826b med chem exam 2
1/187
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
188 Terms
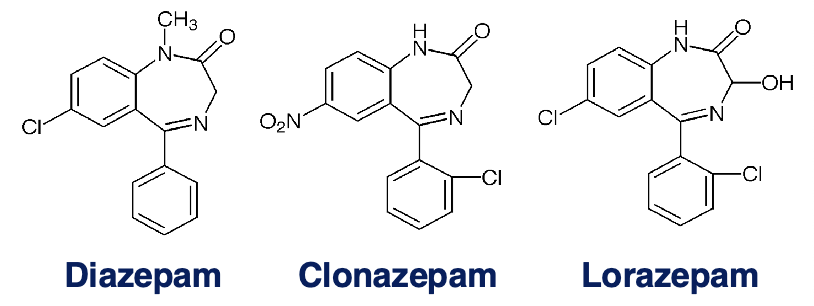
benzodiazepines interaction with alpha 1 subunit chloride ion channel cause
sedation
temazepam, triazolam
benzodiazepines interaction with alpha 2 subunit of chloride ion channel produces
anxiolytic
alprazolam
anti-anxiety
panic disorder
clonazepam
NO2 is EWG on position 7= hypnotic activity
Cl is EWG at R2’ in aryl ring= increase activity
lorazepam
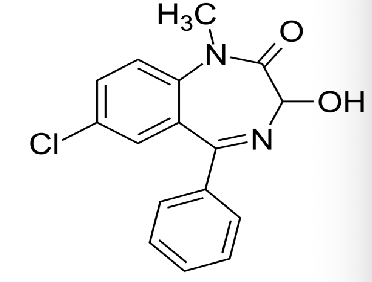
3-hydroxy benzodiazepines
diazepam
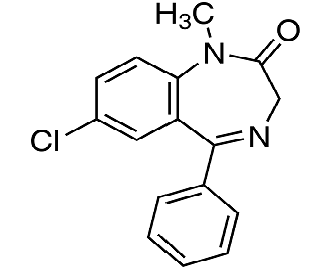
tempazepam
3-hydroxy benzodiazepines
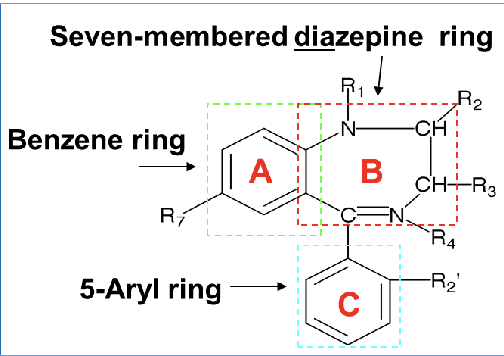
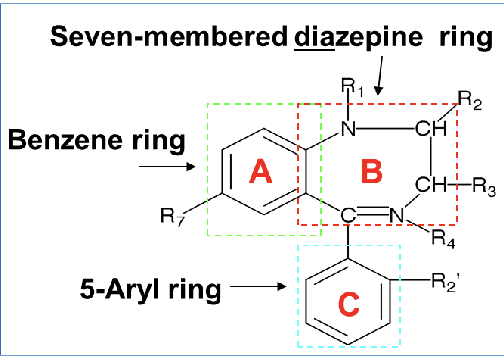
SAR of benzos
R1 on 7 member diazepine ring: substituent here does not affect activity
R2(keto group): O, S, N→ interacts with H-bonding donor groups on receptor
R3: alkyl substitution(methyl,ethyl,etc) decreases activity
R4: usually not substituted(except chlordiazepoxide)
if 4,5 double bond is saturated or moves to 3,4 position→ decreases activity
R5:
5-aryl or 5-cyclohexenyl group = CNS depressant
phenyl group promotes activity
EWG(halogen) at R2’ of aryl ring increase the activity(lorazepam, flurazepam)
R7: EWG(halogen or nitro group) is required for hypnotic activity
flumazenil
antidote for benzo
imidazobenzodiazepine derivative
no aryl substituent at R5
no 4,5 double bond
methyl group at R4
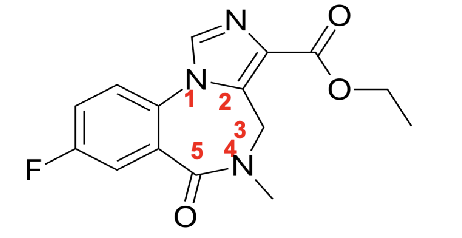
benzodiazepines used for sedation/hypnotics
flurazepam
tempazepam
estazolam
triazolam
quazepam
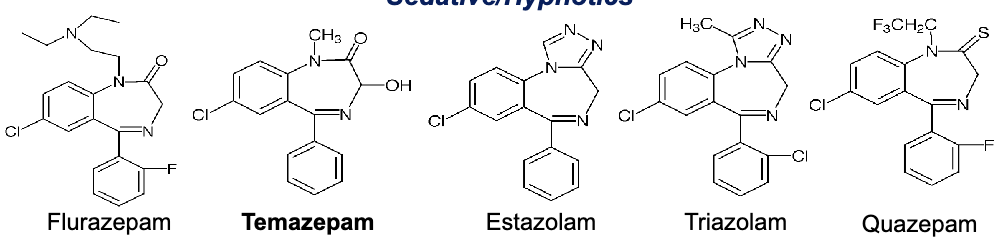
benzodiazepines for panic disorder
alprazolam
clonazepam
benzodiazepines for anti-anxiety
alprazolam
lorazepam
oxazepam
diazepam
chlordiazepoxide
clorazepate
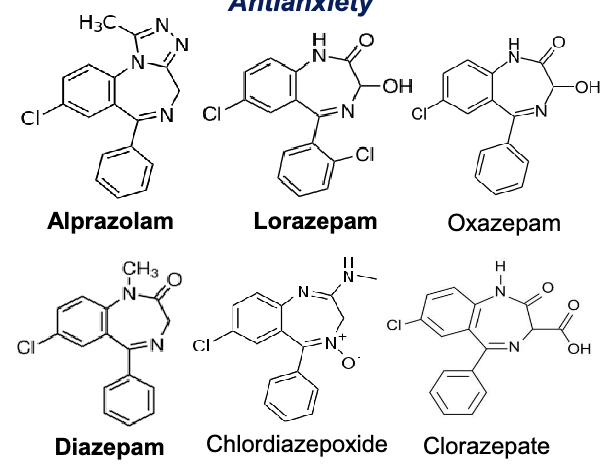
benzodiazepines for anticonvulsants
diazepam
lorazepam
clonazepam
clorazepate
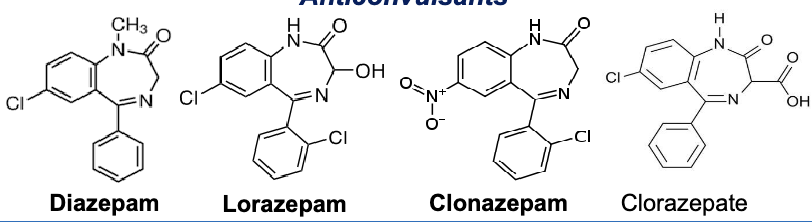
benzodiazepines for alcohol withdrawal
chlordiazepoxide
clorazepate
diazepam
oxazepam
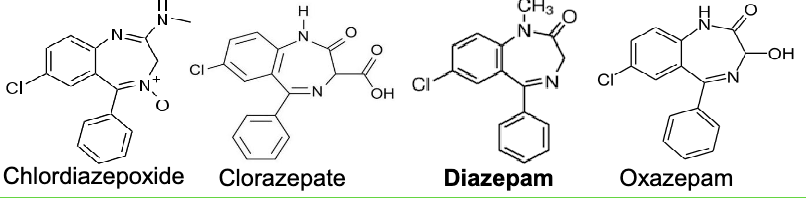
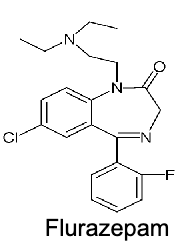
flurazepam
long acting benzodiazepine
partial agonist
short-term treatment of patients with anxiety induced insomnia
residual hangover effects(sleepiness, impaired psychomotor and cognitive function)= increase risk of falls and hip fractures in elderly
CYP3A4 metabolism→ N-1-hydroxylation →N-dealkyation → 3-hydroxylation(inactive metabolite)
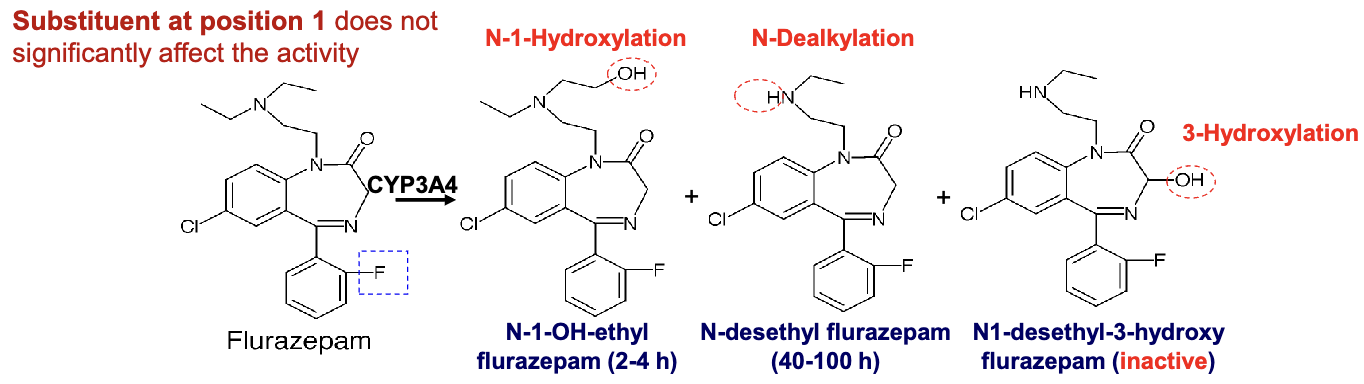
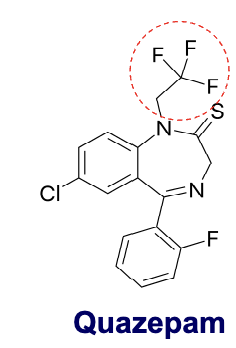
quazepam
triflurorethyl benzodiazepine
selectively targets GABA-A receptor( alpha 1)= little to no muscle relaxant properties
S atom increases lipid solubility
two active metabolites
MOA similar to zolpidem and zaleplon
