lec 12 - PK of monoclonal antibodies (glassman)
1/48
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
49 Terms
zauberkugel - the ‘magic bullet’
concept proposed by paul ehrlich in 1960
hypothesis = the ideal drug will selectively kill a microbe but will NOT harm healthy cells
salvarsam → anti-syphilis drug discovered by ehrlich that had minimal adverse effects (the first magic bullet)
are monoclonal antibodies “magic bullets'“?
very high affinity and selectivity for pharmacologic targets
relatively low distribution into non-eliminating organs
antibodies as drugs - brief history
1890 → kitasato shibasaburo and emil von behring develop ‘antitoxins’ from the serum of immunized animals to treat diphtheria (bacterial infection)
the principle of convalescent plasma therapy
1944 → edwin cohn describes a method to isolate the immunoglobulin fraction of plasma (basis of IVIg therapy)
1975 → george kohler and cesar milstein describe the methodology to produce monoclonal antibodies
1986 → first mAb (orthoclone) approved by the FDA
prevention of kidney rejection in transplant
withdrawn from the market due to toxicities
2000 → first antibody-drug conjugate (mylotarg) approved
treatment of acute myeloid leukemia
withdrawn from the market in 2010 (reintroduced in 2017)
2002 → first fully human mAb (humira) approved
treatment of rheumatoid arthritis
2016 → first biosimilar mAb (inflectra) approved
2021 → 100th mAb product approved by FDA
antibodies are an important therapeutic class
3 of the top 10 selling drugs globally in 2022 were mAbs
mAbs are projected to be 5 of the top 10 selling drug since 2023
antibody therapeutics are a growing drug class
12 antibody therapeutics gained first approval in the Us or EU from jan-nov 2022
24 more were undergoing review as of nov 2022
23 were expected to be submitted for approval in 2023
