PCL201
1/659
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
660 Terms
Pharmacology (lec 1)
science of drugs
study of drug action (pharmacodynamics) and fate (pharmacokinetics) in the body (desirable effects)
basic and clinical biomedical science
Important distinctions:
Pharmacy
Therapeutics
Toxicology: a science focusing on chemicals w/ undesirable biological activity
Pharmacology purposes and aims (lec 1)
Specific purposes:
identify drug targets and understand how drugs work by interacting w/ drug targets
understand how drugs are handled/modified by the living organism
broad aims:
gain insight into normal and abnormal function
improve therapeutic intervention by doctors
Toxicology purpose (lec 1)
looks at how xenobiotics (drugs, chemicals: externally brought into body) are handled and interact w/ targets to cause deleterious effects and reduce these effects (to improve drug)
studying organochlorine contaminants (ex: thalidomide) and pesticides
Pharmacology history: Ancient civilizations (lec 1)
medicinal and toxic plants, preparations from mineral and animal sources
China: Pen Ts’ao (Great Herbal, 2735 BC)
40 volumes of medicinal and toxic plants, antidotes
Egypt: Ebers Papyrus (1500 BC)
>700 drugs in >800 formulas
Pharmacology history: Greek and Roman empires (lec 1)
Hippocratic Corpus (5th c. BC - 2nd c. AD)
~400 drugs, primarily from vegetable origin
rejected divine/spiritual causes and treatment of disease
doctrine of the 4 Humors (blood, phlegm, black bile, and yellow bile)
De Materia Medica (Pedanius Dioscorides, 50-70 AD)
>600 medicinal plants
each disease has a unique cause for which there is a specific remedy
remained in use until ~1600s
considred to be basis of modern medecine
Pharmacology history: Medieval times and islam (lec 1)
The Canon of Medicine (Avicenna, c. 1025)
set the standards for medicine, an authoritative reference as late as the 17th c.
translated to many langs.
list of 800+ drugs/remedies
foundation for experimental testing of drugs
Pharmacology history: late 18th-19th c. (lec 1)
advances in physiology, pathology, and chemistry provided the foundation for pharmacology
focus was on isolating and understanding the effects of natural substances (focus on botanicals)
examples:
Digitalis
Morphine
Salicin
Digitalis from purple foxglove (lec 1)
Digitalis Purpurea = purple foxglove
used to treat “dropsy” (edema from congestive heart failure) (18th c. England)
digitalis extract (Digitoxin and Digoxin) contain cardiac glycosides (used to treat cardiac arrhythmias and heart failure)
Morphine from Opium poppy (lec 1)
Papaver Somniferum = Opium poppy
Opium (sap of poppy) = painkiller
morphine = 1st active alkaloid discovered from opium poppy
morphine used for acute and chronic severe pain
morphine is a precursor to multiple synthetic and semi-synthetic opiates
Salicin from Willow Bark (lec 1)
Salix alba = white willow
willow tree bark and leaves powder used for headache, pain, fever (Hippocrates, 5th c. BC)
1828: salicin isolated from willow bark
1838: salicylic acid purification (active metabolite of acetylsalicylic acid (Aspirin) (Aspirin becomes salicylic acid)
Pharmacology history: mid 19th c. (lec 1)
offical birth of pharmacology as a separate science focused on studying the interaction of chemicals w/ living systems (1847)
fathers of pharmacology: Rudolf Buchheim + Oswald Schmiedeberg
Pharmacology history: Early 20th c.: mech of drug action: receptor theory (lec 1)
Langley and Ehrlich proposed the concept that receptors mediate drug action (1905-1907)
Clark introduced the receptor occupancy model describing the relationship btwn drug concentration and effect (1933)
continued focus on isolating and understanding the effects of natural and endogenous substances, aided by strides in synthetic chem, microbiology, and biochem in the 20th c.
Ach (1906), oxytocin (1906), histamine (1907), insulin (1922), penicillin (1928), streptomycin (1943)
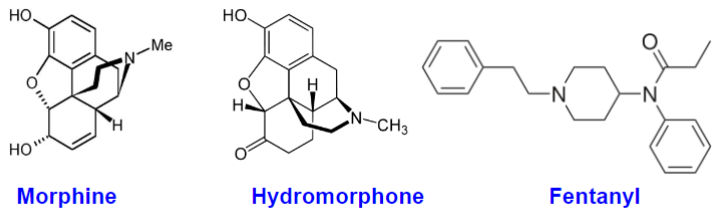
What is a drug? (lec 1)
an external chemical substance (other than those essential for normal function) that can exert a biochem/physiological effect in a living org, whether therapeutic or not
can be natural (morphine), semi-synthetic (hydromorphone) or synthetic (fetanyl)
Official definition by FDA:
drugs defined by their intended use
“intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease”
“intended to affect the structure or any function of the body of man or other animals”
drugs regulated by:
FDA in US
Therapeutic products directorate (TPD) of Health Canada
Things that might be considered drugs (lec 1)
dietary components and/or essential nutrients when given in a high dose used for treatment of disease
endogenous molecules when administered or in high doses
Drug origins (lec 1)
drugs can be classified by origin:
natural compounds (in crude preparations/pure)
semi-synthetic
synthetic
biologics
Natural preparations (crude/galenicals: extract by hot water) (lec 1)