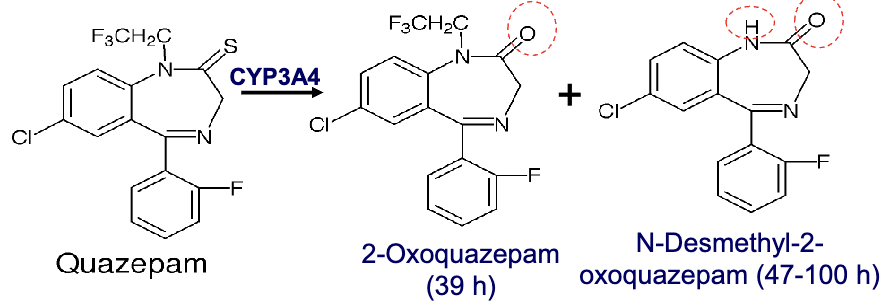
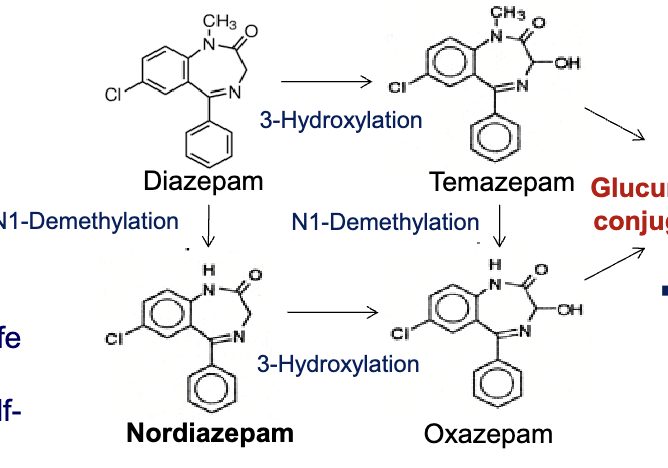
diazepam
3-hydroxylation to Temazepam → N1-demethylation to oxazepam
N1-demethylation to Nordiazepam → 3-hydroxylation to oxazepam
temazepam and oxazepam are largely eliminated by glucuronidaton
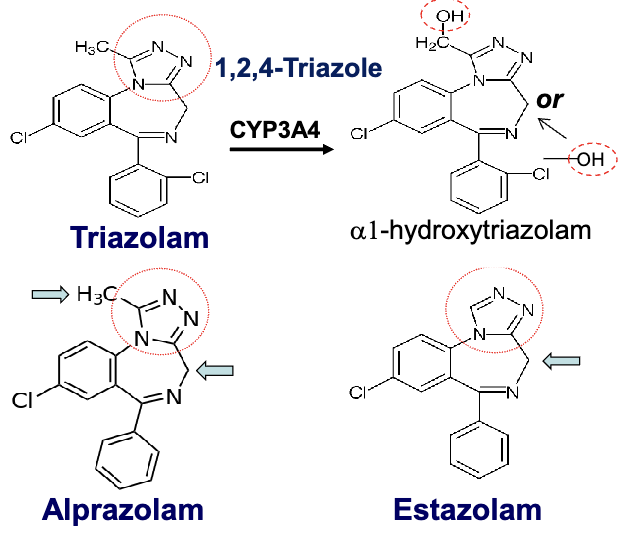
triazolo-benzodiazepines
1,2,4 triazole ring fused to the benzodiazepine core at the 1,2 position= increased stability of the drug
triazolam: short acting
alprazolam: intermediate acting
estazolam: intermediate acting
oxidized metabolites are inactive due to glucoronide conjugation
fused imidazo-benzodiazepine
imidazole ring fused to benzodiazepine core at 1,2 position= increased stability of the drug
midazolam: short acting
metabolized by CYP3A4 to alpha 1 hydroxytriazolam followed by glucuronide conjugation
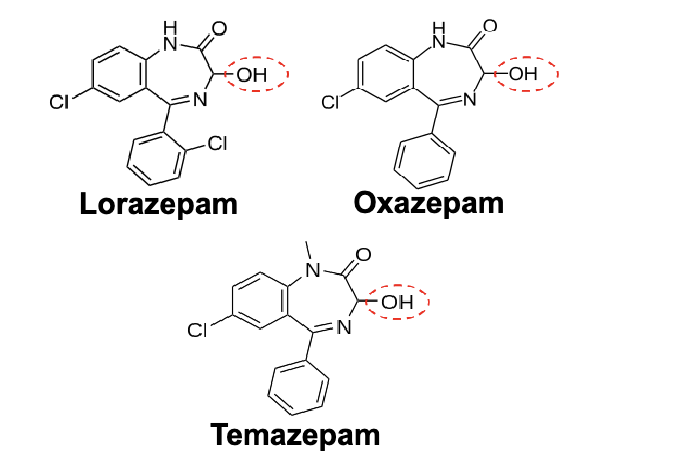
3-hydroxy- 1,4 benzodiazepines
“LOT”- metabolized via direct conjugation with glucuronide, more rapid than oxidation and no active metabolites→ glucuronide conjugation at 3-hydroxyl group
lorazepam
oxazepam
temazepam
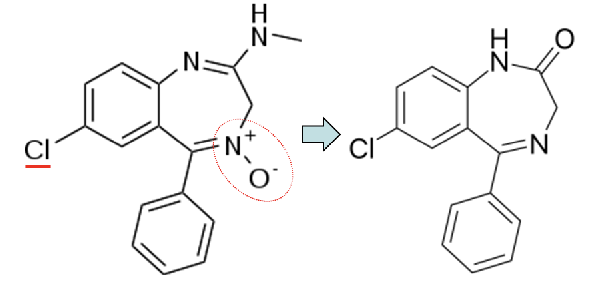
what benzodiazepine has a N-oxide in the R4 position
chlordiazepoxide
has increased activity due to the Cl (EWG) at R7
Z compounds
zaleplon
zolpidem
eszopiclone
show rapid onset of action due to being very lipophilic, rapid absorption and then rapid onset of action
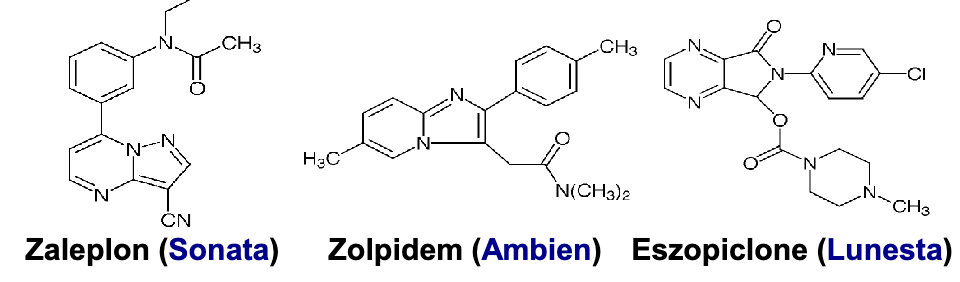
zaleplon pharmacophore and subtype selectivity
pharmacophore: pyrazolopyrimidine?
pyrazolo-pyrimidine heterocycle ring
cyano group(CN) on pyrazole ring
N-ethylacetamide group on upper benzene ring
subtype selectivity
alpha 1» alpha 2=alpha 3
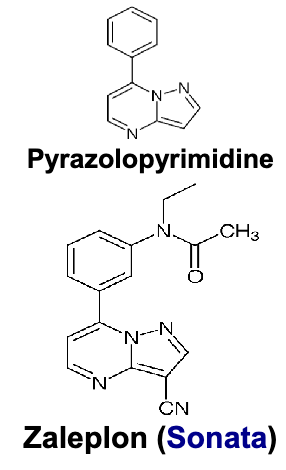
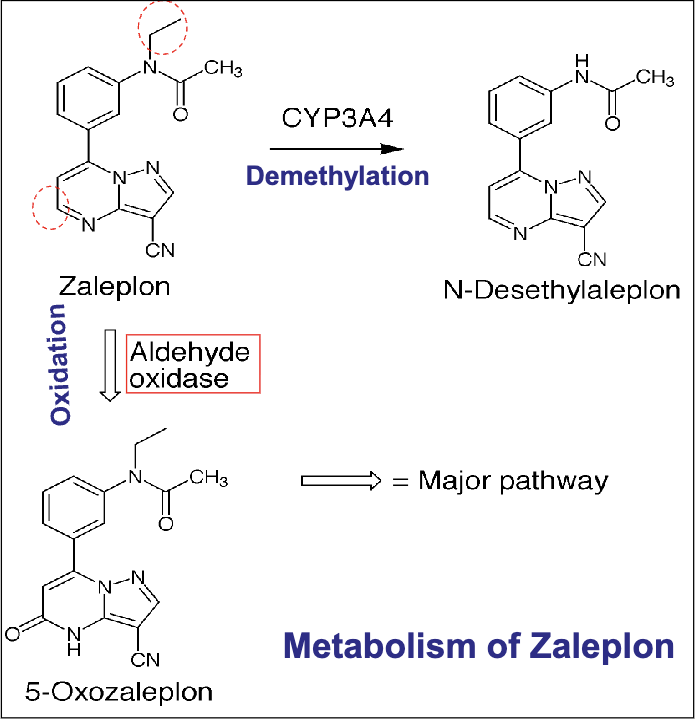
zaleplon
full agonist for type 1 benzo alpha 1 receptor
good at sleep initiation→ not sleep maintenance
no significant rebound insomnia
all metabolites are inactive(demethylation, oxidation(major pathway))
zolpidem pharmacophore and subtype selectivity
pharmacophore
imidazopyridine ring
amide/acetamide group attached to imidazole ring
subtype selectivity
alpha 1»alpha 2=alpha 6
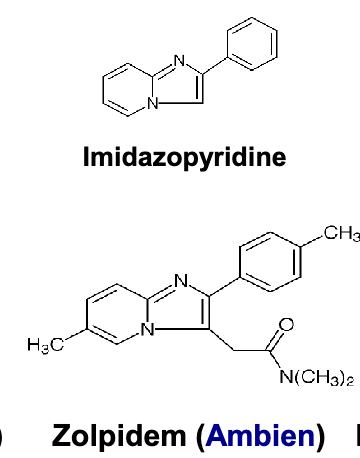
zolpidem
higher affinity to type 1 GABA a receptors containing alpha 1 in the brain→ strong hypnotic activity
rapid onset dur to rapid absorption through GI due to weak base and high lipophilicity
good at sleep maintenance
inactive metabolites
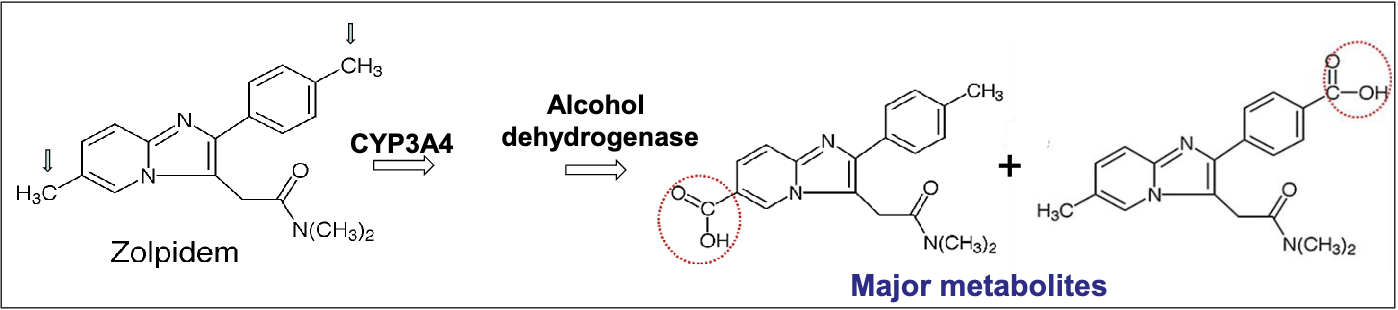
eszopiclone pharmacophore and subtype selectivity
pharmacophore
pyrrolopyrazine derivative
cyclopyrrolone ring
Cl on the pyridine ring
subtype selectivity
not selective for alpha 1
eszopiclone
S- enantiomer of zopiclone
not specific for alpha 1