~140 in late stage clinical trials
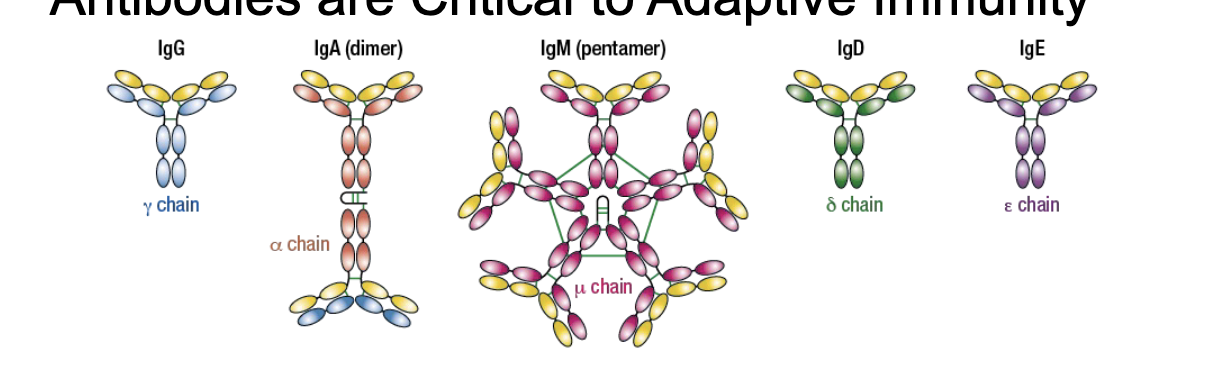
what are antibodies?
types of antibodies
IgG = γ chain
IgA (dimer) = α chain
IgM (pentamer) = μ chain
IgD = δ chain
IgE = ε chain
antibodies are released in response to antigens → part of the memory response
each antibody monomer is a Y shaped molecule, composed of 2 identical heavy chains and 2 identical light chains
some antibodies function as multimers
within a species, the Fc regions of antibodies within a given class are the same
in mammals, antibodies can be distinguished into 5 classes by their heavy chains
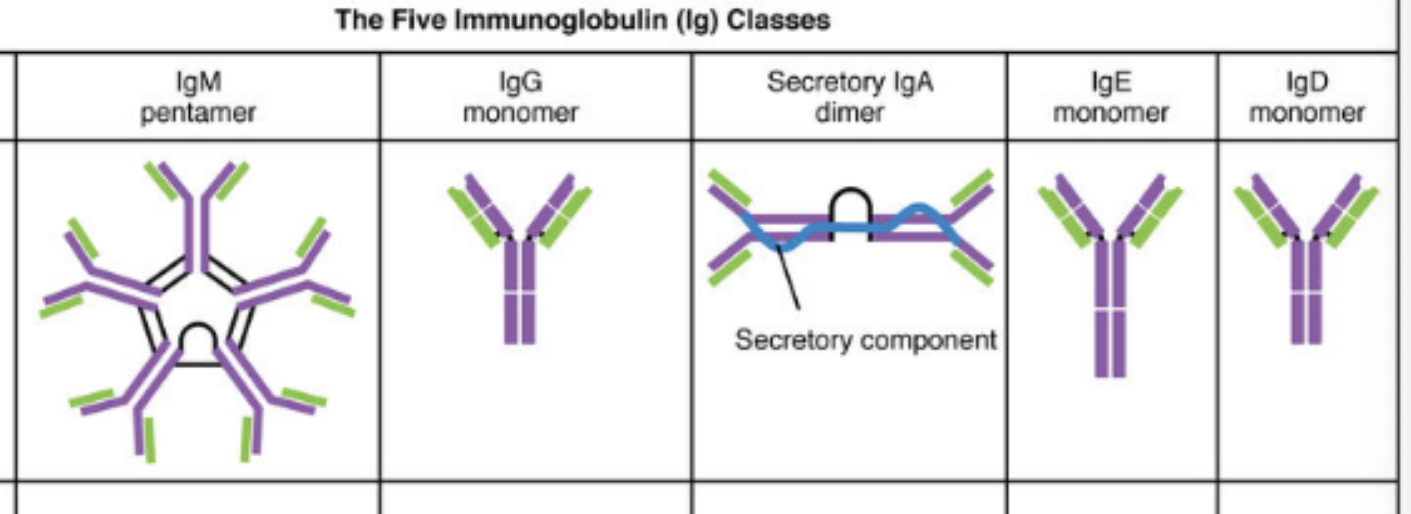
key features of mammalian immunoglobulin
IgG(monomer)
heavy chains = γ
# of antigen binding sites = 2
molecular weight = 150,000 Da
percentage of total antibody in serum = 80%
crosses placenta = yes
fixes complement = yes
Fc binds to = phagocytes
function = main blood antibody of secondary responses, neutralizes toxins, opsonization
IgA (secretory dimer)
heavy chain = α
# of antigen binding sites = 4
molecular weight = 385,000 Da
percentage of total antibody in serum = 13%
crosses placenta = no
fixes complement = no
Fc binds to = nothing
function = secreted into mucus, tears, saliva, colostrum
IgM (pentamer)
heavy chains = μ
# of antigen binding sites = 10
molecular weight = 900,000 Da
percentage of total antibody in serum = 6%
crosses placenta = no
fixes complement = yes
Fc binds to = nothing
function = main antibody of primary responses, best at fixing complement, the monomer form of IgM serves as B cell receptor
IgD (monomer)
heavy chains = δ
# of antigen binding sites = 2
molecular weight = 180,000 Da
percentage of total antibody in serum = 1%
crosses placenta = no
fixes complement = no
Fc binds to = nothing
function = B cell receptor
IgE (monomer)
heavy chains = ε