Plant sources:
Opium
Digitalis
Atropa Belladonna tincture (evening nightshade)
Klamath Weed (St. Johns Wort)
Coffee
Animal sources:
Puffer fish venom (contains Tetrodotoxin)
Desiccated thyroid (contains thyroid hormone)
Extract from pancreatic islet cells (contains insulin)
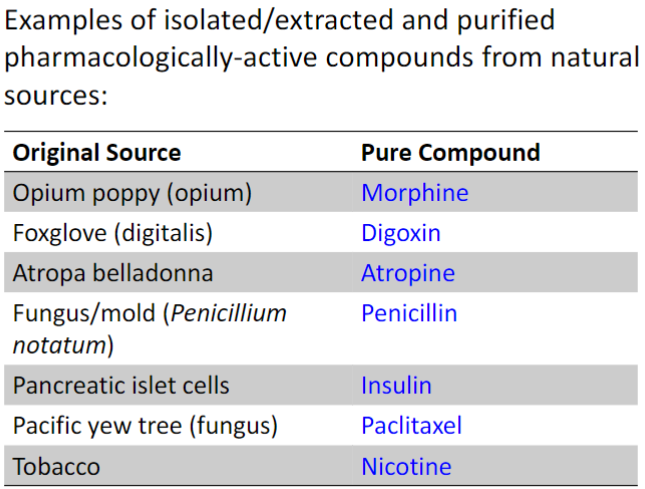
Pure compounds from natural sources (lec 1)
ex: Paclitaxel (Taxol) from bark Pacific Yew Tree in late 1960s
has anti-cancer activity
b4 1990s: almost all paclitaxel was harvested from bark
later discovered to be produced by endophytic fungi in bark
Early 1990s: semi-synthetic prod. from another compound found in Yew tree needles emerged
today: Paclitaxel is directly purified from a plant cell line propagated in fermentation tanks w/ fungi
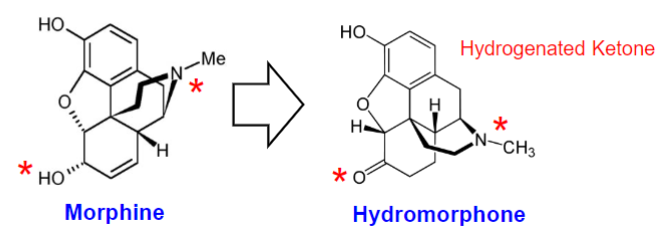
Semi-synthetic compounds (lec 1)
produced by chem modification of pure compounds
improvement of the parent compound w/ respect to potency, specificity, side effects, PK parameters
ex: Hydromorphone (Dilaudid) from morphine
increased potency and lipophilicity
more rapid onset of action, altered adverse effect profile
hydrogenation of ketones
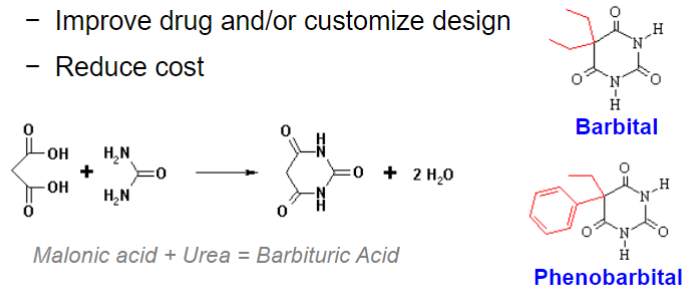
Synthetic compounds (lec 1)
most drugs
range from serendipitous discoveries (ex: barbital) to deliberate synthesis based on predicted chem properties/known molecular features necessary for drug action
improve drug and/or customize design
reduce cost
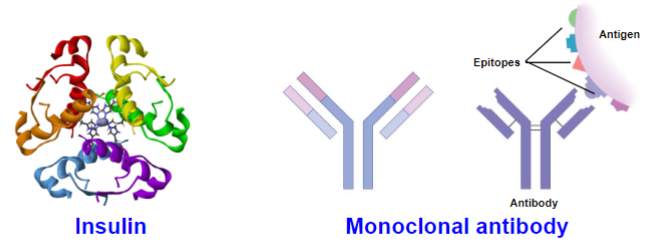
Biologics (lec 1)
monoclonal antibodies, vaccines, recombinant proteins
created by biological processes
may be extracted from human/animal tissues, tissue cultures or produced by recombinant DNA tech
more complex than small-molecule drugs
can be expensive to make
Drug action mechs (lec 1)
drugs can act as/by:
agonists
antagonists
allosteric modulators
enzyme inhibitors
affecting lvls of endogenous compounds
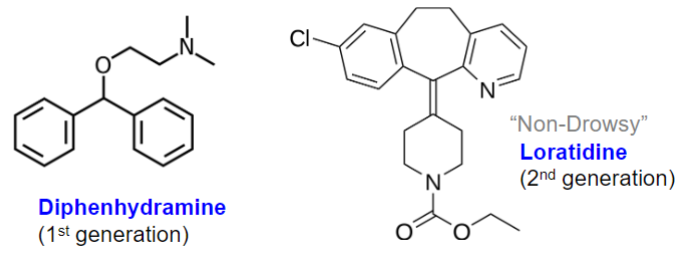
Drug generations (lec 1)
Research + Development continues to improve drug by customizing design
ex: H1 antagonists (antihistamines for allergy)
Loratidine less lipophilic so can’t pass BBB
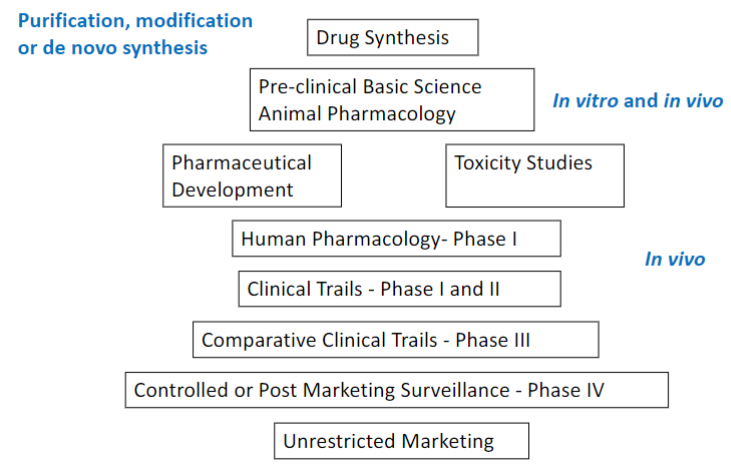
Clinical drug development (lec 1)
cost increases as process moves on (most in clinical trials)
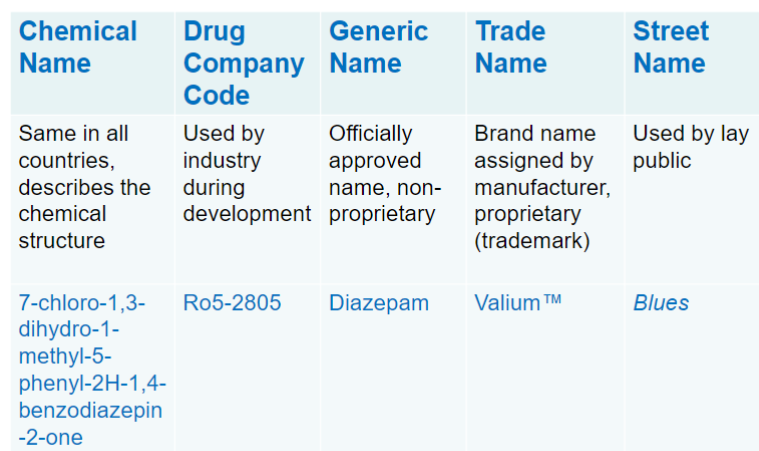
Drug nomenclature (lec 1)
chemical name (chem structure)
drug company code
generic name (non marketed name and consistent across countries and companies)
trade name (trademark)
street name
Patent law (lec 1)
way for companies to make money off drug development
country specific
no other company can market chemical compound for 20 yrs
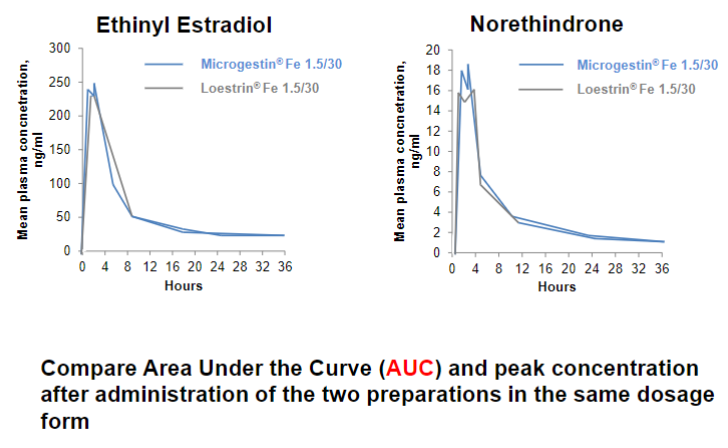
Bioequivalence (lec 1)
2 diff drug preparations w/ equal dosage form produce similar pharmacokinetic measures
ex: Loestrin and Microgestin
pharmacokinetics curve must be 80-120% similar
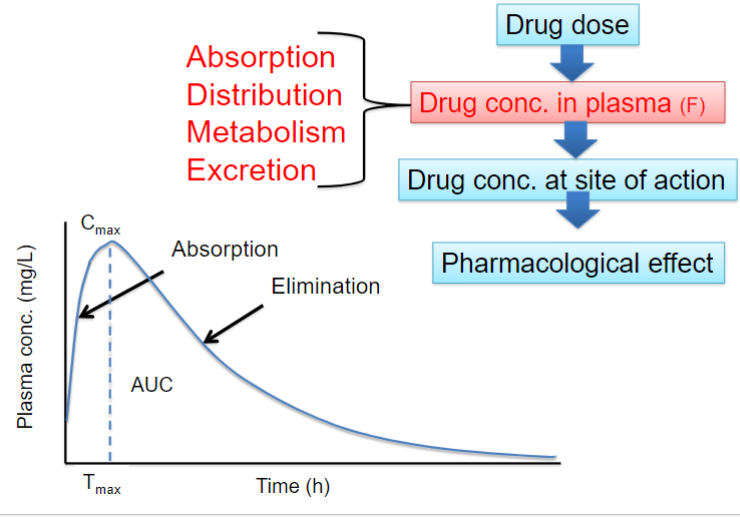
Overview of Pharmacokinetics and pharmacodynamics (lec 2)
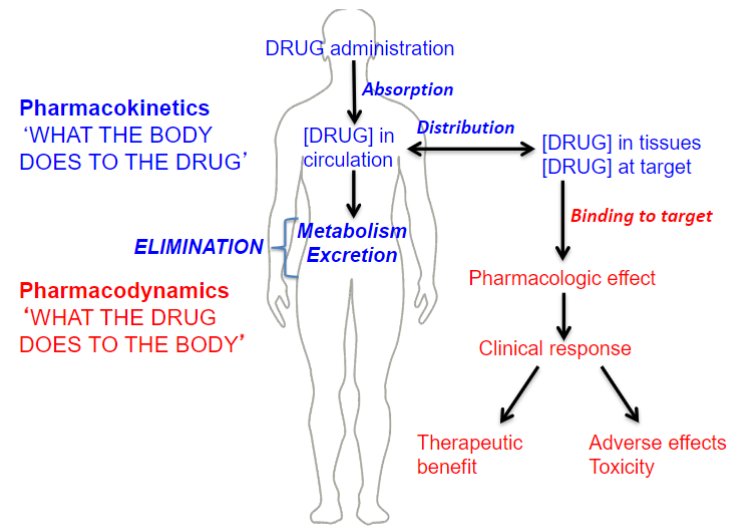
What is Pharmacokinetics (PK) (lec 2)
“what the body does to the drug”
a study of the kinetics (time course) of drug conc and the process involved
mathematical relationsips btwn drug dosing + resulting drug [ ]
Quantitatively describe the various steps of drug disposition in the body: ADME
affected by:
physiochem properties of drug
anatomy and physiology of individual
linked to PD by plasma conc
![<ul><li><p>“what the body does to the drug”</p></li><li><p>a study of the kinetics <strong><u>(time course) of drug conc</u></strong> and the process involved</p><ul><li><p>mathematical relationsips btwn drug dosing + resulting drug [ ]</p></li></ul></li><li><p>Quantitatively describe the various steps of drug disposition in the body: ADME</p></li><li><p>affected by:</p><ul><li><p>physiochem properties of drug</p></li><li><p>anatomy and physiology of individual</p></li></ul></li><li><p>linked to PD by plasma conc</p></li></ul>](https://knowt-user-attachments.s3.amazonaws.com/af70d403-0e15-4049-8335-7f5ccbee383f.jpeg)
Pharmacokinetics: ADME (lec 2)
Absorption:
movement of the drug from its site of administration into the bloodstream
extent of absorption = bioavailability: fraction of a dose that makes it to systemic circulation in its unchanged form
Distribution:
spreading of the drug throughout the body once absorbed
drug distributes from systemic circulation (vasculature) to intracellular and interstitial fluid
Metabolism:
transformation of the drug to more hydrophilic metabolites (primarily in the liver)
Excretion:
removal of drug from the body (primarily in urine + feces)
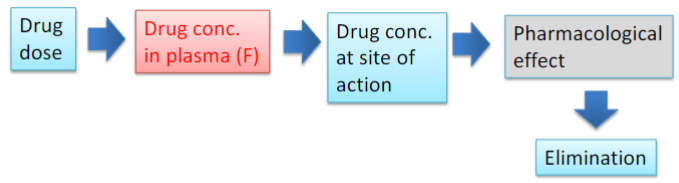
Pharmacokinetic processes ADME (lec 2)
ADME determines the time course of drug conc in blood and tissues following drug administration
PK quantitatively describes the various steps of drug disposition and ADME
used to calculate and understand the relationships btwn drug dosage regimen and resulting drug concs.
