H4 Jarvis- The technology underpinning molecular biology: gene analysis
1/7
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
8 Terms
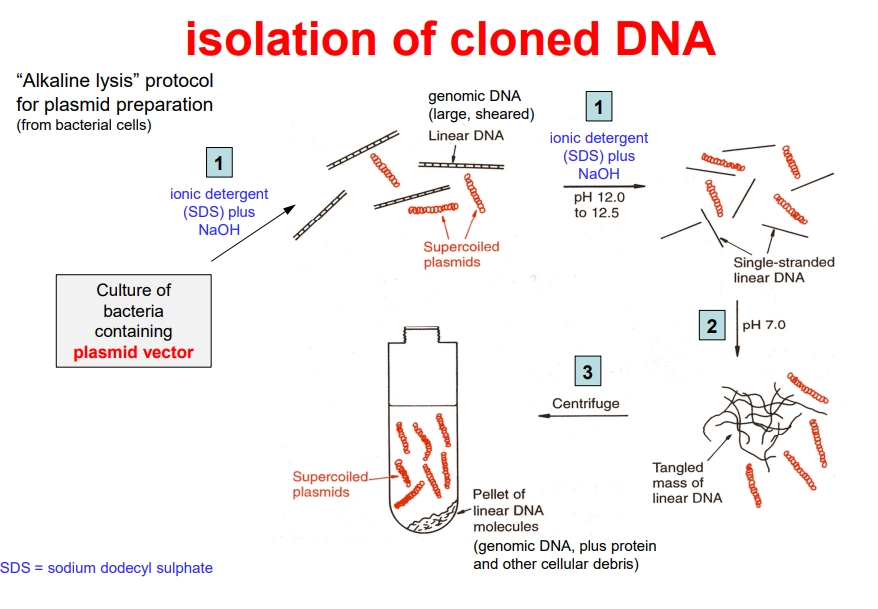
how can we isolate recombinant bacterial plasmids for sequencing?
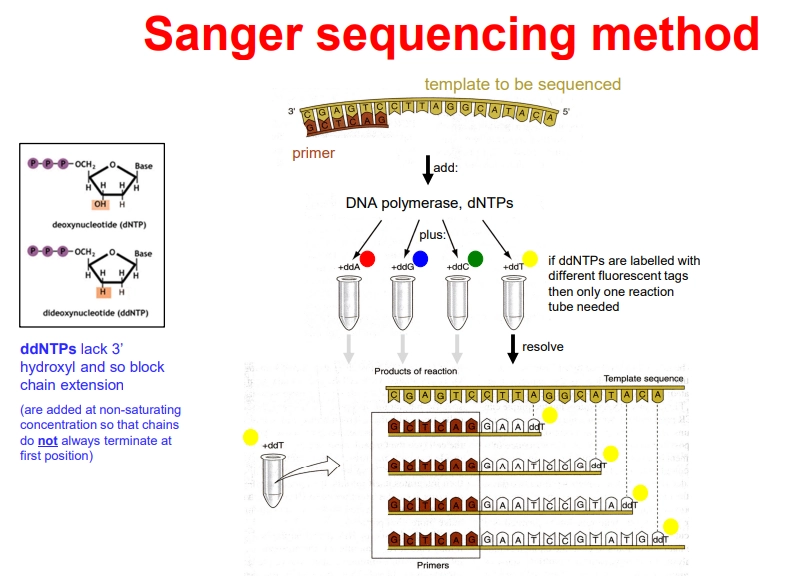
how can we sequence DNA using sanger sequencing?