# of antigen binding sites = 2
molecular weight = 200,000 Da
percentage of total antibody in serum = 0.002%
crosses placenta = no
fixes complement = no
Fc binds to = mast cells and basophils
function = antibody of allergy and anti-parasitic activity
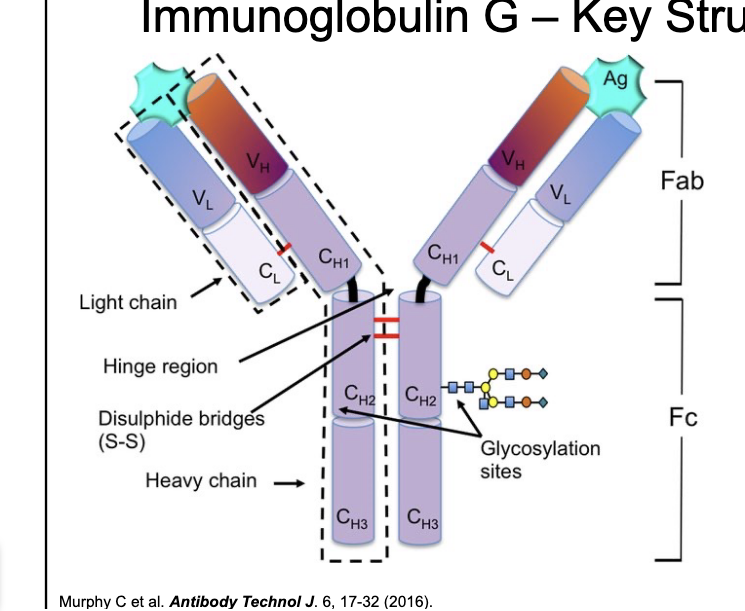
IgG - key structural features
IgG structural features
molecular weight ~150 kDA
consists of 2 heavy and 2 light chains held together by disulfide bonds
IgG key domains
fragment antigen binding
contains CDRs → key site of antigen binding
fragment crystallizable
key site for effector functions
confers long half-life on IgG
IgG - the primary serum Ig
IgG isotypes = different functions in the body
subtle changes in structure correspond to differences in function
development of antibodies as therapeutics
george kohler and cesar milstein won the 1984 noble prize in physiology or medicine for development of monoclonal antibodies
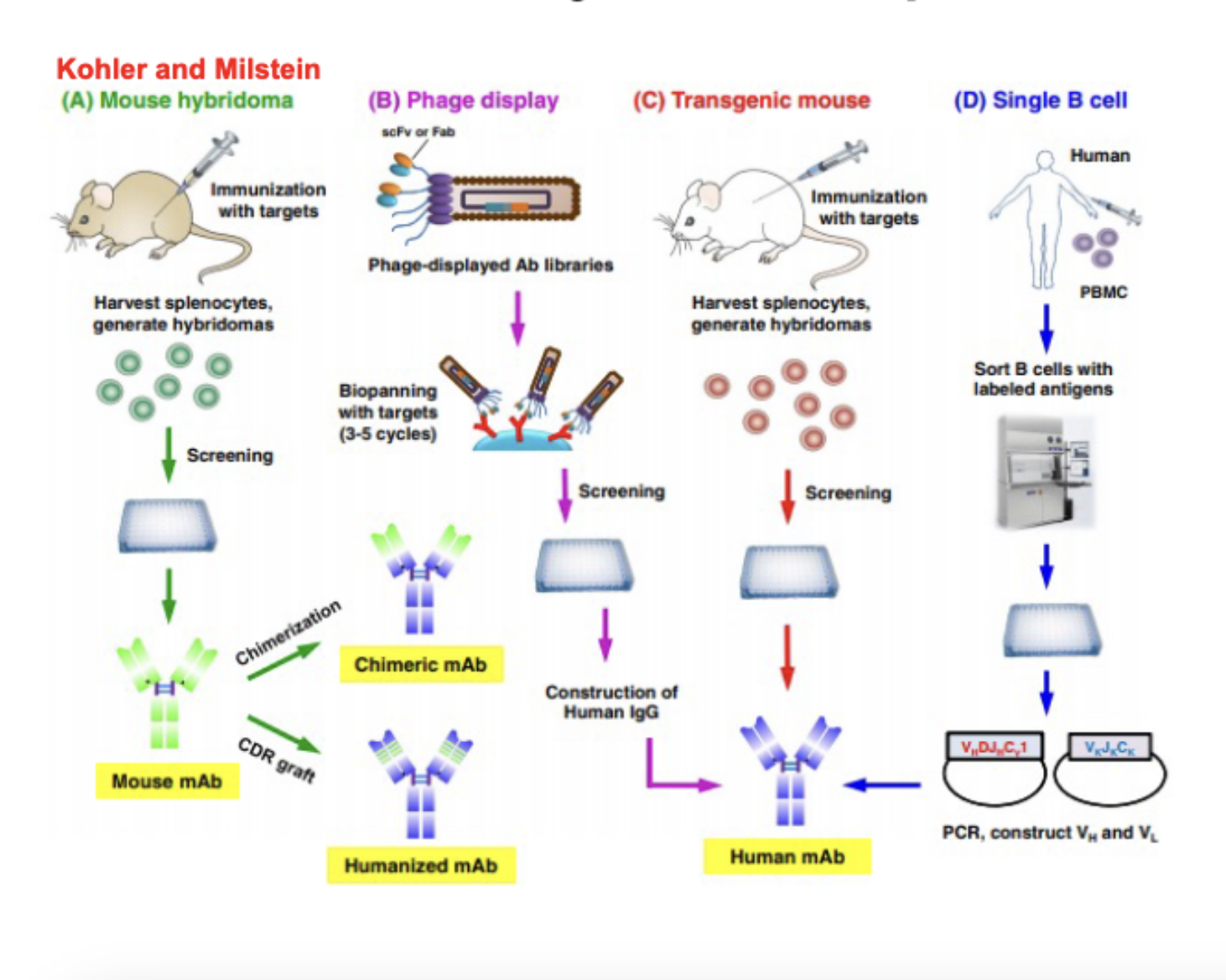
approaches for antibody development
mouse hybridoma
immunize mouse with targets
harvest splenocytes, generate hybridomas
screen the hybridomas
end up with mouse mAb and can either:
chimerization → chimeric mAb
CDR graft → humanized mAb
phage display
phage-displayed Ab libraries
biospanning with targets (3-5 cycles)
screening
construction of human IgG
human mAb
transgenic mouse
immunize mouse with targets
harvest splenocytes
screening
human mAb
single B cell
collect PBMC (which have B cells) from humans
sort B cells with labeled antigens
PCR construct VH and VL
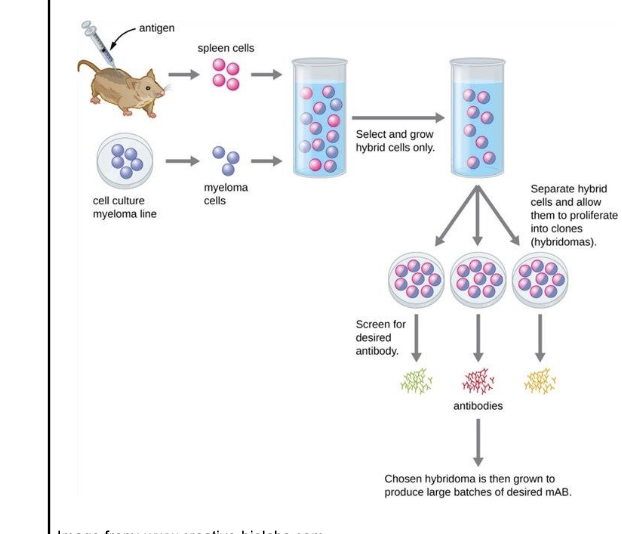
hybridoma technology
animals (mouse, rat, rabbit) are repeatedly immunized with antigen of interest
splenocytes are fused with myeloma cells
apply pressure (HAT media) to select for fused cells
separate fused cells into individual colonies (single cell selection)
screen for binding to target of interest via methods such as ELISA or SPR