effective in chronic insomnia
longer elimination half life than other Z drugs
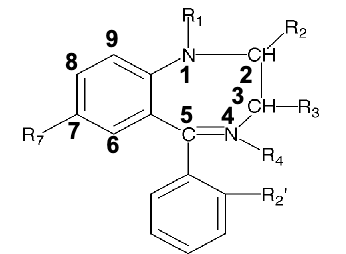
Which of the following statements concerning SAR of benzodiazepines is correct?
A. bulky alky substitutions at R1 increase activity
B. an EWG at R2’ of aryl ring increases activity
C. O, S, or N substituents at R2 increase lipophilicity of benzos
D. substitution at R6 is required for sedative activity
E. the saturation of 4,5 double bond increases sedative activity
B. an EWG at R2’ of aryl ring increases activity

blood coagulation
extrinsic(damage to endothelial tissue), intrinsic(injured endothelium of damaged BV) and common pathways→ interact to form stable clot
lots of tissue factors that meet at activating factor X(common pathway)
platelet activation: changes in morphologic and biochemical state of platelet→ the bind and stick together
fibronolysis
break down of clots→ tPA is protein that helps this
tPA binds to plasminogen→ plasmin
plasmin leads to break down of the clot
anticoagulants
blood thinners→ slow down clotting
reduce fibrin formation and prevent clots from forming and growing
anticoagulant drugs
VKORC1 inhibitor: warfarin
indirect thrombin and Xa inhibitor: heparin, LMWH/enoxaparin, fondaparinux
direct thrombin inhibitors: agatroban, daigatran, bivalirudin
factor Xa inhibitors: rivaroxaban, apixaban, edoxaban, betrixaban
antiplatelets
prevent platelets from clumping→ in turn prevents forming and growing of clots
antiplatelet drugs
COX inhibitor: aspirin
PAR-1 antagonist: vorapaxar
PDE inhibitors: dipyridamole, cilostazol
P2Y12 receptor antagonists: ticlopidine, clopidogrel, prasugrel, cangrelor, ticagrelor
GpIIb/IIIa receptor antagonist: abciximab, eptifibatide, tirofiban
thrombolytics(fibrinolytics)
breakdown/ lyse blood clots already formed within blood vessels
increase plasmin
thrombolytic drugs
tissue plasminogen activator: tPA/Alteplase
Reteplase
tenecteplase
streptokinase, urokinase
dicoumarol
naturally occuring anticoagulant that is derivative of coumadin
replaced by simpler derivative warfarin
is cause of naturally occurring bleeding disease in cattle
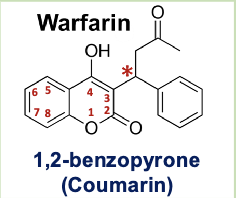
warfarin
coumarin derivative
racemic mixture: S is more potent than R
MOA: competitively inhibits VKORC1 limiting production of carboxylated vitamin K dependent blood clotting factors
pharmacophore: 1,2 benzopyrone
indications for use: treating blood clots, preventing stroke
heparin
sulfated polysaccharide= glycosaminoglycan
potentiates actions of antithrombin (AT)
inhibits fXa and thrombin to similar extent
low molecular weight heparins
enoxaparin, dalteparin, tinzaparin
inhibit factor Xa via AT with much less inhibition of thrombin that heparin