PCR is now carried out in 1 tube, containing the DNA template, a buffer, DNA primers, an excess of dNTPs, DNA Taq polymerase, and differently fluorescently tagged 2’,3’-dideoxynucleotides (ddNTPs)
the primer anneals adjacent to the initiation start and DNA polymerase elongates the strand
when ddNTPs are incorporated, they terminate elongation because they have no 3’ OH to attack the next phosphate and form the phosphodiester bond
they are not added at saturating concentrations, so that termination occurs randomly at every possible position
this produces truncated strands with fluorescent tags that can be automatically detected using laser detection to produce a chromatogram
originally, the reaction had to be carried out in 4 different tubes, one for each ddNTP, because the fragments were separated by electrophoresis and detected using a radioactive primer
this process is limited to ~900 nucleotide sequences due to smaller relative fragment size differences, but is still used for small projects due to convenience and accuracy
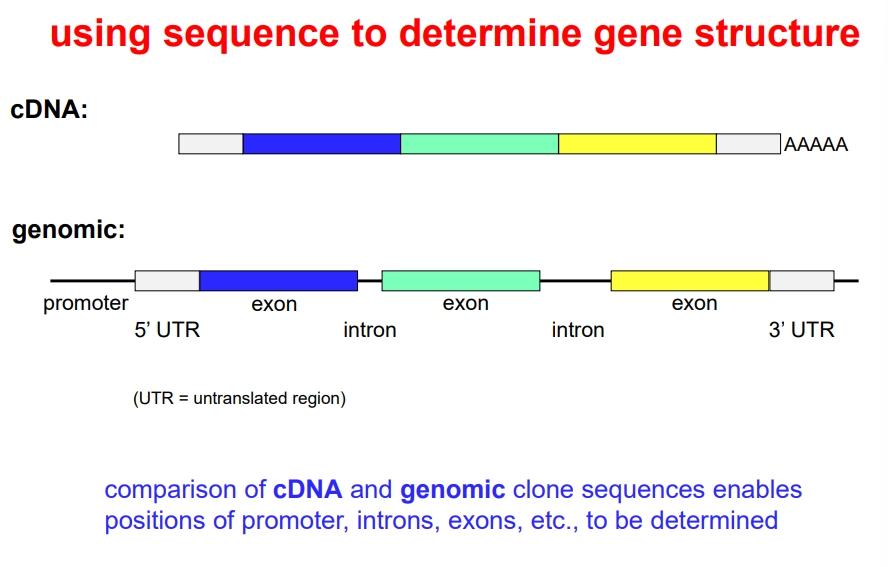
how can we use DNA sequencing to determine gene structure?
sequencing the cDNA and genomic clones can be used to determine the position of exons, introns and UTRs by comparison
how can we estimate the number of copies of a gene that are present in a chromosome?
by southern blotting
when the probes are added to the nitrocellulose they will hybridise to any fragment that contains the gene of interest
this will reveal separate bands due to differing total fragment sizes (after radioactive/fluorescent/enzymatic detection)
the bands can be counted to determine the number of copies of the gene in the chromosome
weaker bands indicates divergent copies of the genes
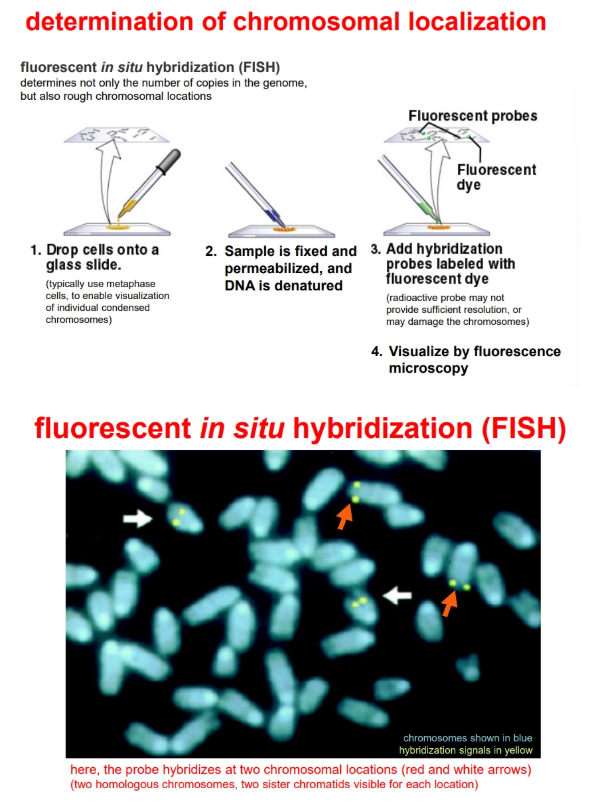
the copy number and rough chromosomal location can be determined by fluorescent in situ hybridisation (FISH)

how can we determine chromosome localisation of a gene?