drawbacks for clinical development
NOT amenable to development of human antibodies
no direct path to engineer antibodies for optimal behavior
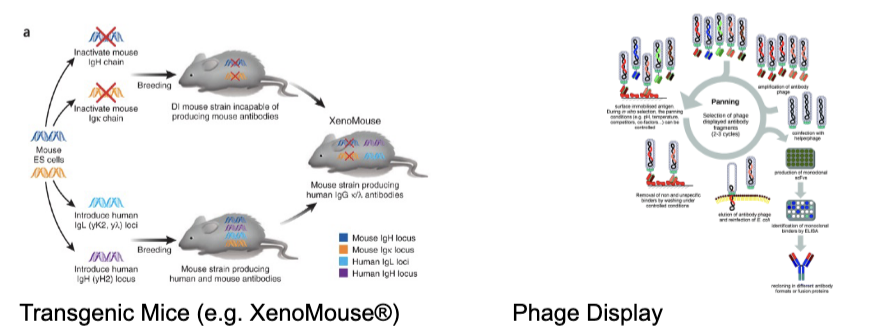
humanization of therapeutic antibodies
molecular biology has allowed movement from fully mouse mAbs → fully human
increasing “human” content correlates with decreased immunogenicity (reduction in the ability of a substance to stimulate an immune response; so the immune system wont recognize the antibody as something ‘bad’) and increased half life
recent developments in mAb
transgenic mice
human immunoglobulin genes knocked in, rodent genes knocked out
allows production of fully human antibodies in rodents
ex. vectibix (anti-EGFR)
phage display
in vitro selection of candidates and affinity maturation
can produce antibodies derives from numerous species
ex. humira (anti-TNF-alpha)
george smith and gregory winter won the 2018 nobel prize in chemistry for development of phage display
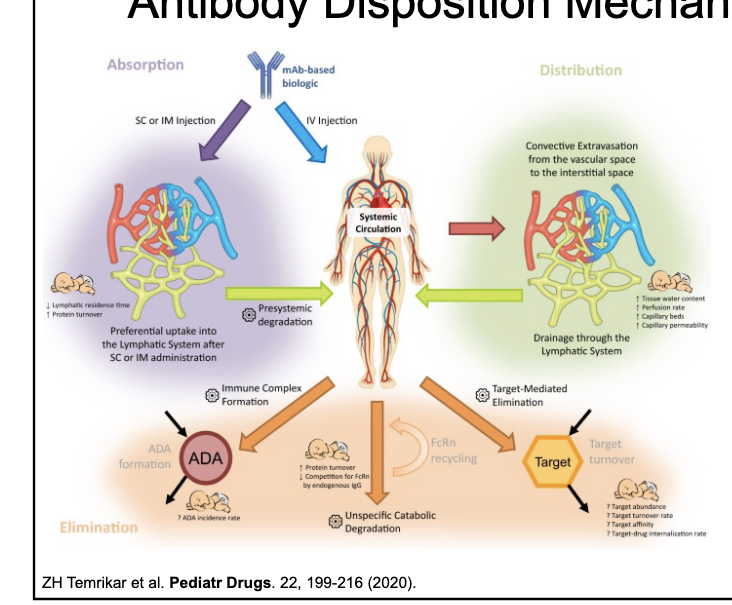
antibody disposition mechanisms
antibodies are NOT
orally bioavailable
extensively protein bound
metabolized by CYP450s (or other DME)
renally filtered
substrates for ABC or SLC transporters
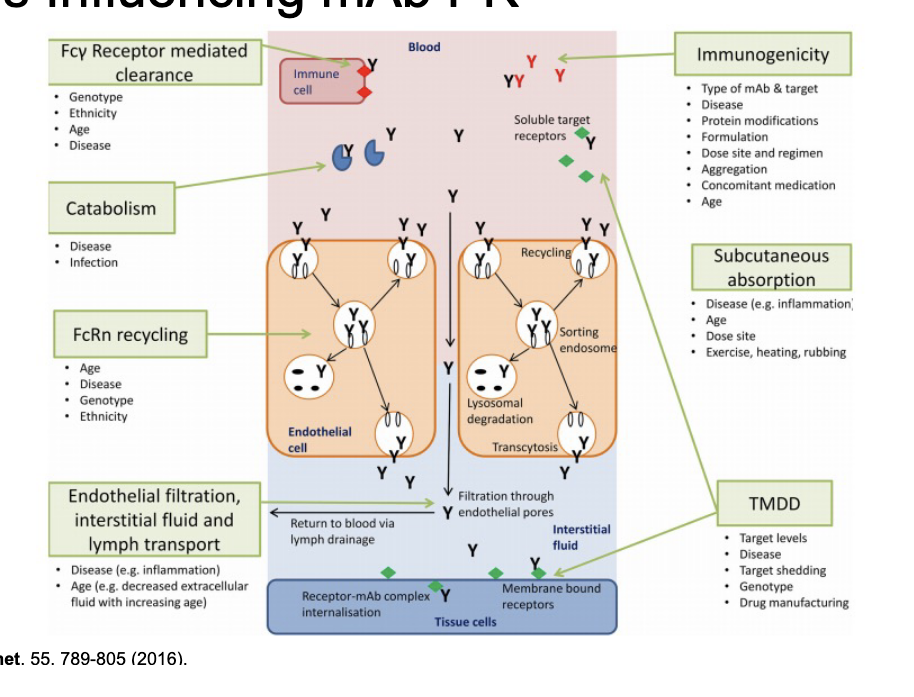
factors influencing mAb PK
Fcy receptor mediated clearance
genotype
ethnicity
age
disease
catabolism
disease
infection
FcRn recycling
age
disease
genotype
ethnicity
immunogenicity
type of mAb & target
disease
protein modifications
formulation
dose site and regimen
aggregation
concomitant medication
age
subcutaneous absorption
disease
age
dose site
exercise, heating, rubbing
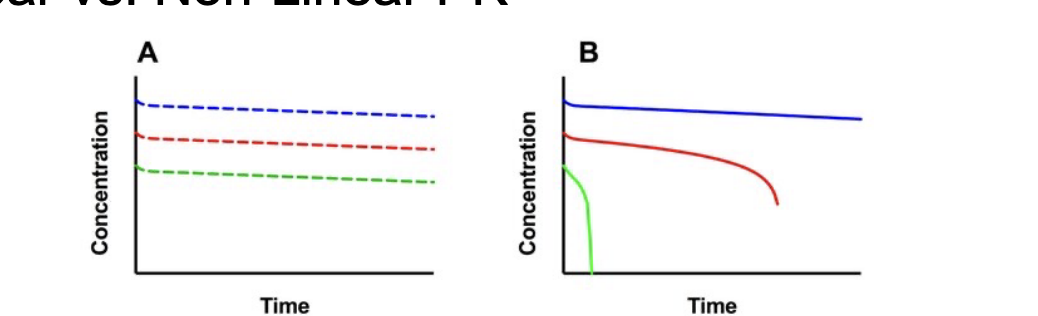
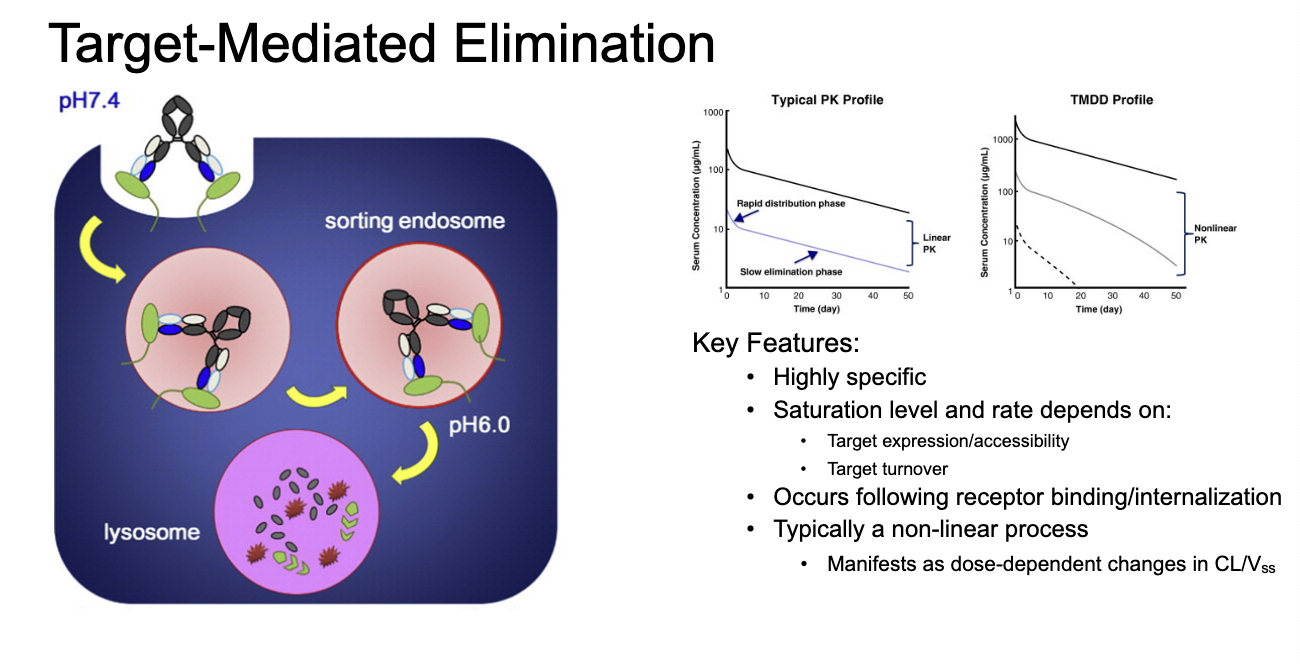
linear vs non-linear PK
linear PK: all ADME processes can be characterized by processes that are dose/concentration-independent