greater inhibitory action of fXa
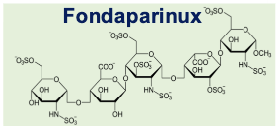
fondaparinux
synthetic anticoagulant→ based on pentasaccharide sequence making up the minimal AT binding region of heparin
antithrombin-dependent pentasaccharide
higher anti-Xa activity
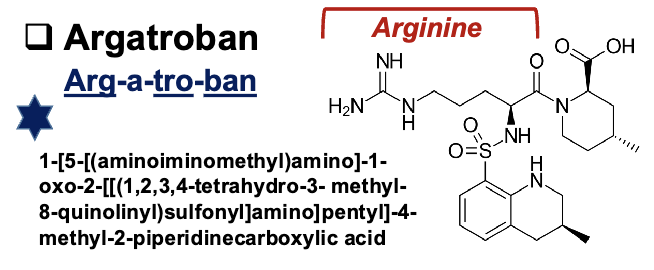
direct thrombin inhibitor drugs
agatroban
dabigatran
bivalirudin
hirudin
agatroban
synthetic direct inhibitor derived from amino acid Arginine of fibrinogen
binds into thrombin active site
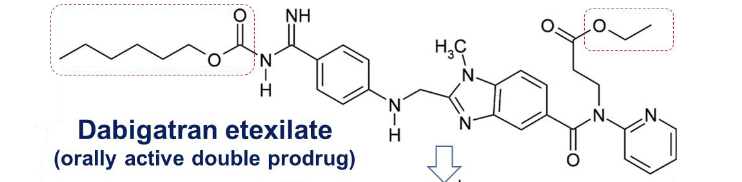
dabigatran etexilate
orally active doube prodrug due to ethyl ester and hexyloxycarbonyl carbamide side chains
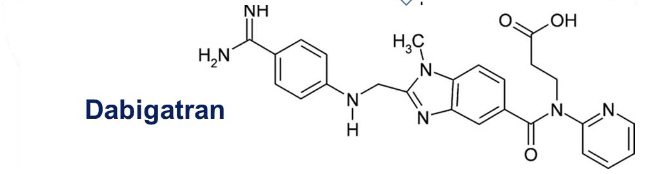
dabigatran
contains alpha-NAPAP
serine proteases inhibitor
bivalirudin
20 AA peptide containing active site thrombin inhibitor (D-Phe-Pro-Arg)
competitive and reversible inhibitor of thrombin→ prevents major bleeding events
function of factor Xa
activated fX assembles with Va to form prothrombinase→ cleave prothrombin to produce active thrombin
inhibition of Xa activity is believed to be more efficient approach to anticoag than direct
wider therapeutic window and decrease risk of rebound thrombosis
direct Xa inhibitors
L shaped molecules
rivaroxaban
apixaban
edoxaban
betrixaban
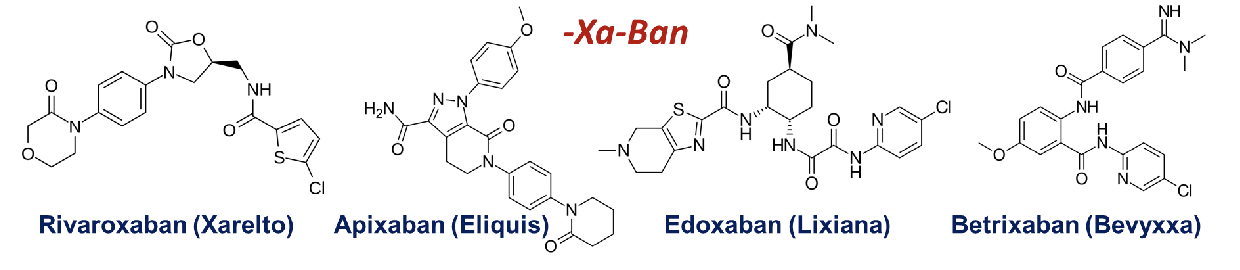
rivaroxaban
high affinity to factor Xa and good oral bioavailability
no substitutions on benzene ring=highest potency
morpholinone group= important role in binding affinity and bioavailability
chiral center (S)- in oxazolidine ring
S enantiomer is only with pharmacological activity
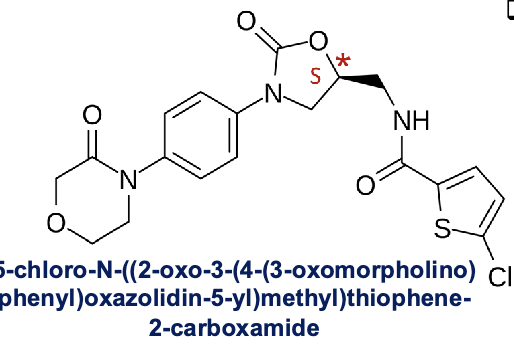
SAR of apixaban
pyrazolopyridine w/ these groups:
S1 pocket: (P1)p-methoxyphenyl stablizes complex through interaction with non-active site, (P3)carbonyl oxygen of pyrazolopyridine interacts with Gly-216 as well as H2O molecule, (P2) carboxamide group contributes to greatest binding and similar clotting activity with favorable PK among others
S4 pocket: (P3)-pyrazole N-2 nitrogen atom interacts with AA Glutamine-192, (P4) lactam analogues have very high binding affinity in this pocket and selectivity vs other proteases
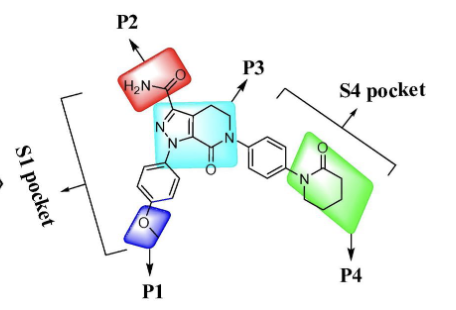
molecular targets for antiplatelet agents
cyclooxygenases
P2Y12- clopidogrel, prasugrel, ticlopidine, ticagrelor, cangrelor
Gp alpha-IIb beta3- abciximab, tirofiban, eptifibatide
phosphodiesterases- dipyridamole, cilostazol
PAR1- vorapaxar
thromboxane A2(TXA2)
produced by activated platelets
has prothrombotic properties→ stimulates activation of new platelets and increasing platelet aggregation
action is mediated through binding to TP receptors
aspirin