Pharmacodynamics (lec 2)
“what the drug does to the body”
study of effects and mechs of therapeutic and toxic action of a drug
Binding: interaction w/ target (receptors, enzymes, molecules)
Mech of action: changes in signaling (cascades/alteration of pathways)
effect: physiological/biochemical changes
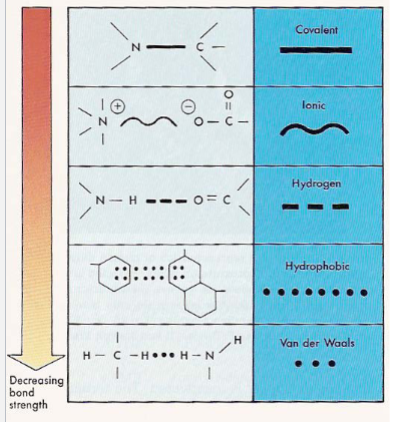
Drug target interactions (lec 2)
irreversible drug-receptor interactions (cov/ionic) aren’t common and occur via strong chem bonds (aspirion/anti-tumor drugs)
not always desirable
most drug-receptor interactions are reversible via weak chem bonds (hydrophobic/van der waals)
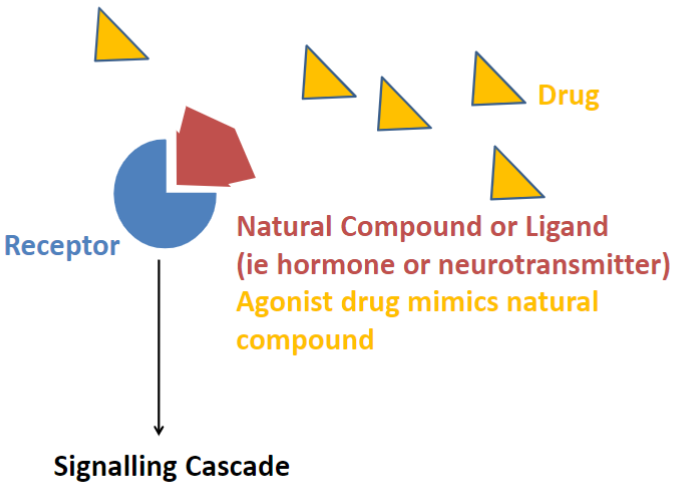
The “lock and key” (lec 2)
agonists: mimics natural compound
Nature of the interaction (lec 2)
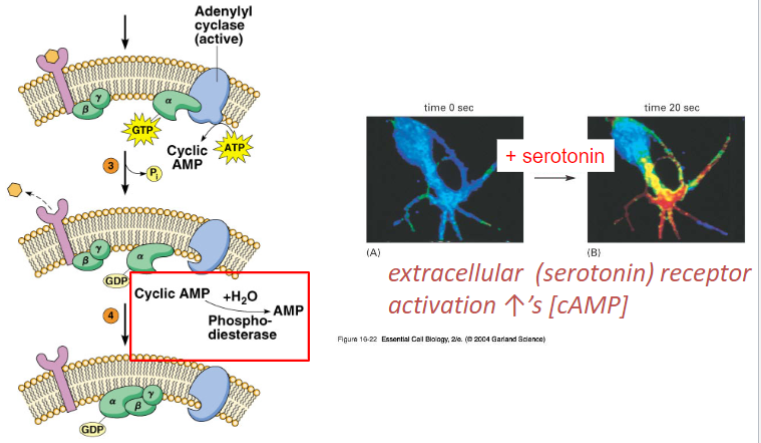
receptors are proteins/enzymes that participate in intracellular communication via chem signals
ligands: signaling molecules, can be endo/exo -genous (neurotransmitter/hormone/drug)
Effector molecules: those that are activated by the signaling cascade and begin biological response (G-Protein)
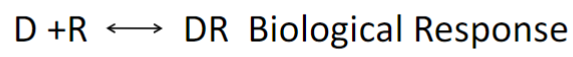
Effector activating adenylyl cyclase or extracellular (serotonin) receptor (lec 2)
increase in cAMP
Nature of Drug-target interaction (lec 2)
has an effective conc range (saturation)
has desired biological specificity
chem specificity (enantiomers)
racemic mixtures
could lead to higher doses
could lead to unintended side effects
BUT are cheaper to make
can be inhibited/antagonized/blocked
Characterization of D+R binding (lec 2)
rxn is “on and off” like enzyme kinetics
an acute response can vary w/ dose (shown w/ picture)
affinity measures the strength of the D-R interaction
the Dose-Response curve measures the interaction of D-R
Classical theory: Response is relative to drug conc and the # of receptors activated
increase dose leads to increased response (bc more receptors activated)

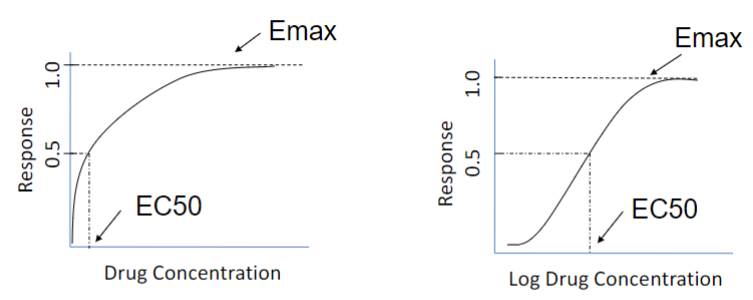
Dose response-curve (lec 2)
semi-logarithmic transformation
expands conc scale at low [ ] (binding changes rapidly)
compresses conc scale at high [ ] (binding changes slowly)
doesn’t change value of Kd (affinity)
![<ul><li><p>semi-logarithmic transformation</p></li><li><p>expands conc scale at low [ ] <strong><u>(binding changes rapidly)</u></strong></p></li><li><p>compresses conc scale at high [ ] <strong><u>(binding changes slowly)</u></strong></p></li><li><p>doesn’t change value of Kd (affinity)</p></li></ul>](https://knowt-user-attachments.s3.amazonaws.com/6ecdf7f1-b3ae-4917-86cf-44539edb1bb6.jpeg)
Dose-response curve: Efficacy (lec 2)
Emax: maximal response achieved by an agonist (efficacy)
EC50: drug conc at 50% of Emax (potency)
ED50: 50% of max effective dose
affinity (strength of interaction btwn target and drug)
Kd
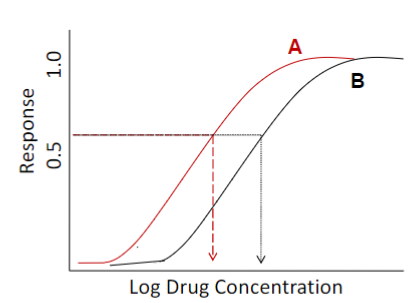
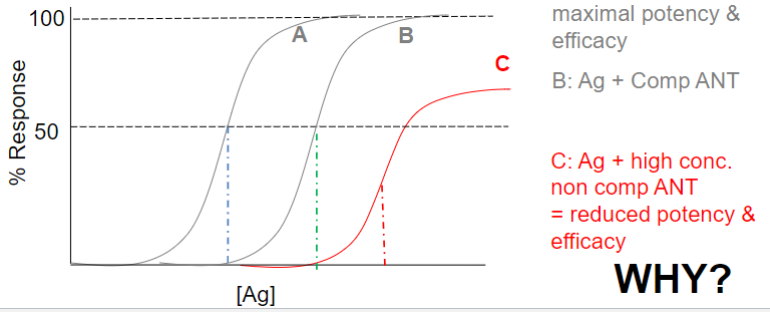
Dose-response curve: potency (lec 2)
A is more potent because response comes w/ lower drug conc
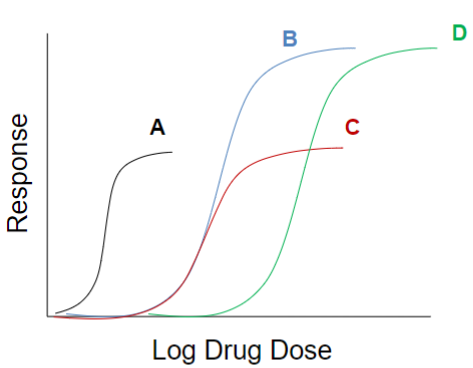
Efficacy vs. Potency (lec 2)
looking at multiple drugs interacting w/ target via log dose response curve, we can compare efficacy and potency
A more potent
B and D highest efficacy
clinical relevance of a drug depends on the maximal efficacy and ability to activate receptors more than the drug’s potency
Application of D-R curve info (lec 2)
Y axis: determines the “measurement” (effective vs. lethal vs. toxic response)
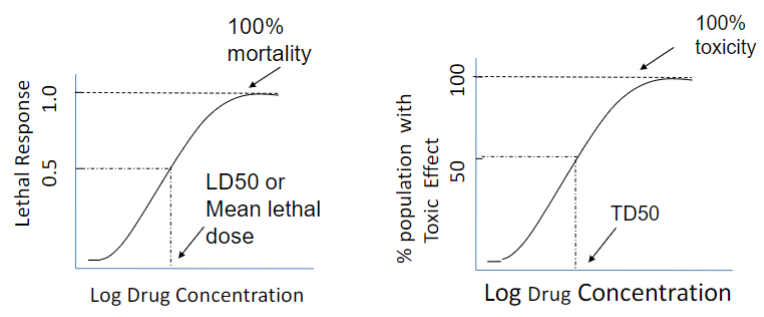
Applications of D-R curve (lec 2)
larger T.I (therapeutic index) = safer drug
~833 = very safe
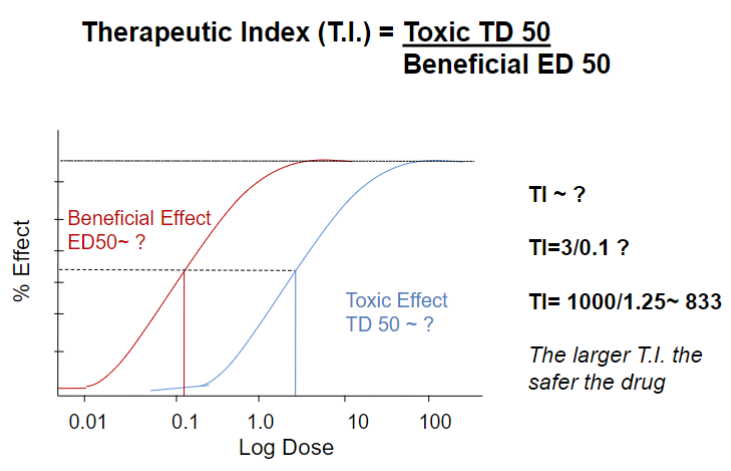
Drug classification (lec 2)
drugs can be classified by their interaction w/ the target and subsequent “response”
Agonist:
full agonist (100% max efficacy)
partial agonist
positive allosteric modulator (affects agonist activity)
Antagonist:
competitive
