Unit 5 - Reactivity
1/98
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
99 Terms
Enthalpy
Heat energy exchange between reaction and its surroundings (thermodynamics)
Breaking Bonds
Absorbs energy
Homolytic Cleavage
Each atom gets an electron when EN is similar
Heterolytic Cleavage
1 atom gets both electrons when EN is different
BDE
Bond breaking corresponds to homolytic bond cleavage
Exothermic
Energy gained by bonds exceeds energy needed for bonds broken, products more stable than reactants
Endothermic
Energy needed for bonds broken exceeds stability gained by bonds formed, products less stable than reactants
Entropy
Molecular disorder
Delta S is positive when
More moles of product than reactant
Cyclic becomes acyclic
If H and S is negative
Spontaneous at low temperatures
If H and S is positive
Spontaneous at high temperatures
Reaction Rates are based on
Concentration of rate law substances
Ea
Temperature
Geometry, orientation, steric
Catalysts
Transition States
Have lifetime bond vibration, high energy state
Intermediates
Have lifetime longer than bond vibration, actually exists
Nucleophile
Electron rich species, lewis bases
Electrophile
Electron deficient, lewis acid
Electron Movement Patterns
Nucleophile attack
Loss of leaving group
Protein transfer
Rearrangement
Nucleophile Attack
Nucleophile (even pi bond) attacks electrophile, sometimes followed by resonance arrows
Loss of Leaving Group
Heterolytic bond cleavage
Proton Transfers
Deprotonation or protonation
Hyperconjugation Level 2
Sigma and p orbital overlap from substitution, increasing stability of carbocation from adjacent sigma bond with empty p orbital
1,2-Hydride Shift
Overlap causes electron to jump when stabilization is better
1,3-Alkyl Shift
Methyl group will jump to other carbon
Carbocation Rearrangement Scenarios
4-5 Ring Expansion
Increase conjugation
Increase degree of carbocations
Elimination Reaction
Reaction with base and pi bond forms
Substitution Reaction
Nucleophile replaces halogen
Concerted Mechanism
Breaking bond and bond to nucleophile happens at the same time
Stepwise Mechanism
Leaving group leaves, then nucleophile attacks
Sn2 Inversion
Stereo centres invert from nucleophile attacks from the backside to get proper arrangement of the nucleophile’s HUMO with electrophile LUMO
More stable transition state
Slow reaction rate
Alpha and Beta Carbons hinder
Backside attack, making it slower
Polarizable
Size of electron cloud, larger cloud = better pulling and nucleophiles
E2 Elimination
Concerted reaction where strong base removes beta proton causing loss of leaving group and double bond
Trans isomers are more
Stable than cis isomers
More substitution in alkenes is more
Stable from hyperconjugation
Regiochemistry
Determines which region reacts
Zaitsev Product
More substituted (unhindered bases)
Hoffman Product
Less substituted alkene (bulky bases)
Stereospecific
Substrate has stereoisomerism and has one stereoisomers as the product
Alkyne and Na NH3
Partial trans alkene
When alpha and beta are chiral
Must consider co planar arrangement using newman projection
Anti Periplanar
Around 180 degrees for E2
If there are 2 beta hydrogens and a chiral alpha centre
2 products will form
Stereoselective
Substrate can make two stereoisomers as products, where 1 is the major product
Sn2 Favours
Primary halides, unhindered neighbours
E2 Favours
Tertiary Halides
Sn1
Loss of leaving group, then weak nucleophile
Solvolysis
Nucleophile is the solvent
E1
Leaving group followed by pi bond formation
Sn1 Reactions produce two different
Configuration, inversion and retention
Sn1 and E1 favours
Tertiary Halides
Sliver in Sn1 and E1 allows
Secondary Halides to go forward
Steps to Predict Mechanism
Determine Reagent
Analyze the substrate and temperature
Consider regio and stereochemistry
Alkyl Sulfonates
Alternatives to alkyl halides with sulphur but more stable and resonance stabilized