irreversible inhibitor of COX activity→ acetylation of serine residue in active site(ser 529 in cox1, ser 516 in cox2)
“suicide inhibitor”
works by the same mechanism to block cox2 in other tissues
vorapaxar
PAR-1 antagonist
PAR-1 GPCR which is activated by cleavage of part of their extracellular domain by thrombin in the platelet → activation of platelet
drug works by binding to PAR1’s ectodomain→ antagonizes and doesn’t allow for cleavage
synthetic derivative of himbacine(piperdine alkaloid)
anticoagulants that target PDE can -
increase the concentration of cAMP or cGMP in platelets
PDE normally catalyzed and decreased the level→ regulating platelet function, this inhibits PDE allowing for more cAMP and cGMP to be available to inhibit platelet functions
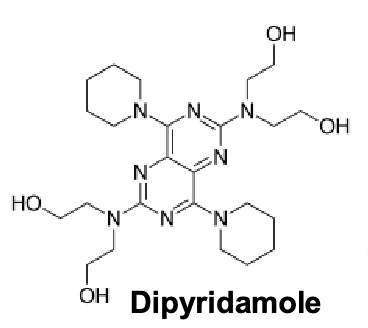
dipyridamole
inhibits PDE5 and PDE3
member of piperdines, pyrimidopyrimidine, tertiary amino compound and tetrol
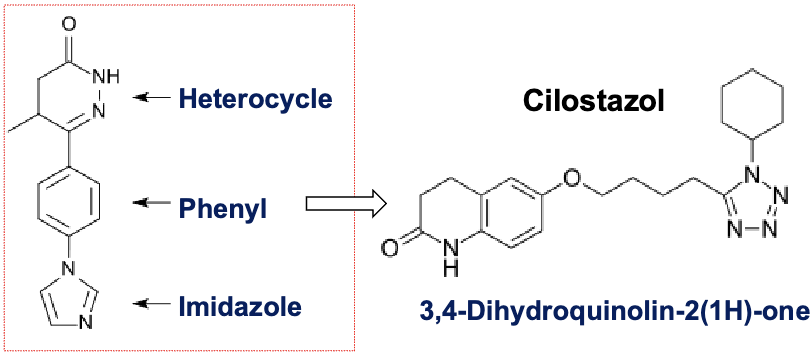
cilostazol
selective PDE3 inhibitor
increase in cAMP increase active form of protein kinase A(PKA)→ directly related to inhibition of platelet aggregation
contains heterocycle, phenyl and imidazole ring
role of ADP receptor (P2Y12) in platelet activation
when platelets activated by collagen or thrombin→ release ADP from dense granules
ADP binds to P2Y12 receptor and activated receptors on the platelet membrane→ leads to change in platelet shape and results in amplified and sustained aggregation of platelets
thienopyridine derivatives
selective, irreversible ADP receptor/P2Y12 inhibitors
ticlopidine
clopidogrel
prasugrel
all prodrugs that are activated by cytochrome p450
ticlopidine
clopidogrel
prasugrel
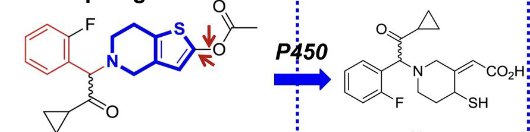
developed by replace the ester group of clopidogrel with a metabolically stable ketone and adding a ester group at the thiophene 5-position
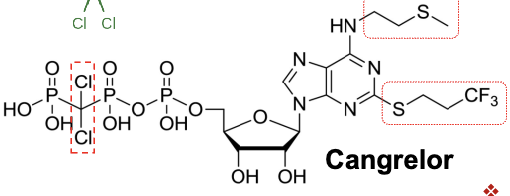
cangrelor
nucleotide analogue→ chemical structure that resembles ATP→ natural antagonist at P2Y12 receptor
IV non prodrug
reversible antagonist, superior to the irreversible P2Y12 antagonist
replacement of anhydride oxygen between Ph-beta and Ph-gamma w/ dichloro methylene to keep potency of ATP with similar pKa to avoid metabolism to proaggretory ADP
addition of S-propyl at the purine moiety- enhanced affinity
methylsulfanylethylamino group at C6 position can lead to a tenfold increase in activity
trifluoropropylsulfanyl group at C2 enhance activity
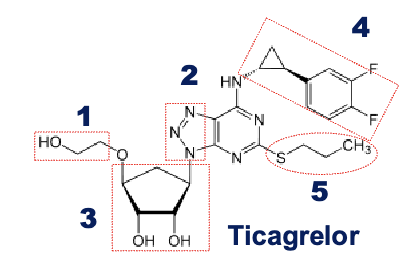
ticagrelor
triazolopyrimidine- adenosine isostere
cyclopentane ring is similar to ribose and the 1,2,3 triazolo- 4,5-d pyrimidine moiety resembles nucleobase adenine
PO non prodrug
SAR of ticagrelor
replacement of Ph chain with a 2-hydroxyethoxy group to make the drug reversible and orally available→ 300 fold reduction in potency