all rate processes can be described by first-order kinetics
results in PK profiles that are dose-proportional
non-linear PK: saturable processes (non-linear rates) are requires to describe some or all ADME processes
results in PK profiles that are NOT dose-proportional
how to identify non-linear PK
systematic trends observed in dose-normalized PK profile and NCA parameters with non-linear PK
greater than dose-proportional increases in AUC with dose suggests saturable clearance mechanism
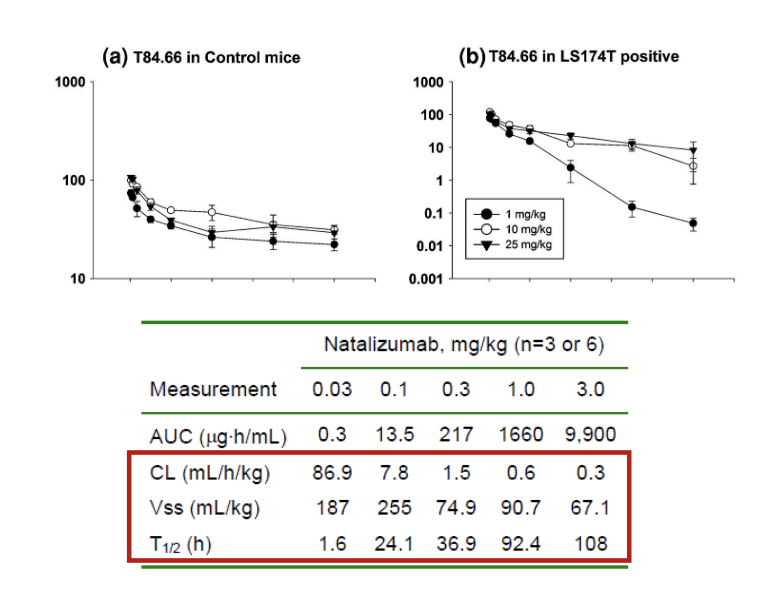
linear antibody PK-
general expectations
biexponential PK profile typically observed
long terminal half life (2-3 weeks)
low Vd
characteristics of mAbs with ‘typical’ PK
NO target-mediated drug disposition
typically bind to soluble targets
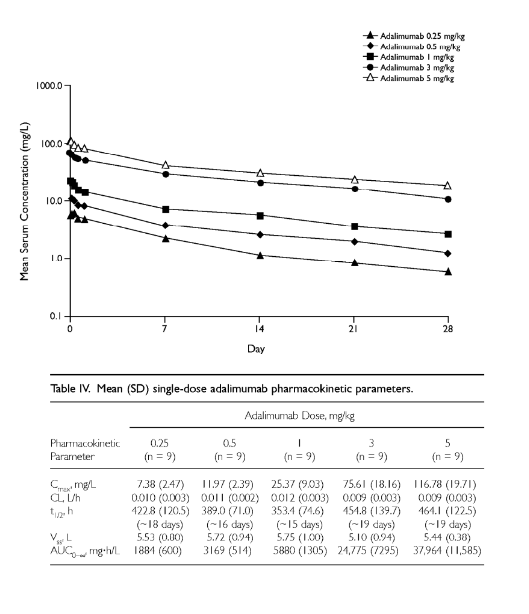
non-linear antibody PK
general expectations
greater than proportional increases in AUC
dose-dependent changes in CL, Vd, t1/2
characteristics of mAbs with non-linear PK
often due to target-mediated drug disposition
typical targets are either:
membrane bound - HER2, EGFR, CD20
soluble and form immune complexes - IgE
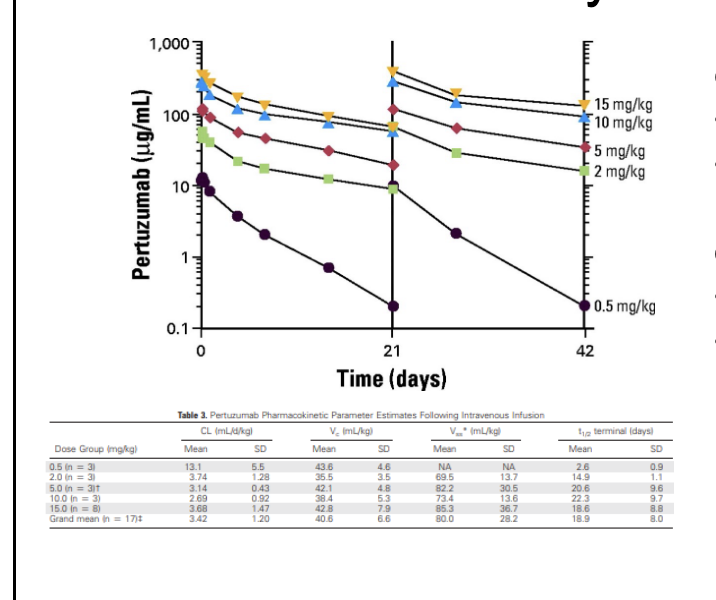
IgG elimination
is concentration dependent
inverse relationship between serum IgG concentrations and half life
shorter half life → greater IgG concentrations and vice versa
brambell proposed that this was due to the presence of salvage receptor for IgG
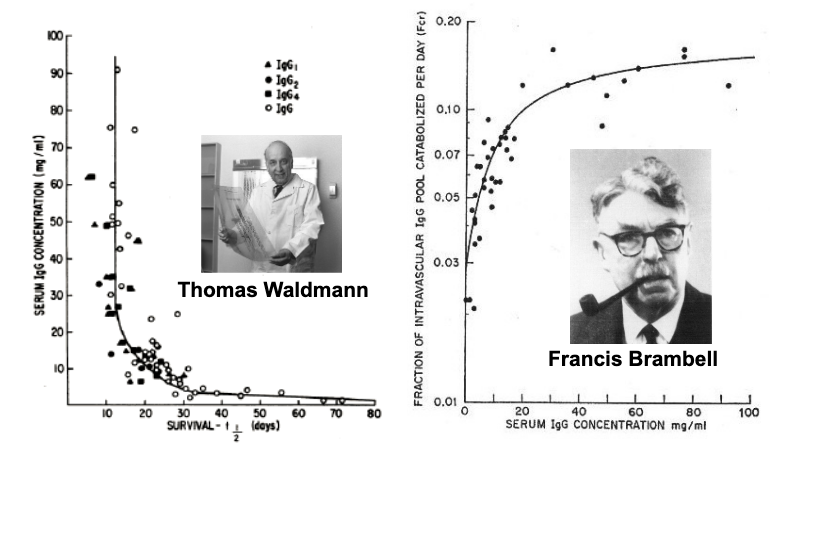
neonatal Gc receptor - FcRn
identified as the protection receptor for IgG by 3 groups in 1996
functions as a 65 kDA heterodimer
15 kDa light chain - β-2-microglobulin
50 kDa heavy chain - MHC1-like protein
expressed throughout the body
endogenous roles
IgG/albumin homeostasis
maternal transfer of IgG to fetus/neonate
how does FcRn protect IgG from catabolism?