by fluorescent in situ hybridisation (FISH)
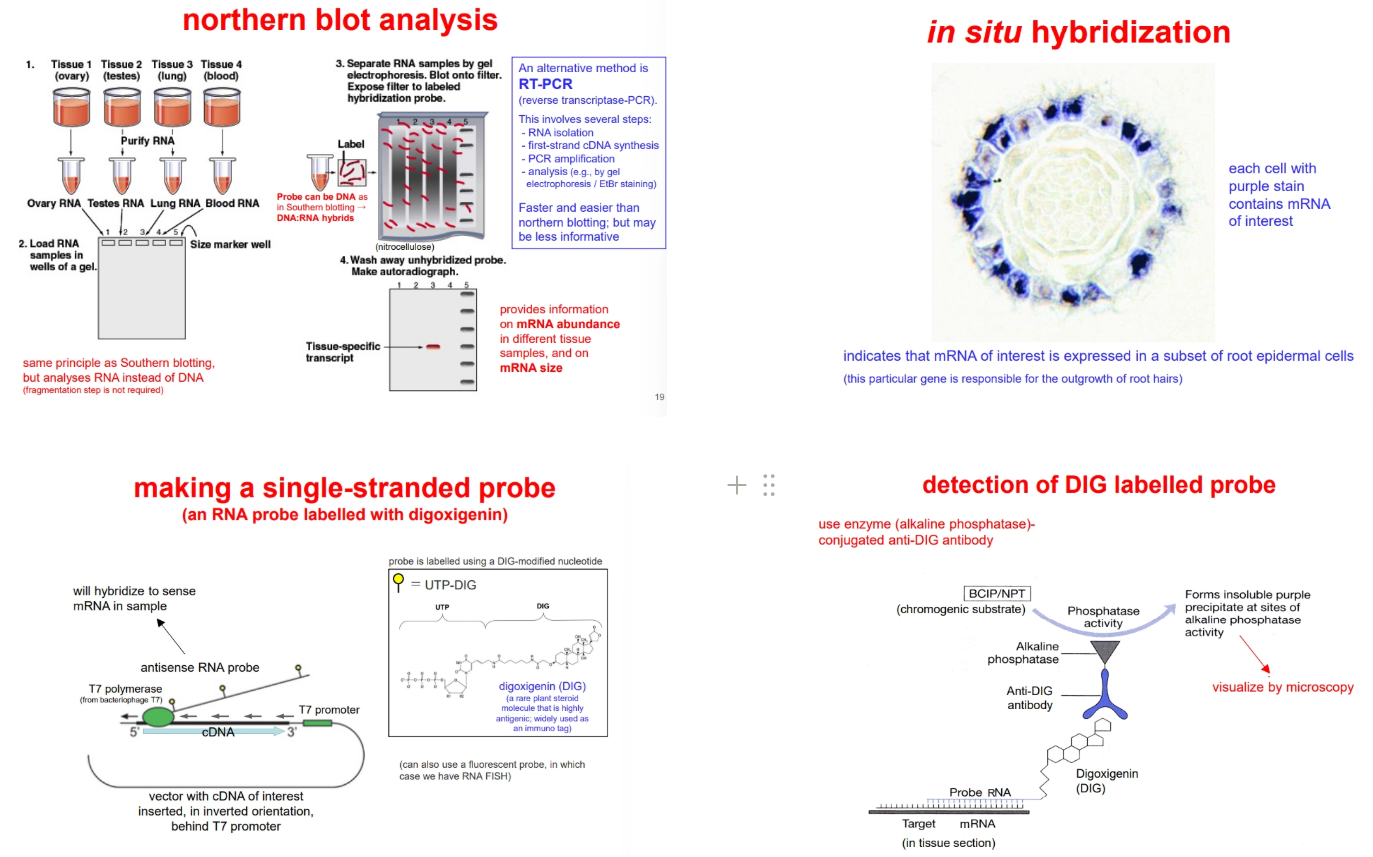
how can we determine when and where a gene is expressed?
to determine what tissues produce a gene (including at different points of development/under different conditions), we can use:
northern blotting (derived from southern blotting, but using the mRNA instead of fragmented chromosomal DNA, radioactive detection is used instead of fluorescence)
reverse transcriptase-PCR (RT-PCR) and electrophoresis
to determine what cells within a tissue express a gene, we can use:
in situ hybridisation of a single-stranded probe (more sensitivity, because it won’t re-anneal to itself after denaturation) to fixed thin sections of tissue
to produce the probes, the cDNA for our gene of interest is inserted into a plasmid vector in an inverted orientation, such that transcription of the vector produces an antisense RNA probe (no sense probe strand present = no re-annealing)
during transcription of this probe, UTP-DIG is incorporated (UTP bound to digoxigenin)- this is detected in the tissue section using an antibody attached to alkaline phosphatase, which converts a substrate from colourless to purple (detected under a microscope)
this isn’t the same as detecting where the protein itself is produced, as genes are often regulated post-transcriptionally
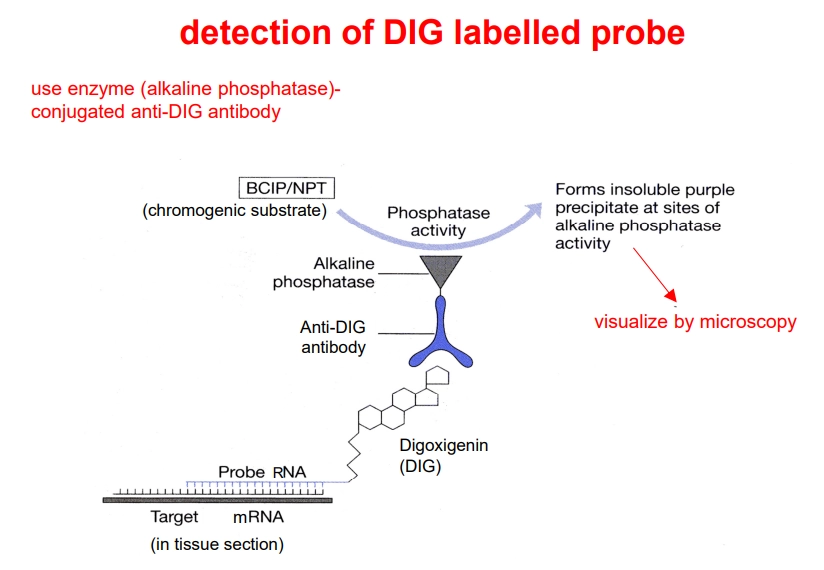
how can we produce single-stranded RNA probes?