changing the purine with triazolopyrimide to bring potency to the same level
replacement of ribose with cyclopentyl group to avoid chemical instability of the glycosidic bond
addition of phenyl cyclopropylamine substituents in the 6 position to offer high affienities to P2Y12 receptor, introduction of fluorines at the phenyl ring leads to further improved metabolic stability
variation of C2 minor impact on activity, improves PK properties→ ex. thioether alkylchain
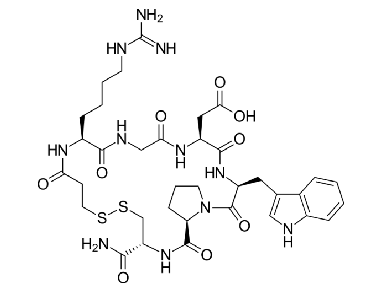
eptifibatide
glycoprotein IIb/IIIa inhibitor
cyclic heptapeptide derived from disintegrin protein found in snake venom (Barbourin)
disintergrins work by inhibiting the clumping of platelets
tirofiban
small molecule inhibitor of the protein-protein interactin between fibrinogen and GpIIb/IIIa→ developed from pharmacophore based virtual screen lead
it is an arginylglycylaspartic acid (RGD) mimetic
fibrin-specific thrombolytics
alteplase- recombinant form of tPA(serine protease found on endothelial cells)
reteplase- recombinant plasminogen activator
tenecteplase(TNKase)- recombinant tPA w/ high fibrin selectivity
tPA- alteplase
substrate is plasminogen
tPA specifically acts on plasminogen to convert it into the active enzyme plasmin→ crucial for breaking down blood clots
reteplase
recombinant non-glycosylated form of human tissue plasminogen activator
also binds to fibrin with lower affinity than alteplase→ improving ability to penetrate into clots without retention within fibrin= enhanced fibrinolytic activity will be achieved
tenecteplase
tetra-alanine substitution makes it resistant to inhibition by PAI-1
administered IV as a single bolus
greater fibrin specificity than alteplase
urokinase
non-fibrin-specific agent to catalyze systemic fibrinolysis
present in blood and in extracellular matrix of many tissues
not tPA derivative
Which of following statements is not correct regarding heparin and its derivatives?
A. Enoxaparin has a preferential and longer lasting effect on factor Xa compared to heparin.
B. Heparin shows more inter-patient variability than others.
C. Enoxaparin is a synthetic anticoagulant based on the pentasaccharide sequence.
D. Fondaparinux has no direct effect on thrombin.
C. Enoxaparin is a synthetic anticoagulant based on the pentasaccharide sequence.
Which of following drugs is not a direct factor Xa inhibitor?
A. Rivaroxaban
B. Apixaban
C. Edoxaban
D. Agatroban
D. Agatroban
Which of following ADP receptor antagonists is not a prodrug?
A. Clopidogrel
B. Cangrelor
C. Prasugrel
D. Ticlopidine
B. Cangrelor
Which of following thrombolytic drugs is not a derivative of Tissue plasminogen activator (tPA)?
