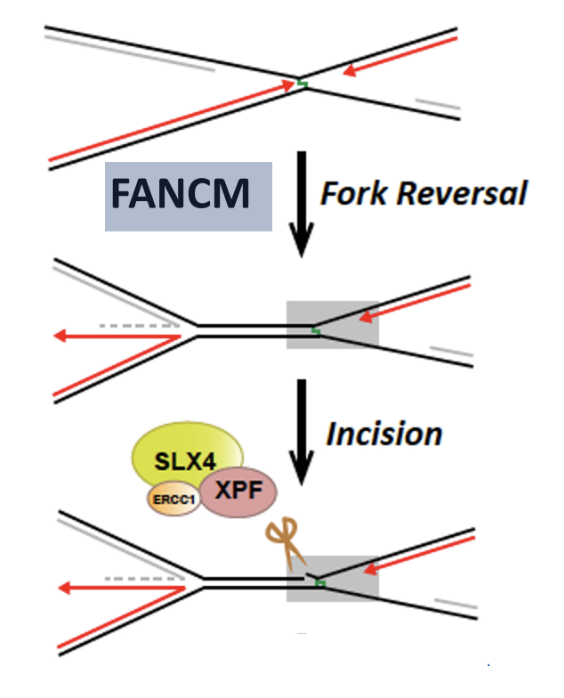
Disassembly of the replisome (CMG unloading)
FANCI-FANCD2 monoubiquitylation by FANKL
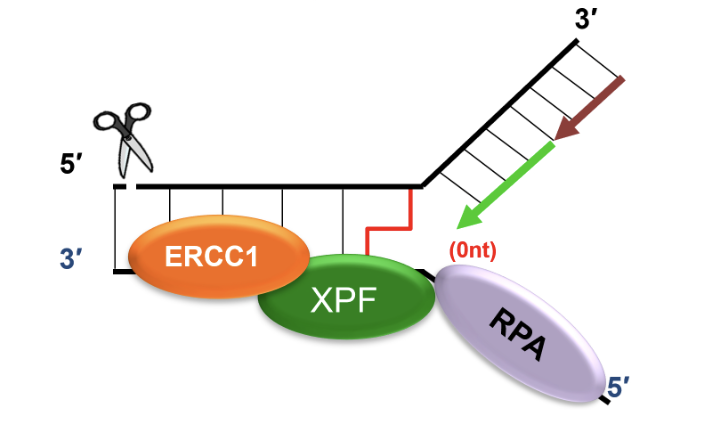
Recruits SLX4 and ERCC1-XPF to cleave DNA crosslink, releasing one of the damaged chromatids
dsDNA adjacent to crosslink produced by FANCM is an excellent substrate for SLX4 and ERCC1-XPF mediated cleavage
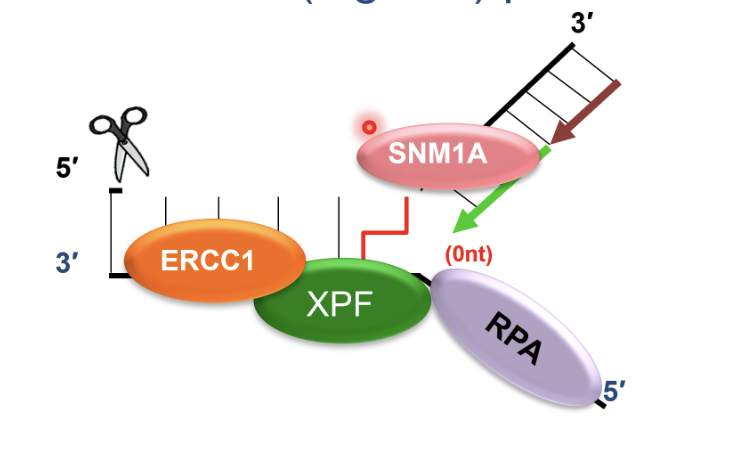
SNM1A recruited to ERCC1-XPF incised intermediate by interacting with PCNA using its PIP motif
This is a damage tolerant 5’-to-3’ exonuclease that digests through and past the incised intermediate, leaving a single nucleotide and providing a downstream template for subsequent TLS complete repair of crosslink chromatid
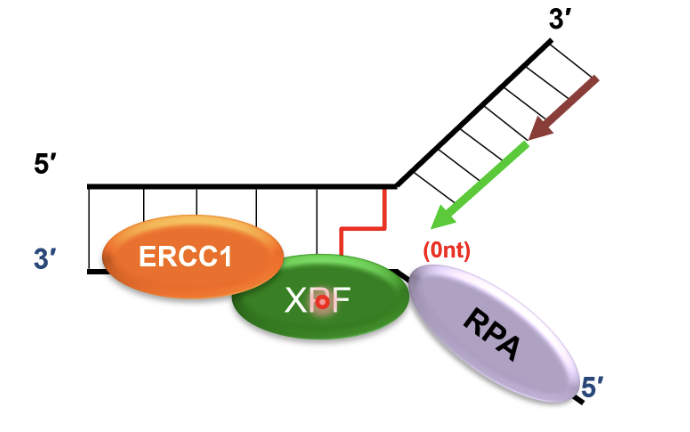
TLS polymerases take the stalled fork and extend it past the cross link in an error prone manner
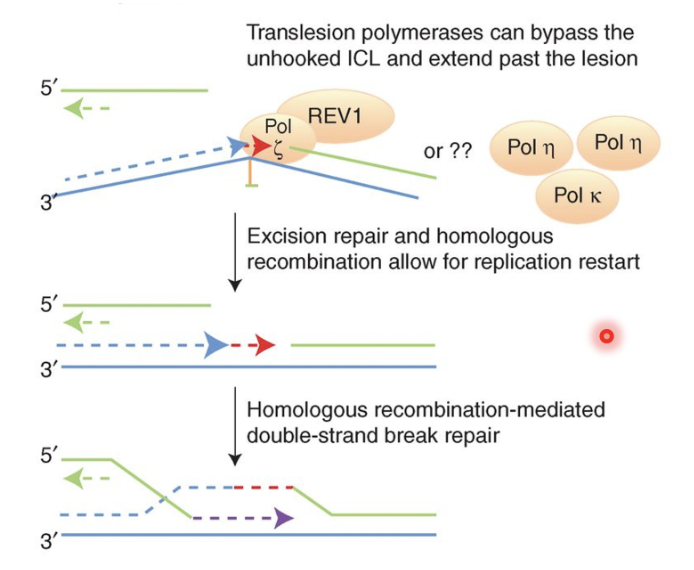
TLS pol ζ recruited to ERCC-1 incised and SNM1A-resented ICL and synthesises DNA past ICL, repairing first chromatid
This produces an intact chromatid and a broken chromatid (DNA DSB) which is repaired by BRCA2 and other HR factors using the intact chromatid as a template
Remnant of the initial crosslink may be removed by NER