noncompetitive: Irreversible or negative allosteric modulator (affects agonist activity)
Agonists (lec 2)
a drug can mimic (agonist) or enhance (positive allosteric modulator) the action of an endogenous compound at its site of action
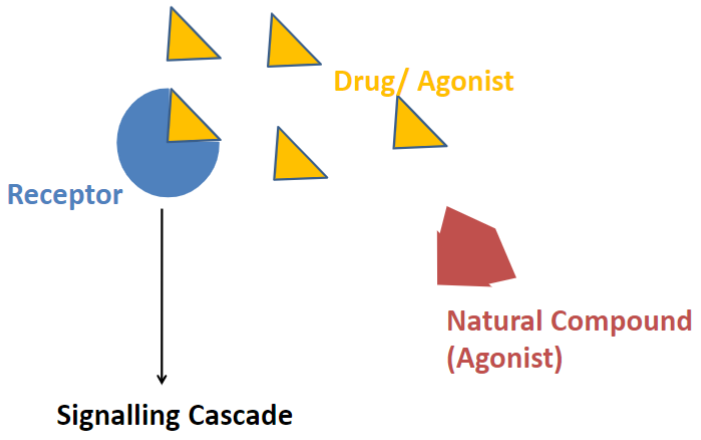
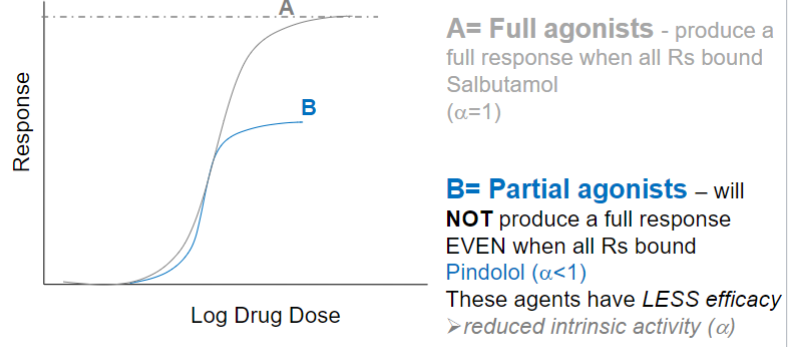
Full agonist (lec 2)
mimics effects of endogenous ligand
GENERALLY binds at site that endogenous ligand bins
initiate biological response upon binding R
produce a full response when all Rs bound
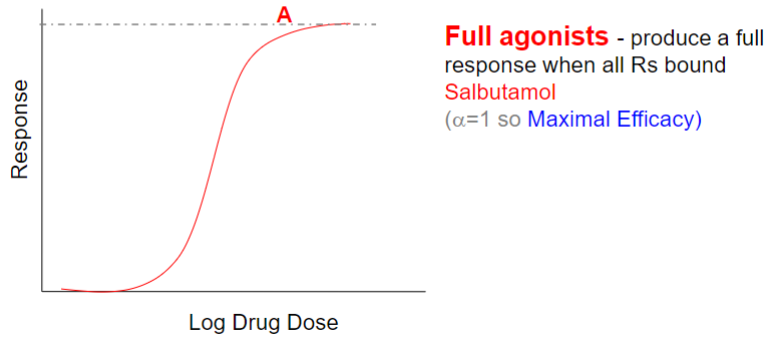
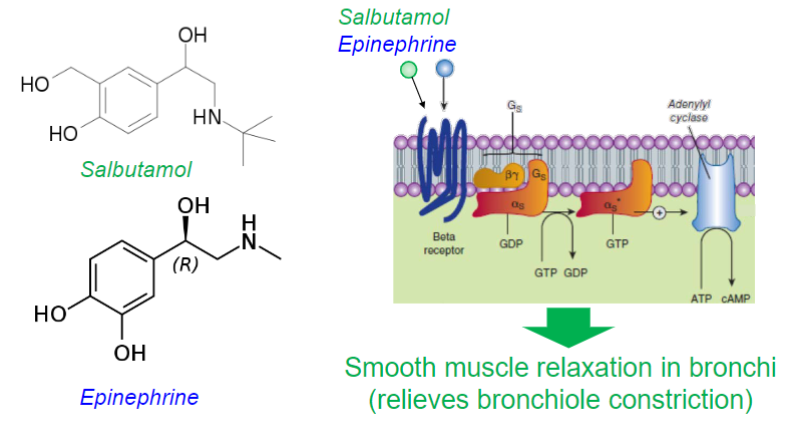
Salbutamol (lec 2)
selective β2 adrenergic receptor agonist (competes w/ epinephrine)
for relief of bronchospasm in asthma and COPD
causes smooth muscle relaxation in bronchi
Partial agonist (lec 2)
mimics effects of endogenous ligand
GENERALLY binds at site that endogenous ligand bins
initiate biological response upon binding R
will NOT produce a full response even when all Rs bound
these agents have less efficacy (reduced intrinsic activity)
ex: Pindolol
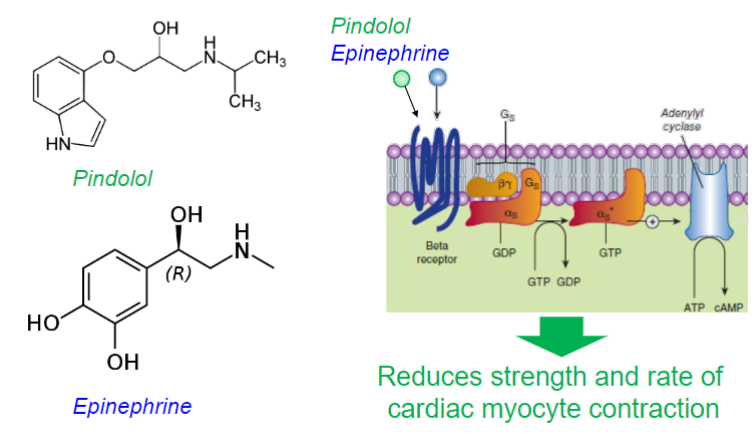
Pindolol (lec 2)
selective β1 adrenergic receptor partial agonist
used to treat hypertension by “blunting” the β adrenergic signaling at the heart
reducing strength and speed of cardiac contraction

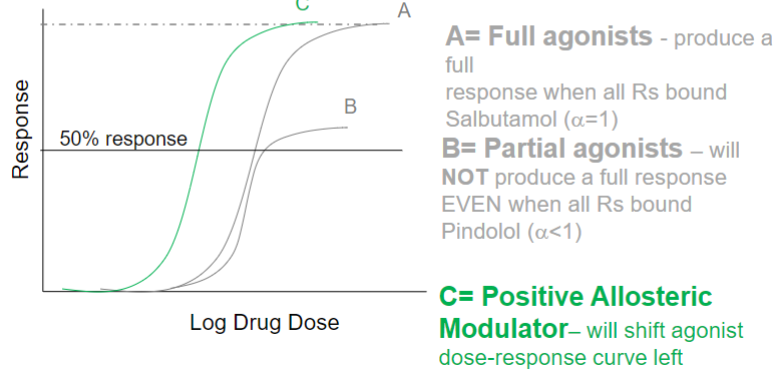
Positive allosteric modulators (lec 2)
enhances effect of endogenous ligand
GENERALLY binds at site distinct from endogenous ligand binding site
will shift agonist dose-response curve left (less agonist required for response)
ex: Diazepam
Diazepham (ValiumTM) (lec 2)
Positive allosteric modulator of GABAA receptor (an inhibitory channel that decreases neuronal activity)
used for treatment of anxiety, insomnia, and seizures
increases frequency of ion channel opening when GABA binds to GABAA receptor
leads to more chloride ions entering the neuron
CNS depressant effects
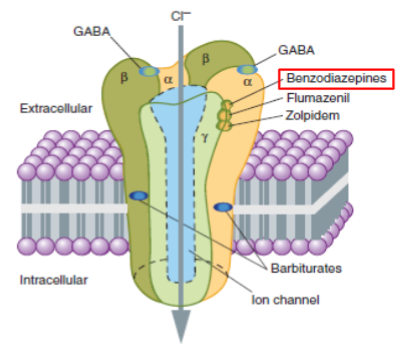
Antagonists, inhibitors and blockers in diff fields (lec 2)
pharmacological: blocks ability of a D-R interaction by another ligand
Cimetidine binds H2 R and blocks ability of histamine to bind therefore lowers gastric acid secretion
chemical: interaction of 2 compounds in solution such that effect of active drug lost
Metal chelator + toxic metal
Physiological: interaction of 2 drugs w/ opposing physiological actions
histamine decreases BP via H1 R and epinephrine increases BP via β-Adr R
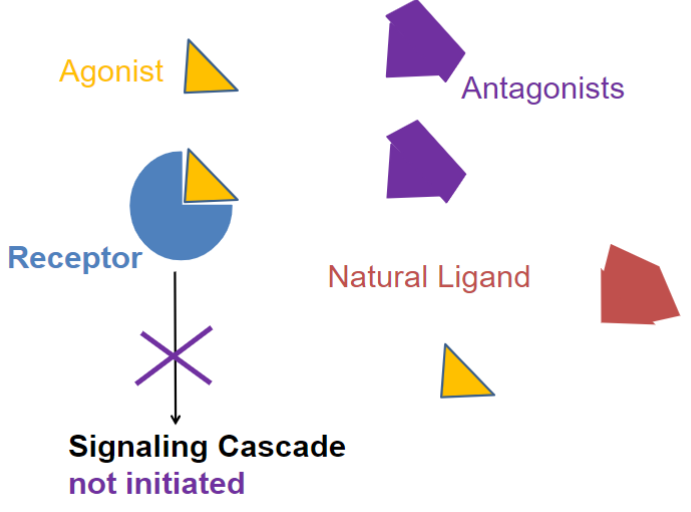
Antagonists (lec 2)
binds R but doesn’t initiate biological activity/signaling cascade
biological effects from “preventing” natural or endogenous agonist from binding and R activation
0 efficacy on its own

Competitive antagonist (lec 2)
a drug that can block the action of endogenous and other compounds at their site of action
inhibition can be overcome by increase in [agonist]
affects Agonist potency (shifts D-R curve right)
clinically useful
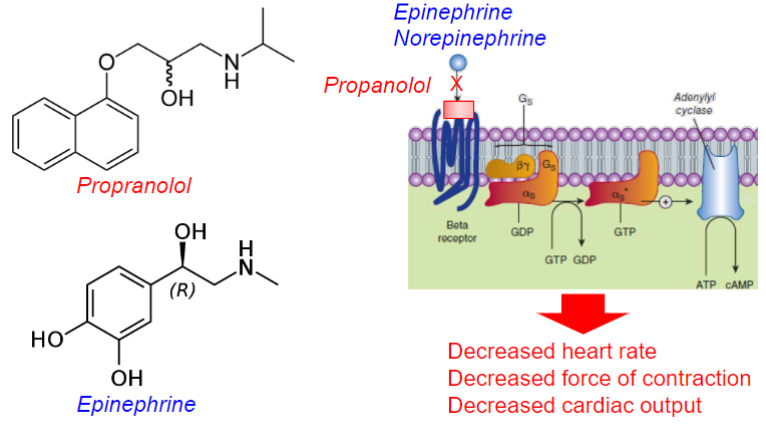
ex: Propranolol
![<ul><li><p>a drug that can block the action of endogenous and other compounds at their site of action</p></li><li><p>inhibition can be overcome by increase in [agonist]</p></li><li><p>affects Agonist potency <strong><u>(shifts D-R curve right)</u></strong></p><ul><li><p>clinically useful</p></li></ul></li><li><p>ex: Propranolol</p></li></ul>](https://knowt-user-attachments.s3.amazonaws.com/1600165d-76e5-43a4-b03b-1d72936a9316.jpeg)
Propranolol (lec 2)
Competitive, non-selective beta antagonist at the beta adrenergic receptor (beta-blocker)
used for treatment of hypertension (high bp)
decreased heart rate, force of contraction and cardiac output
Non-competitive antagonist (lec 2)
irreversible or negative allosteric modulator
may not be at same site on R as agonist (neg. allosteric modulator)
may be covalent bonding (irreversible)
affects Ag potency and Efficacy (shifts D-R curve right + down)