proteins are internalized via fluid-phase pinocytosis (NO IgG-FcRn binding)
endosomal acidication causes protonation of key histidines in IgG and FcRn
protonation leads to favorable IgG-FcRn binding
IgG-FcRn complexes are recycled to the cell surface
before this, non-receptor bound proteins are degraded in the lysosome
following exposures to extracellular pH, IgG-FcRn complexes dissociate
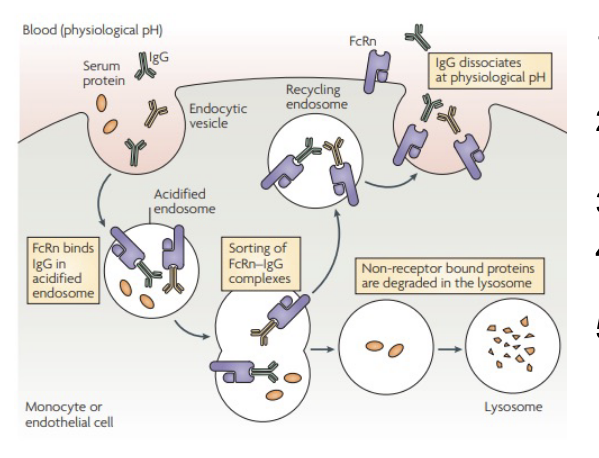
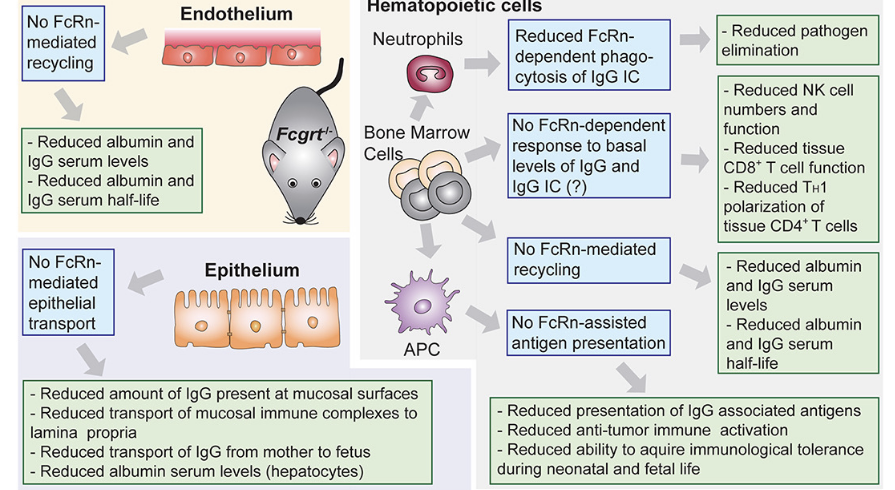
where does FcRn function?
main focus: protection of IgG from catabolism
endothelium
NO FcRn-mediated recycling leads to:
reduced albumin and IgG serum levels
reduced albumin and IgG serum half-life
epithelium
NO Fc-Rn-mediated epithelial transport leads to:
reduced amount of IgG present at mucosal surfaces
reduced transport of mucosal immune complexes to lamina propia
reduced transport of IgG from mother → fetus
reduced albumin serum levels
hematopoietic cells
bone marrow cells
NO FcRn-dependent response to basal levels of IgG and IgC leads to:
reduced NK cell numbers and function
reduced tissue CD8T cell function
reduced TH1 polarization of tissue CD4*T cells
NO FcRn-mediated recycling
reduced albumin and IgG serum levels
reduced albumin and IgG serum half-life
neutrophils (which come from bone marrow cells)
reduced FcRn-dependent phagocytosis of IgG leads to
reduced pathogen elimination
APC (which comes from bone marrow cells)
NO FcRn-assisted antigen presentation leads to
reduced presentation of IgG associated antigens
reduced anti-tumor immune activation
reduced ability to acquire immunological tolerance during neonatal and fetal life
routes of admin - mAb therapeutics
oral
bioavailability = negligible
tmax = N/A
barriers
low gastric pH
GI tract enzymes
Gi epithelium
IV
bioavailability = 100%
tmax = immediately
barriers = N/A
Subq and IM
bioavailability = 52-80%
tmax = 6-8 days
barriers
lymphatics
immune cells
SC admin is very attractive for pharma development
absorption is largely via the lymphatics
resonable bioavailability
less pain compared to IM injections
more convenient to pts compared to IV
significant investment into absorption enhancing strategies
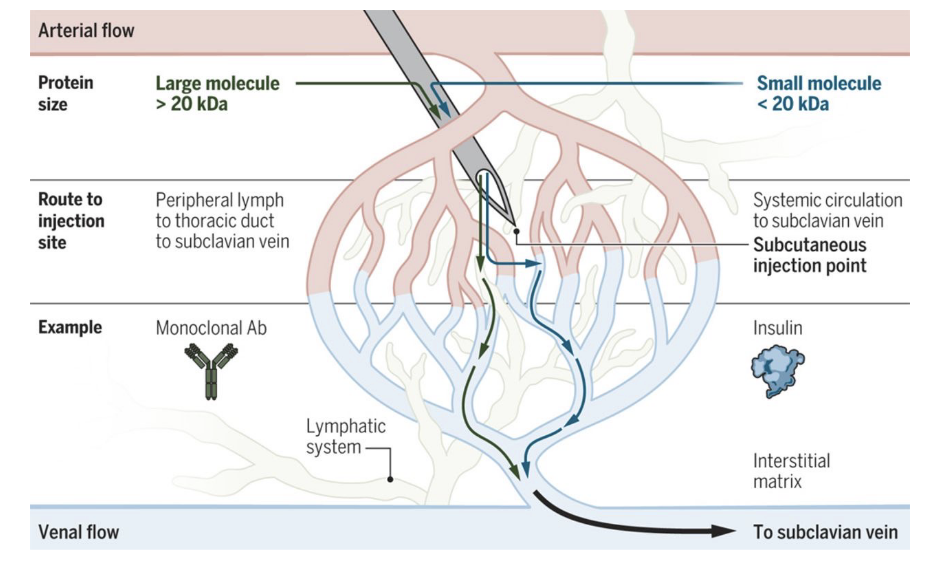
SC absorption of mAbs
for large molecule >20 kDa
route to injection site = peripheral lymph → thoracic duct → subclavian vein
for small molecule <20 kDa
route to injection site = systemic circulation to subclavian vein
PK following SC dosing
relatively slow absorption → tmax ~1 week
reasonable bioavailability (50-90%)
often only see monoexponential PK