A. Reteplase
B. Urokinase
C. Tenecteplase
D. All of them
B. Urokinase
drugs approved for spasticity
diazepam: benzo
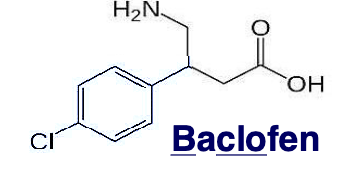
baclofen: GABA derivative
tizanidine: sympatholytic
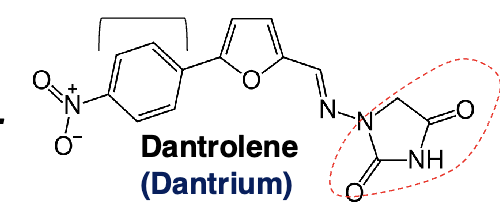
dantrolene: direct skeletal muscle relaxant
gabapentin: antiepileptic
botulinum toxin
drugs approved for spasm
cyclobenzaprine: 5HT2 receptor antagonist
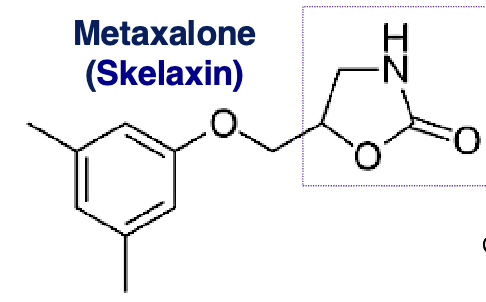
metaxalone: 2-oxazolidone
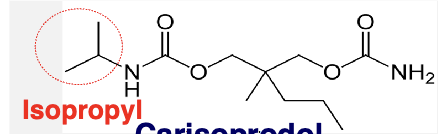
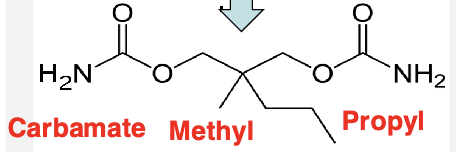
carisoprodol: carbamate derivatives
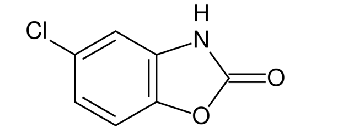
chlorzoxazone: 2-benzoxazolinone
ophenadrine: muscarinic antagonist
methocarbamol: glycerol monoether
diazepam: benzo
antispastic drugs targeting GABA receptors
baclofen
benzodiazepines
antispastic drugs targeting alpha 2 adnergic receptors
tizanidine
antispastic drug act directly on skeletal muscle cells
dantrolene
benzodiazepines
inhibit excessive muscle tone and hyperactive stretch reflexes by potentiating GABA activity
relieve skeletal muscle spasticity and accompanying pain in variety of neurological disorders
pharmacologic effect mediated via positive modulation of four different subtypes of GABA a receptors (chloride channels)
diazepam
clonazepam
lorazepam
baclofen
specific agonist at GABA B receptors (potassium channels)
binding opens the potassium channel and hyperpolarizes the neuron= reduction of the release of excitatory NT in both brain and spinal cord
relieves muscle tightness, muscle spasms and stabbing nerve pain related to spine injuries and MS
dantrolene
directly acting skeletal muscle relaxant
hydrantoin derivative→ without antiepileptic activity
lessens excitation-contraction coupling in muscle cells by binding to ryanodine receptor as an antagonist and inhibiting Ca efflux into cytoplasm from sacoplasmic reticulum
tizanidine
centrally acting alpha2 adrenergic agonist
sympathetic antagonist by inhibiting nonadrenergic neurotransmission→ originates from Locus Ceruleus
1,2,5- thiadiazole→ 2-amioninidazoline
slows excitatory action in the brain and nervous system→ allows muscles to relax with much less hypotensive effects than clonidine
MOA: bind to alpha 2 adrenergic→ dissociation of an alpha subunit from inhibitory Gi protein→ inactivation of adenylate cyclase through the association of an alpha subunit→ decrease of intracellular cAMP→ inactivation of protein kinase A(PKA)→ decrease of noradrenaline release
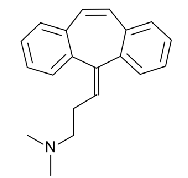
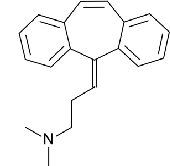
cyclobenzaprine
5-HT2 receptor antagonist→ inhibits neuronal excitation originating from raphe nuclei that travel to alpha motor neurons
influences both gamma and alpha motor neurons→ leads to reduction in muscle spasms
contains dibenzo(a,d)cycloheptene, methylidene group substituted by 2-(dimethylamino)-ethyl group
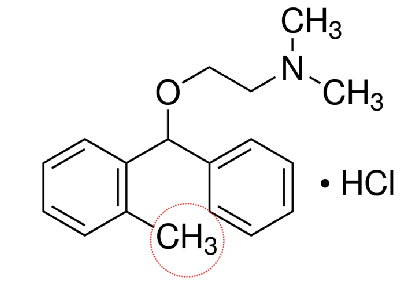
orphenadrine
centrally acting- nonopiate analgesic and muscle relaxant
an amino-alcohol ether structurally related to diphenhydramine
NMDA-type glutamate antagonist (phencyclidine binding site) to reduce excitatory neurotransmission in brain stem
histamine H1 receptor antagonist with anticholinergic activity as well
skeletal muscle relaxant for shhort term treatment of muscle discomfort cause by health problems or injuries such as sprains or strains
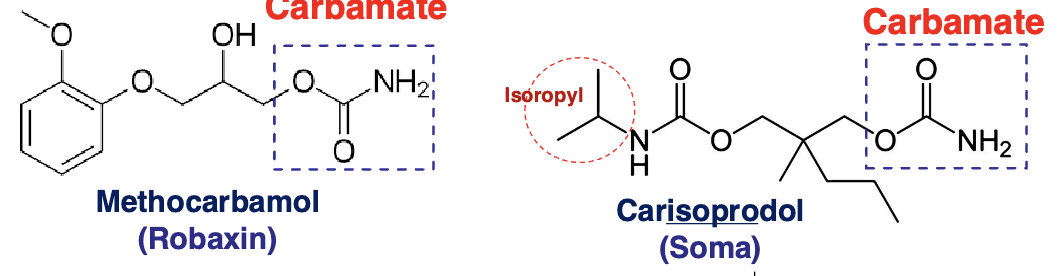
carbamate ester muscle relaxants
methocarbamol
carisoprodol
meprobamate→ 2 carbamates on each side (metabolite of carisoprodol from N-dealkylation)
metaxalone
general CNS depressant with no significant anticholinergic effects
lack of abuse, few ADE and relatively low degree of sedation
contains 2-oxazolidinone
aromatic ether
carisoprodol
effects mutliple sites in the CNS including the thalamus and limbic system
meprobamate
in the absence of GABA→ directly activates GABA a receptors
chlorzoxazone
inhibits muscle spasm by exerting an effect primarily at the level of the spinal cord and subcortical areas of the brain
less effective than others
Which of the following statements about Cyclobenzaprine is NOT correct?