certain amt of receptors are changed so it can’t be used
Neg. allosteric modulator (lec 2)
impede the action of endogenous and other compounds at diff site of action
ex: β-carboline
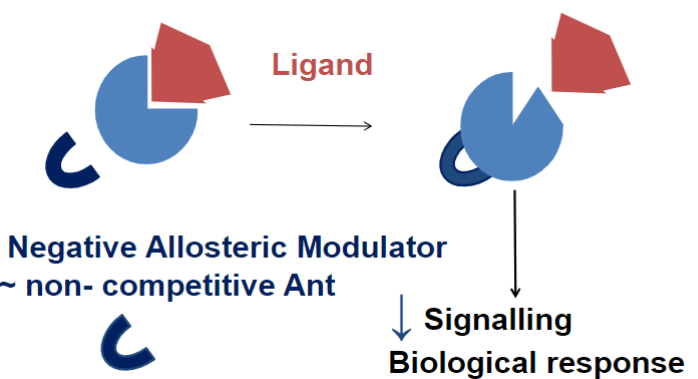
β-carboline (lec 2)
Alkaloid found in plants and animals
acts as a negative allosteric modulator of the GABAA receptor
induce convulsions and increase anxiety (anxiogenic)
binds at the benzodiazepine site
decreases GABA stimulated chloride movement into cell (less chloride ions entering neuron)
less CNS depression and more excitable cells
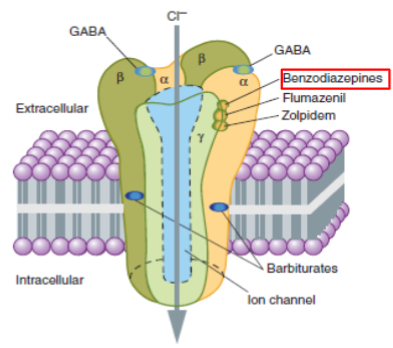
Antagonists, blockers, and inhibitors (lec 2)
Antagonists: reserved for drugs acting on receptors
bind to agonist site
may be “blockers” (block agonists from binding and activating R)
Blockers and inhibitors: MAY refer to antagonists at Rs (beta blockers) BUT also to drugs that act via other methods to reduce signaling cascades
block transporters (Fluoxetine blocks transporter for serotonin)
inhibit enzymes (ibuprofen inhibits COX enzyme)
act as “sink” for protein (Etanercept is an antibody for TNFα)
Allosteric modulators (Diltiazem is a Ca2+ channel blocker)
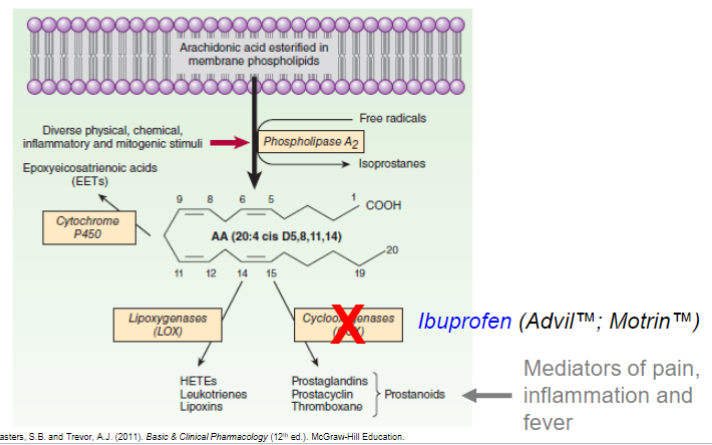
Ibuprofen (lec 2)
non-steroidal anti-inflammatory drug (NSAID)
inhibits the enzyme cyclooxygenase (COX)
stops release of Prostanoids (mediators of pain, inflammation and fever
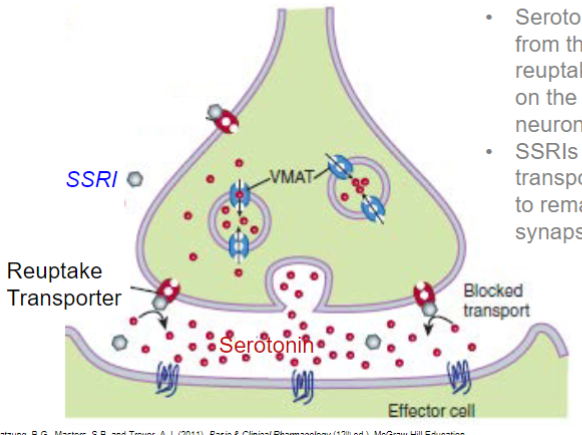
Fluoxetine (lec 2)
selective Serotonin reuptake inhibitor (SSRI)
serotonin is removed from the synapse by reuptake transporters on the presynaptic neuron
SSRIs block transporters, allowing it to remain active in the synapse longer
Other mechs of drug action (lec 2)
create osmotic load (Laxatives like mannitol) (puts H2O into GI tract)
change pH (ammonium chloride to acidify the urine) (traps drug)
chelation (EDTA to bind divalent metal cations)
disruption of membranes (Polymixin antibiotics disrupt bacterial cell membranes)
damage DNA (chemotherapeutic agents like antimetabolites)
Drug biological action (lec 3)
for drug to have biological action is MUST be soluble in bodily fluids and able to cross biological membranes
ability to permeate biological membranes dependent on:
drug’s physicochemical properties
membrane/tissue physiological and anatomical characteristics
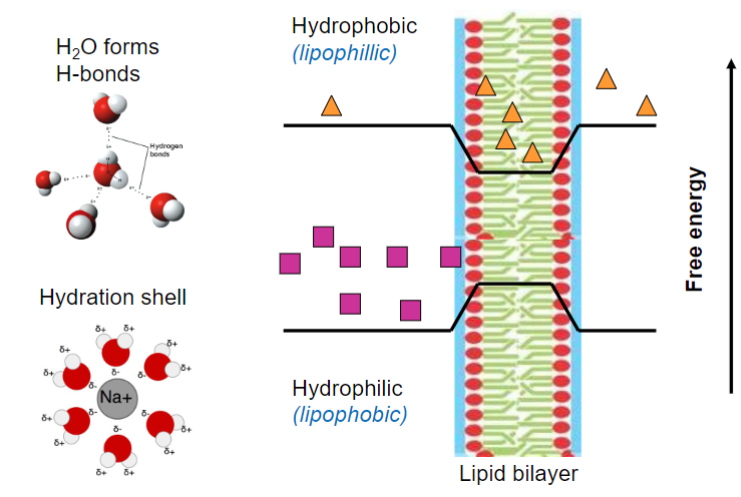
Biological environment: membranes (lec 3)
phospholipid bilayer:
amphipathic lipids (hydrophilic head/lipophilic tails)
embedded proteins (some form channels/pores)
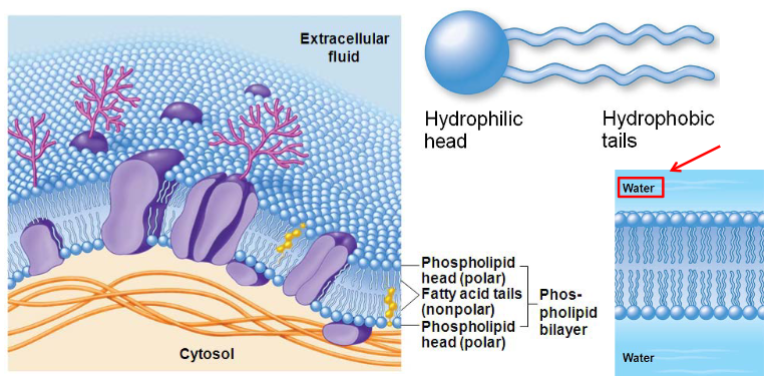
Characteristics of biological membranes (lec 3)
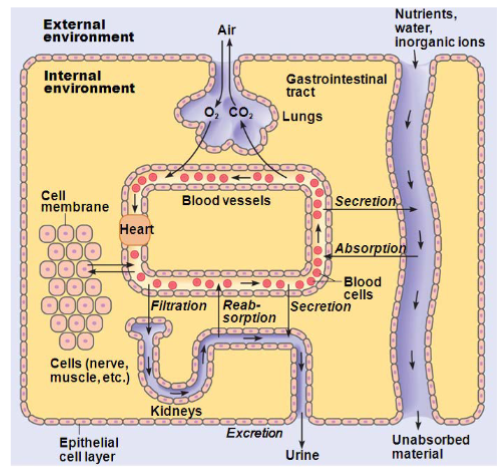
drugs must pass across biological membranes to be absorbed, distribute and reach their site of action, and be eliminated from the body
BUT drugs must also be able to traverse water (we are mostly made of H2O)
Physicochemical drug properties (lec 3)
physiochem properties of chem agent affect drug movement in biological environments
molecular size
solubility in water and lipid phases
partition coefficient or Pow
total polar surface area (TPSA)
extent of ionization (charge)
Molecular size (lec 3)
expressed in daltons (Da or kDA)
most drugs are 200-500 Da (reasons for drugs = small molecule chemicals)
proteins are much larger (must be injected, can’t be taken orally)
insulin: 6000 Da
albumin: 65000 Da
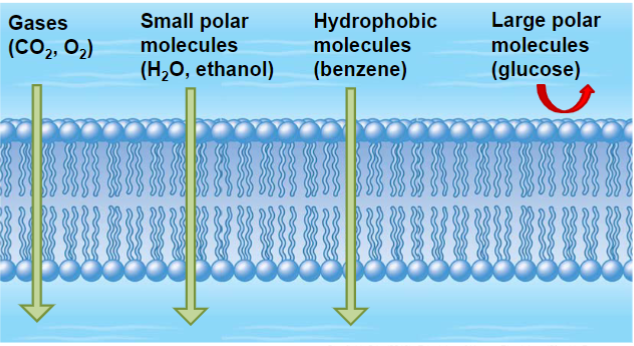
Molecular weight and hydrophilicity (lec 3)
smaller molecules generally cross membranes and distribute more readily
more important for hydrophilic drugs (larger size limits passive diffusion, must be transported w/ channels)
Solubility (lec 3)
ability of a solute to dissolve in a solvent to form a homogenous solution
nature of solvent and solute (polarity) (“like dissolves like”)
hydrophilicity:
a drug must be soluble in aq body fluids in order to distribute or reach its site of action
polar or ionic compounds (bc of H-bonds)