first phase may be masked by absorption
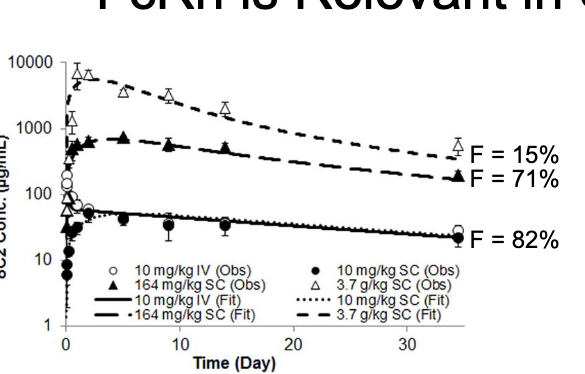
FcRn is relevant in SC absorption
bioavailability of mAbs decrease at high dose levels
genetic knockout of FcRn reduces bioavailability of mAbs to ~30%
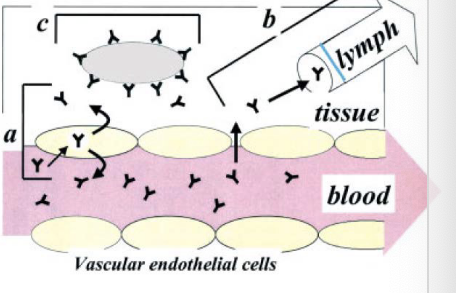
IgG distribution
pathway: diffusion
efficiency: negligible
barriers: cell membrane
pathway: bulk fluid flow
efficiency: tissue-dependent
barriers
endothelial pores
interstitial pressure
pathway: pinocytosis
efficiency: relatively low
barriers: endocytic rate
pathway: receptor-mediated uptake
efficiency: target dependent
barriers
target expression
target accessibility
endocytic rate
binding affinity
tissue concentrations « plasma concentrations
slow extravasation into tissues
relatively rapid drainage via lymphatics
small Vss from NCA analysis
typically close to plasma volume
true Vss measurement requires tissue concentrations of mAbs
IgG elimination
intracellular catabolism
following fluid-phase endocytosis (non-specific)
limited by interactions with FcRn
target mediated elimination (specific)
cell surface receptor: internalization
soluble target: formation of large complexes - phagocytosis
receptor-mediated elimination (Fcy receptors)
may trigger endocytosis and catabolism
receptor-mediated protection: FcRn
level of plasma concentration of IgG
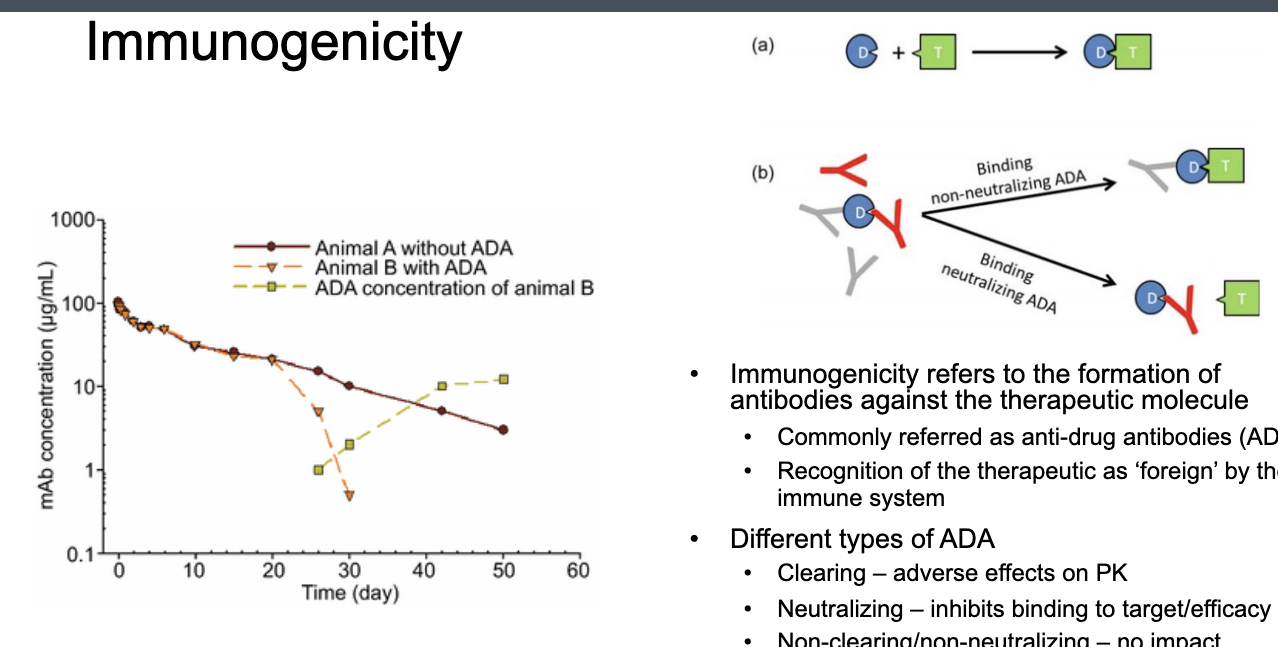
immunogenicity
formation of anti-drug antibodies (ADA) resulting in accelerated clearance or loss of activity
renal clearance of IgG
generally accepted molecular weight cutoff for glomeular filtration ~60 kDa
in healthy individuals, <0.01% of IgG is expected to pass thru glomerulus
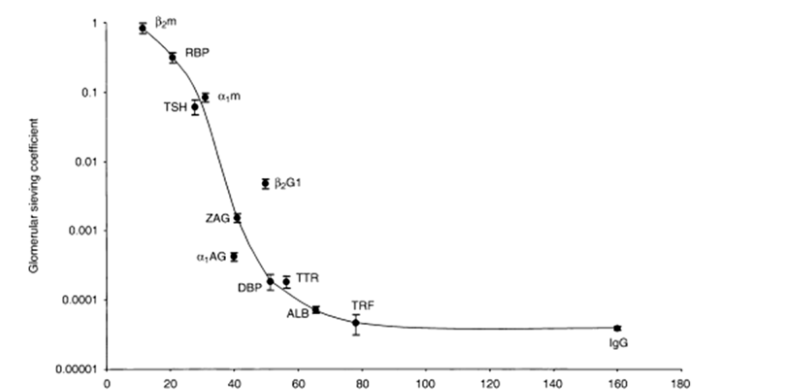
elimination following pinocytosis
non-specific
can occur in any cell
results in catabolism → component amino acids
degradation products = non-toxic
efficiency is blunted by FcRn recycling
requires extremely high dose of IgG to saturate
at typical mAb doses, linear process
target-mediated elimination
highly specific
saturation level and rate depends on
target expression/accessibility
target turnover
occurs following receptor binding/internalization
typically in non-linear process
manifests as dose-dependent changes in CL/Vss
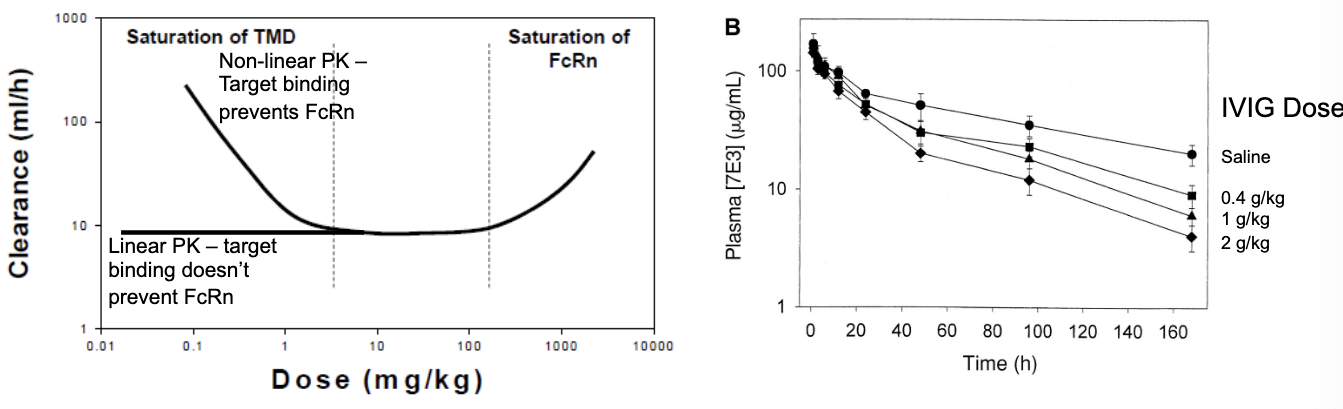
IgG clearance balances 2 saturable processes
at low doses, any target mediated processes will dominate the profile
dose-dependent decreases in CL until saturation
at low doses = plenty of target receptors available so high CL → as dose increases, more IgG is in the system but but the # of target receptors is the same so CL goes down → once targets are saturated, CL does not change