these are used during in situ hybridisation (to determine what cells within a tissue express a certain gene)
the cDNA for our gene of interest is inserted into a plasmid vector in an inverted orientation, such that transcription of the vector produces an antisense RNA probe
this will hybridise to the sense mRNA, without the need for denaturation, like with a double-stranded probe (absenc2e of sense probe strand prevents the probe re-annealing to itself, decreasing noise)
during transcription of this probe, UTP-DIG is incorporated (UTP bound to digoxigenin)
this can be detected using an anti-DIG antibody conjugated to alkaline phosphatase, which can convert a colourless chromogenic substrate into a purple precipitate which can be seen through a microscope
how can we determine when and where a protein is produced?
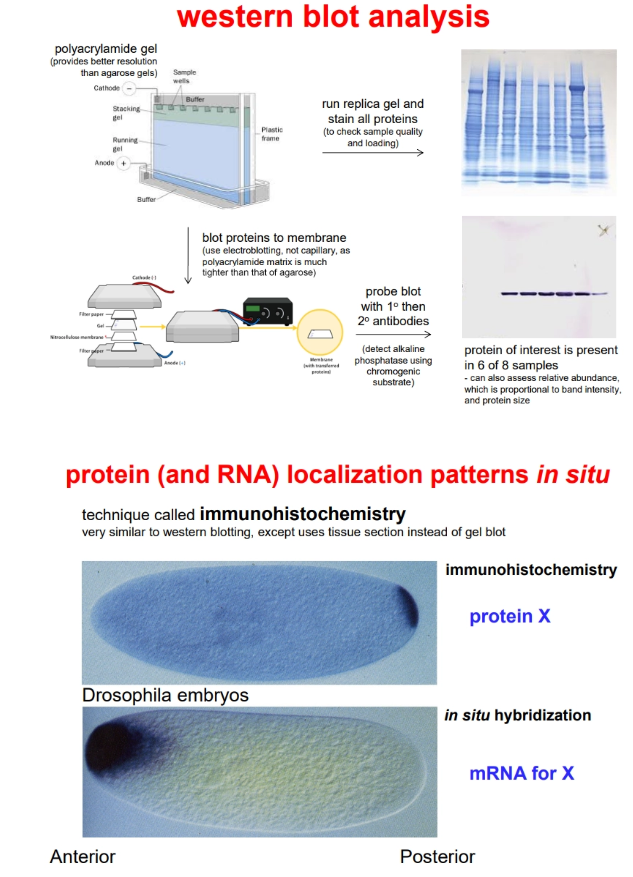
to determine what tissues accumulate a protein, we can use western blotting (derived from southern blotting, but using the protein instead of DNA, polyacrylamide gel instead of agarose, and electroblotting instead of capillary blotting)
to determine where a protein accumulates within a cell/tissue, we can use immunohistochemistry (similar to in situ hybridisation)
detection requires antibodies for the protein of interest:
a primary antibody detects the protein- this is produced by immunising an animal using the bacteria-produced protein (with a hexahistidine tag attached for purification)
a secondary antibody, conjugated to alkaline phosphatase, detects the primary antibody (this allows for signal amplification/sensitivity and use of a single reagent for multiple protein targets)
alkaline phosphatase converts a colourless chromogenic substrate to a purple precipitate (detected under a microscope)