lipophilicity:
key physical property determining membrane permeability
vital for drug to pass through lipid bilayer (A, D, E)
potential for bioaccumulation (drug concentrates in adipose and lipid compartments)
non polar compounds
Passage across membranes indicates permeability (lec 3)
more nrg needed for more hydrophilic molecule
Ideal drug (lec 3)
not only polar/non-polar, best to be amphipathic
drugs have both polar + non-polar groups, combo of these determine solubility in vivo

Measuring solubility (Lec 3)
determined through partition coefficient (Pow or Kow)
Pow can be determined experimentally or in silico
Log P = log of octanol: water partition coefficient
Pow at equilibrium
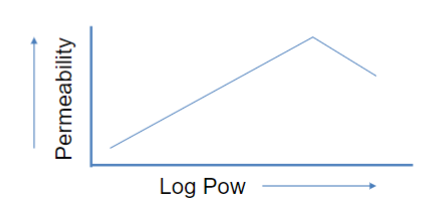
“drug-like” molecules have log P = 0.5 - 5
log P > 5 leads to less permeability bc can’t pass through small water layer on membrane
high Kow = high lipid solubility (good permeation across membranes)
low Kow = low lipid solubility (poor permeation across membranes)
Fick’s law of diffusion (lec 3)
a drug will flow from area of high conc to low conc, w/ rate of flow being higher w/ larger conc grads
Lipinski’s rule of 5 (lec 3)
MOST ORAL drug molecules should have:
log P </= 5
molecular weight </= 500 g/mol (Da)
# of H-bond acceptors </= 10
# of H-bond donors </= 5
molecules violating more than one of these rules “may have probs w/ ORAL bioavailability”
doesn’t predict drugs biological activity
Role of TPSA: “Topological Polar Surface Area” (lec 3)
contribution to the outer electron shell “surface area” of the molecule from electronegative atoms/atoms w/ unpaired electrons (ex: N, O) and from groups containing these atoms
polar surface area of >140Å2 tend to be poor at permeating cell membranes
to access brain (cross BBB), TPSA = <90Å2

Ionization (lec 3)
many drugs are weak acids/bases containing functional R groups that can be ionized (charged) or unionized (uncharged) depending on pH of the surrounding medium
due to protonation/deprotonation of the compound as a result of the drug’s interaction w/ aq medium
unionized forms of a drug can more readily diffuse across a membrane
ionized forms of a drug will have lower Kow and thus a reduced ability to permeate the membrane
charged are more hydrophilic due to their ability to interact w/ H2O dipoles through H-bonds

General principles (lec 3)
drugs in aq solution exist at an equilibrium btwn ionized/non-ionized forms (equilibrium can be shifted by varying the pH of a medium ([H+])
whether a drug will be mostly ionized/unionized depends on:
pH of medium
pKa of drug
whether drug is an acid/base
acidic drugs ionize by losing a proton
tend to ionize in more basic medium (high pH, low [H+])
protonated form = neutral (more lipid soluble)
basic drugs ionize by accepting a proton
tend to ionize in more acidic medium (low pH, high [H+])
unprotonated form = neutral (more lipid soluble)
![<ul><li><p>drugs in aq solution exist at an equilibrium btwn ionized/non-ionized forms (equilibrium can be shifted by varying the pH of a medium ([H+])</p></li><li><p>whether a drug will be mostly ionized/unionized depends on:</p><ul><li><p>pH of medium</p></li><li><p>pKa of drug</p></li><li><p>whether drug is an acid/base</p></li></ul></li><li><p>acidic drugs ionize by losing a proton</p><ul><li><p>tend to ionize in more basic medium (high pH, low [H+])</p></li><li><p>protonated form = neutral (more lipid soluble)</p></li></ul></li><li><p>basic drugs ionize by accepting a proton</p><ul><li><p>tend to ionize in more acidic medium (low pH, high [H+])</p></li><li><p>unprotonated form = neutral (more lipid soluble)</p></li></ul></li></ul>](https://knowt-user-attachments.s3.amazonaws.com/620a1122-4c4a-4dea-9fab-d7789245e366.jpeg)
Acids (lec 3)
proton donors (ionize after losing H+)
at high pH, reaction is driven right to give more H+, more drug is deprotonated and therefore ionized (less lipid souble)