Alcohols and Acids
OH reacts to make H2O and CB replaces it
Aprotic Solvents speed up
Sn2 reactions because aprotic molecules have a buried positive charge, decreasing stabilization of nucleophile and less Ea
Protic Solvents speed up
Sn1 because protic solvents have exposed positive charge which stabilizes the nucleophile and carbocation but create a greater Ea
If base and nucleophile is strong
At cold, secondary Sn2
At cold/rt primary Sn2
At hot, tertiary E2
If base is weak but nucleophile strong
Sn2
If base and nucleophile weak
In hot, tertiary E1
In cold/rt, tertiary Sn1
In sliver, secondary E1 or Sn1
If strong acid
Primary Sn2
If hot, poor nucleophile secondary or tertiary E1
If good nucleophile, secondary or tertiary Sn1
Addition and Elimination Reactions are
Opposite equilibrium of each other
At low temperatures
Enthalpy dominates and addition reactions are favoured
At high temperatures
Entropy dominates, and elimination reactions are favoured
Hydrohalogenation
Addition of H-X to alkene and the major Markovnikov product is formed from carbocation stability
Markovnikov Addition
The more substituted carbon gets the nucleophile
Anti Markovnikov Addition
Less substituted carbon gets the nucleophile in the presence of oxygen
Why can hydrohalogenation create both chiral centres
Nucleophile can attack either p orbital
Acid Catalyzed Hydration
Water is added across alkene following Markovnikov regioselectivity of 50/50 arrangement
If making alcohols
Use excess water
If making alkene
Use concentrated acid with no water added
Oxymercuration Demercuration
Follows Markovnikov addition of OH, but no rearrangement occurs due to HgAcO
Oxymercuration Demercuration Steps
HgAcO bounds to least substituted alkene
Binds to both carbons and water, replaced the more subbed carbon
It gets deprotonated and HgAcO reduced
Halogenation
Stereoselective anti addition of Cl or Br across C=C bond
Anti Addition makes
Trans products
Syn Addition makes
Cis products
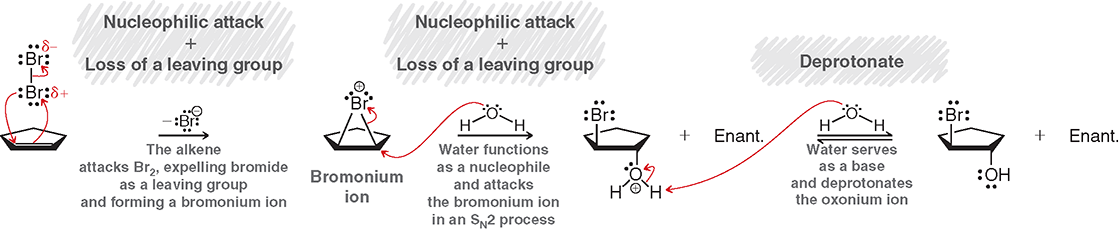
Halohydrin
Halogenation in water creates an anti addition where the OH is added to the more substituted carbon and halide the less
Halohydrin Steps
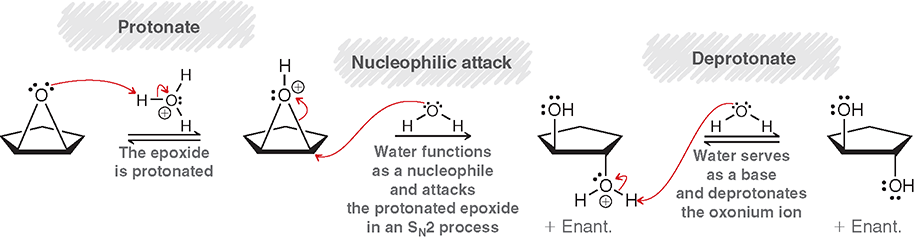
Dihydroxylation
Anti addition of OH and OH across pi bond in peroxyacid and O2 acts like an electrophile
Dihydroxylation Steps
If carbocation
Markovnikov addition
If no carbocation
Anti markovnikov addition
Hydroboration Oxidation
Adds H and OH with anti Markovnikov regioselectivity but syn addition
Alkene Catalytic Hydrogenation
Addition of H2 on an alkene with a metal catalyst through syn addition because metal surface delivers H on the same side
Syn Dihydroxylation
Adds 2 OH in a concerted syn fashion with OsO4 and NMO as co-oxidant to redox OsO4
Ozonolysis
Concerted process with O3 and DMS to cleave alkene into two carbonyls
Geminal Dihalide
Halides are bonded to same carbon
Vicinal Dihalide
Halides are bonded to different carbons
NaNH4 used to
Deprotonate and shift the equilibrium of alkyne to neutralize them
Alkyne Acid Catalyzed Hydration
Alkyne goes through an enol through Markovnikov addition, then turned into a carbonyl with H2O
Tautomerization
Different location of pi and H bonds
Alkyne Hydroboration Oxidation
Anti Markovnikov addition for enol, then undergoes resonance
Enol
Alkene with an alcohol
Alkyne Catalytic Hydrogenation
Alkyne turns into alkane with metal to make a cis alkene first then trans
Lindlar’s Catalyst
Deactivated catalyst that only lowers EA for first H2 addition to make an alkene only
Dissolving Metal Reduction
Stereoselectively reduces alkyne into a trans alkene with Na and NH3
Alkylation of Terminal Alkynes
Alkynes lengthened with NH2 to deprotonate and a halide added on
For Terminal Alkynes, Markovnikov hydration makes a
Ketone
For Terminal Alkynes, Anti Markovnikov hydration makes an
Aldehyde