A. It is a directly acting muscle relaxant.
B. It is a 5-HT2 receptor antagonist.
C. It is structurally similar to amitriptyline.
D. It has anticholinergic effects.
E. It decreases the activity of descending serotonergic neurons in the spinal cord.
A. It is a directly acting muscle relaxant.
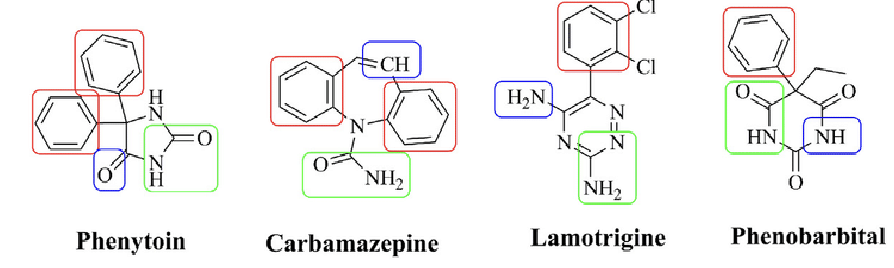
pharmacophoric pattern of AEDs
red rectangle: hydrophobic domain
green rectangle: hydrogen bond acceptor/donor
blue rectangle: electron donor moiety
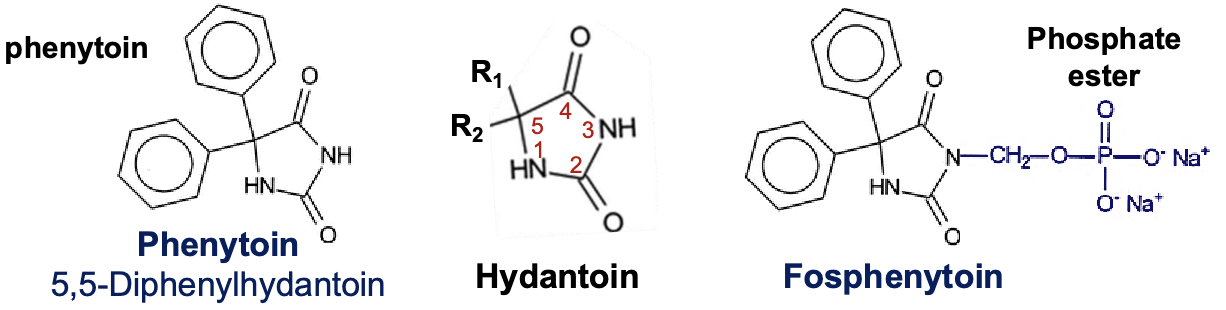
phenytoin/ fosphenytoin
hydantoin derivatives (imidazolidine)
substituting a phenyl group position 5 of the hydantoin molecule
2 phenyl groups at position 5 produces most anticonvulsant activity
fosphenytoin(IV/IM)→ phenytoin(IV) + a phosphate ester
conversion occurs by phosphates in the liver and RBC
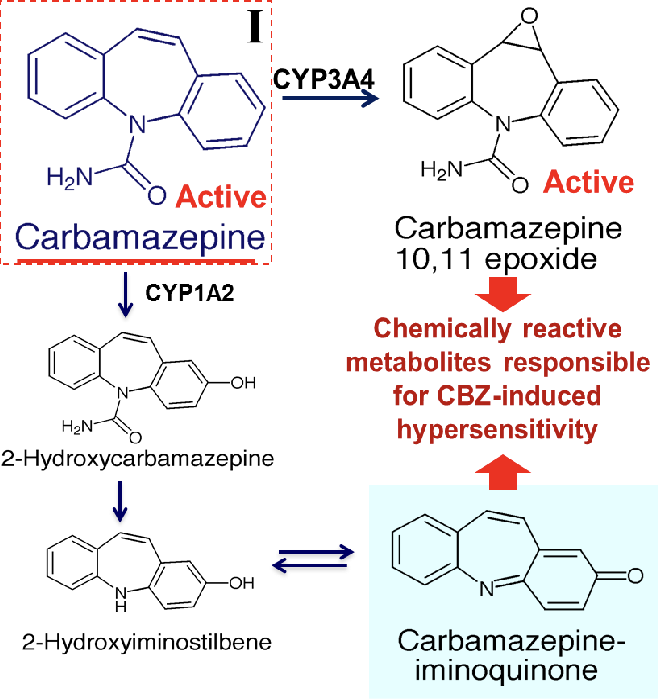
dibenzazepine 5-carboxamide derivatives
carbamazepine, oxcarbazepine, eslicarbazepine
bind to a stabilize the inactive state of voltage-gated sodium channels
carbamazepine
CI in patients with hx of hypersensitivity to TCAs
chemically reactive metabolites cause CBZ induced hypersensitivity
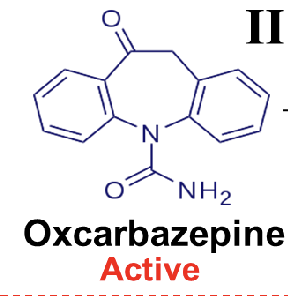
oxcarbazepine
less potent with less serious SE due to absence of epoxide or iminoquinone metabolites
keto group in 10 position
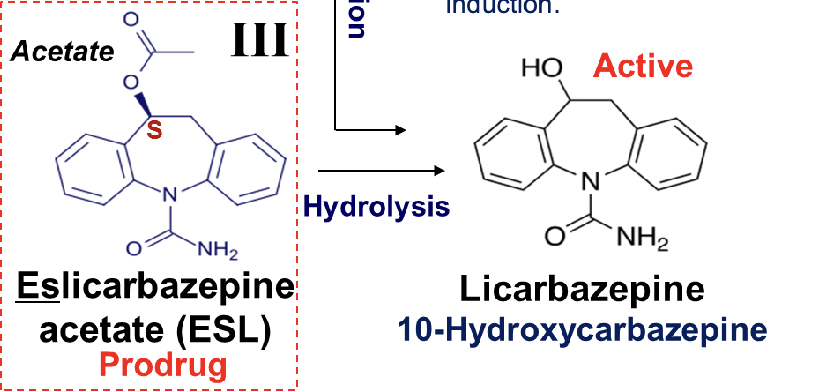
eslicarbazepine acetate
prodrug, acetate ester with S configuration
less potent with less serious SE due to absence of epoxide of iminoquinone metabolites
hydrolyzed into active metabolite→ licarbazepine
hydroxy group in the 10 position of ring minimizes the enzymatic induction of the cytochrome P450 and autoinduction
rufinamide
triazole derivative(5 membered nitrogen heterocycle) that binds to inactivated form of voltage gated sodium channels
lennox gastaut syndrome