at low pH, reaction is driven to the left, more drug is protonated and therefore unionized (more lipid soluble)

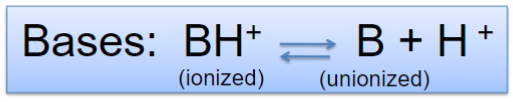
Bases (lec 3)
proton acceptors (ionize after gaining H+)
at high pH, reaction is driven right to give more H+, more drug is deprotonated and therefore unionized (more lipid soluble)
at low pH, reaction is driven left, more drug is protonated and therefore ionized (less lipid soluble)
How much drug is ionized at given pH? (lec 3)
depends on the drug’s pKa
use Henderson-Hasselbach equation: pH - pKa = log [unprotonated]/[protonated]
pka < pH = deprotonated
pka > pH = protonated
![<ul><li><p>depends on the drug’s pKa</p></li><li><p>use Henderson-Hasselbach equation: pH - pKa = log [unprotonated]/[protonated] </p></li><li><p>pka < pH = deprotonated</p></li><li><p>pka > pH = protonated</p></li></ul>](https://knowt-user-attachments.s3.amazonaws.com/163a65b7-1379-4c7f-b3af-136561698fed.jpeg)
pKa (lec 3)
pKa reflects pH which [protonated] = [deprotonated]
doesn’t determine whether drug is acid or base
depends on functional group
Ionization plays major role in determining… (lec 3)
solubility and absorption of the compound (bioavailability)
cell/membrane permeation and distribution to site of action, plasma-protein binding and volume of distribution
elimination of compound
binding of a compound at its site of action
because pH is diff physiological compartments varies
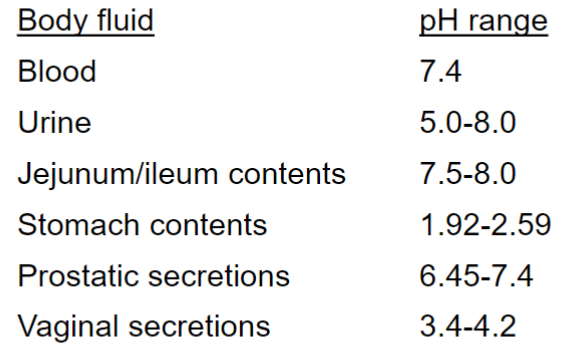
Weakly basic drug w/ pka ~ 9 (lec 3)
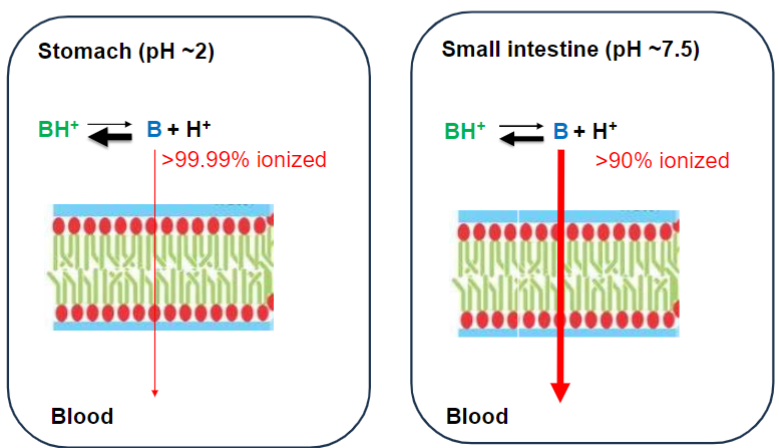
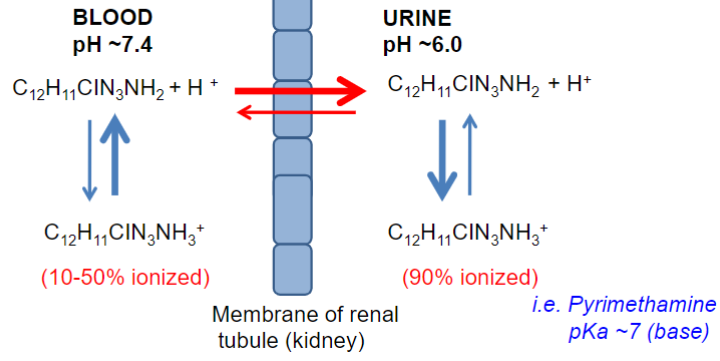
Elimination affected by “trapping” drug in compartment due to pH diffs (lec 3)
diffs in pH of fluid “compartments” can be important in excretion
BUT can also intentionally alter urinary pH to enhance renal excretion
Barriers to drug movement w/in body (lec 3)
Physical/anatomical barriers:
cell membranes, which preclude cellular passage
links formed btwn cells, which preclude intercellular passage of molecules
Functional barriers:
transport systems that can carry the drug out of cells, thereby lowering drug conc in tissues/compartments w/ those systems
Partition Coefficient (lec 3)
major factor w/ regards to drug permeation
higher Pow = more rapidly absorbed
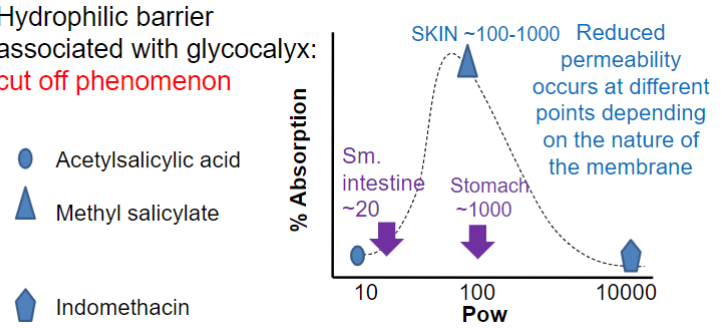
hydrophilic barrier associated w/ glycocalyx: cut off phenomenon
reduced permeability at diff pts depending on nature of membranes
small intestine has cut off ~20 bc microvilli leads to more of the small water layer on membranes
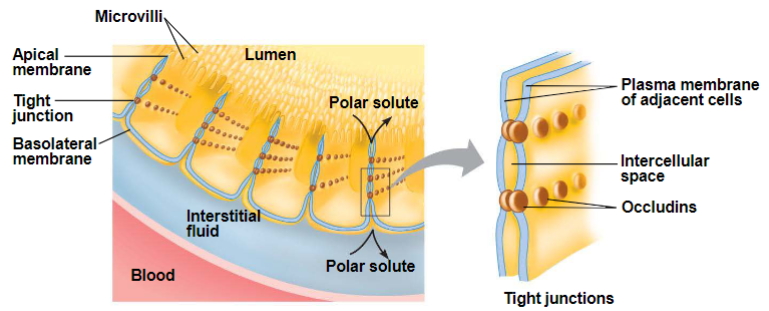
Physical barriers: epithelial cells (lec 3)
epithelial cells of skin, GI, bladder, cornea are joined by tight junctions/occluding zonulae
intercell spaces are completely blocked
drugs must pass through cells
Endothelial cells (lec 3)
capillaries w/ Maculae
most transient holes
intercell spaces
pinocytic vesicles
capillaries w/ fenestrations
excretory/secretory organs
long lasting
those <45kDa may pass through basement membrane
non protein bound molecules
capillaries w/ occluding zonula (BBB)
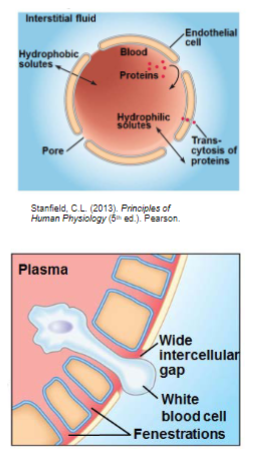
Physical barriers: BBB (lec 3)
Brain capillaries
endothelial cells joined by tight junctions/occluding zonulae
NO intercellular spaces
few transient fenestrae
major issue in drug targeting
even w/ exceptions to occluding zonulae of capillaries w/in brain, it is IMPERATIVE to note that MANY COMPOUNDS are excluded from CNS via BBB
drugs w/ desired CNS action should be lipophilic/utilize “help”
Physical barriers: Placenta (lec 3)
assume all drugs cross placenta
limited maternal blood flow into placenta therefore equilibration of drug btnw mom and fetus takes at least 10-15 mins
but physiochem properties of the drug will determine its relative ability to cross placenta (bio membrane)
degree of lipophilicity
drug size
extent of ionization
extent of plasma protein binding
drug transporters/efflux pumps
Functional barriers: the Transporters (lec 3)
transport systems that can carry the drug out of cells, thereby lowering drug conc in tissues/compartments w/ those systems
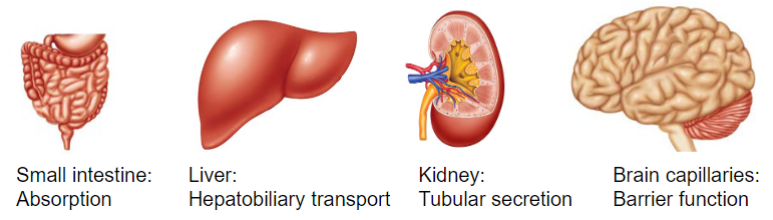
Sites of drug transport (lec 3)
some are located throughout the body (ubiquitous) and others have specific tissue expression
commonly found in enterocytes, hepatocytes, renal tubular cells and BBB epithelial cells
small intestine: absorption
liver: hepatobiliary transport
Kidney: tubular secretion
Brain capillaries: brain function
Polarization terminology (lec 3)
enterocytes (intestine): basolateral (blood), apical (gut lumen)
Hepatocytes (liver): sinusoidal (blood), Canalicular (bile)
Renal tubular cells (kidney): basolateral, apical (kidney lumen, urine)
Brain capillary endothelial cells (BBB): abluminal (brain), luminal (blood capillary)
location of transporters can affect time course of drug in body (absorption vs elimination)
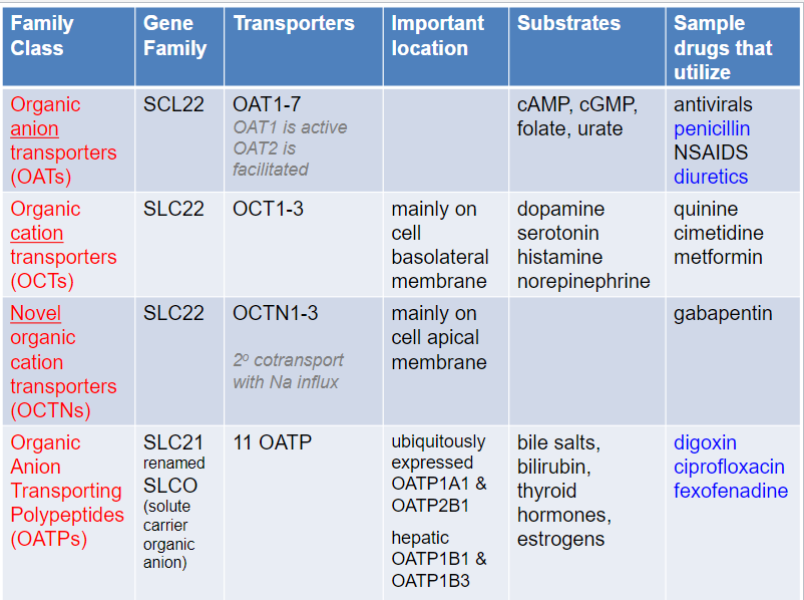
Solute Carrier (SLC) transporters (lec 3)
distinct from primary active transporters and ion channels, both structurally and functionally
localized to cellular membranes as well as organelle membranes (ex: SLC25 fam of mitochondrial transporters)
most are transport specific molecules, but some are broad-range (ex: SLC21 and SLC22 fams)
important drug targets (ex: SLC 6 + 12 fams)
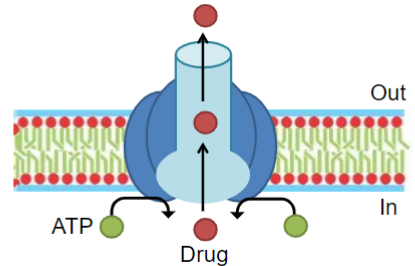
ATP binding cassette (ABC) transporters (lec 3)
substrates undergo primary active transport
hydrolysis of bound ATP powers unidirectional transport of substrates against their conc grad (usually out of cells)
important for limiting cellular exposure to drugs and toxins
beneficial when preventing unwanted exposure to environment toxins
can limit therapeutic efficacy of cytotoxic drugs such as chemotherapeutics and antibiotics
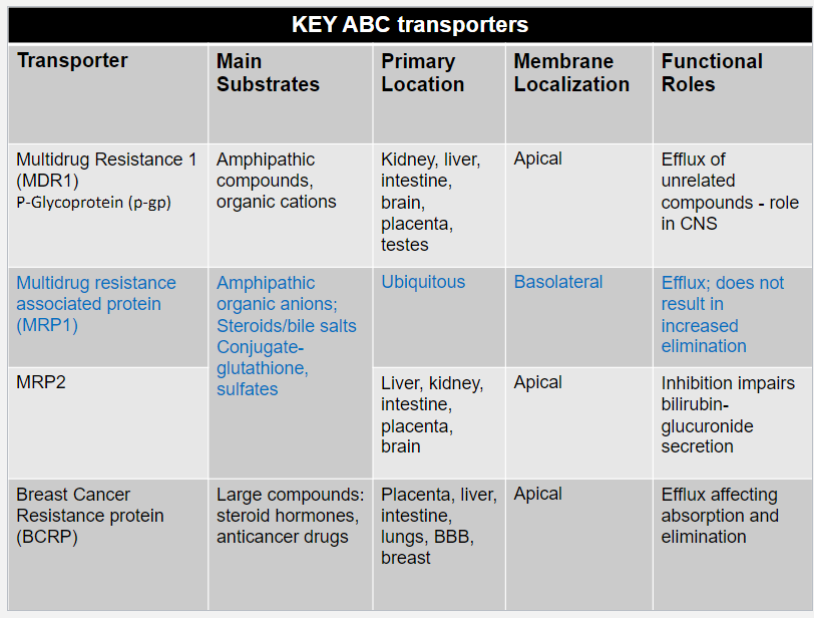
P-gp structure and binding (lec 3)
substrate spans membrane and becomes locked in a p-gp drug-binding pocket near the intracell leaf of membrane
2 ATP molecules bind to the intracellular ATP-binding sites
hydrolysis of ATP to ADP promotes conformational change in p-gp
substrate is released into extracell environment
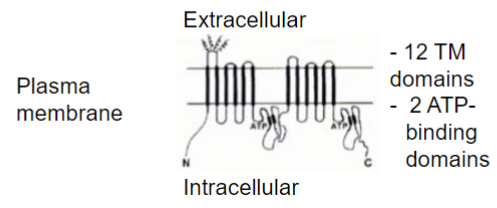
P-gp localization and direction of efflux (lec 3)
P-gp localization favours preferential transport of substrates out of tissues and into blood, feces, mucus or urine
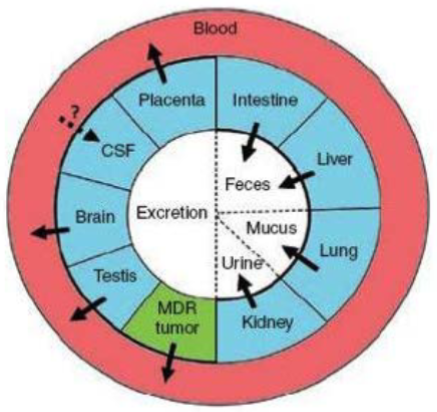
Multidrug resistance (lec 3)
ABC transporters w/ broad substrate specificity can affect therapeutic actions of various chemotherapeutics and antibiotics when expressed in tumor/bacterial cells
these transporters contribute to multidrug resistance: ability of microorgs/tumor cells to resist the action of a wide variety of chems
main multidrug resistance transporters are p-gp, MRPs, and BCRP
inhibiting these transporters is common practice alongside standard chemotherapy
Transporters can lead to… (lec 3)
therapeutic failure
significant in infectious disease and cancer drug therapy
anti-infectious disease agents: several genetic polymorphisms P-glycoprotein, anionic + cationic and other transporters
multiple cancers: ABC transports (P-gp, MRP1, and BCRP) up-regulated in diff tumors + over-expressed in various cancer cells
increased AE
improve efficacy and safety
remove drugs from “locations” (placental transfer)
digoxin, glyberide
increase distribution (deliver drugs to targets)
statins
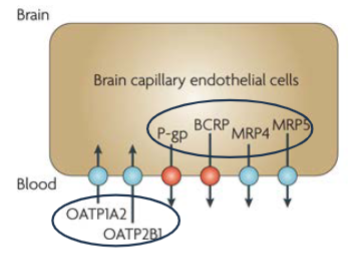
Ex: BBB (lec 3)
Epithelial occluding zonulae (tight junctions) and luminal expression of p-gp, BCRP and MRP efflux transporters all heavily limit drug transport to the brain (protects brain)
OATP1A2 is one of the few luminal influx transporters at the blood brain barrier, and substrates of this transporter can achieve significant brain concentrations