target binding competes with FcRn → target binding prevents FcRn
target is usually not saturated
@ low doses, IgG binds to its target receptors → bound complexes are degraded → high clearance → as dose increases, target receptors become saturated, meaning not enough receptor to bind IgG so more of it remains in circulation so clearance decreases
target binding prevents FcRn
at linear PK the clearance is the same b/c target binding does NOT apply
target binding is saturated so NO degradation so only FcRn is active so its constant CL
at extremely high doses, FcRn will be saturated
dose dependent increases in CL
greater clearance because all FcRn is saturated so it can’t protect IgG anymore so CL goes up
therapeutic mAbs - target types
soluble
target is generally present in circulation
ex. vascular endothelial growth factor
membrane, non-internalizable
target is expressed on cell surface but does NOT endocytose
ex. CD20
membrane, internalizable
target is expressed on the cell surface but endocytoses (either naturally or stimulated by binding)
ex. epidermal growth factor receptor
membrane, shaddable
target is expressed on the cell surface but may be released into extracellular space
ex. human epidermal growth factor recepetor 2
target properties influence PK of mAbs
immunogenicity
immunogenicity refers to the formation of antibodies against the therapeutic molecule
commonly referred as anti-drug antibodies (ADA)
recognition of the therapeutic as ‘foreign’ by immune system
different types of ADA
clearing → adverse effects on PK
neutralization → inhibits binding to target/efficacy
non-clearing/non-neutralization → NO impact
factors associated with immunogenicity
duration of therapy
incidence typically increases with treatment time
ex. infliximab
ADA rarely appears prior to 2 months
after 12 months, >90% incidence
dose
ADA more frequently detected at lower doses of mAb
NB: may be due to assay interference
route of admin
ADA often found more frequently with SC/IM dosing
frequency/onset of ADA CANNOT be predicted
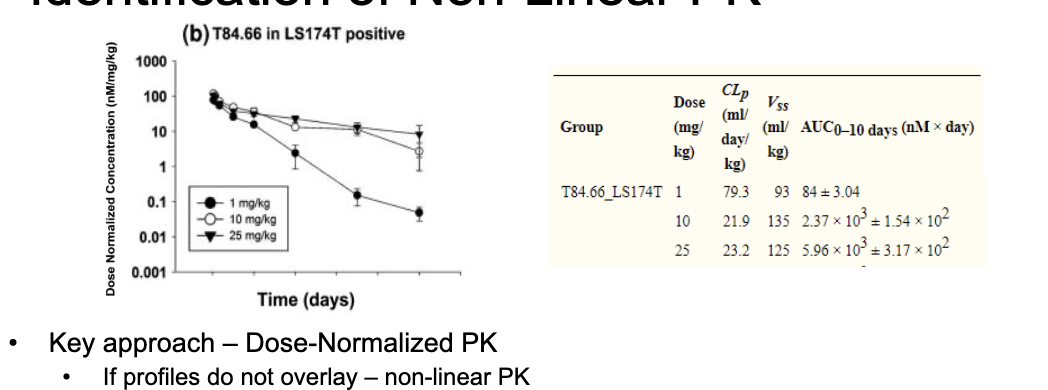
identification of non-linear PK
key approach - dose-normalized PK
if profiles do NOT overlay = non-linear PK
in example, lower relative exposure at 1 mg/kg dose compared to 10 and 25 mg/kg
other metrics - NCA derived parameters
check if CL and Vss have any trends with dose
in example, CL decreases and Vss increases with increasing dose
for mAbs, non-linear PK is often attributable to target-mediated disposition
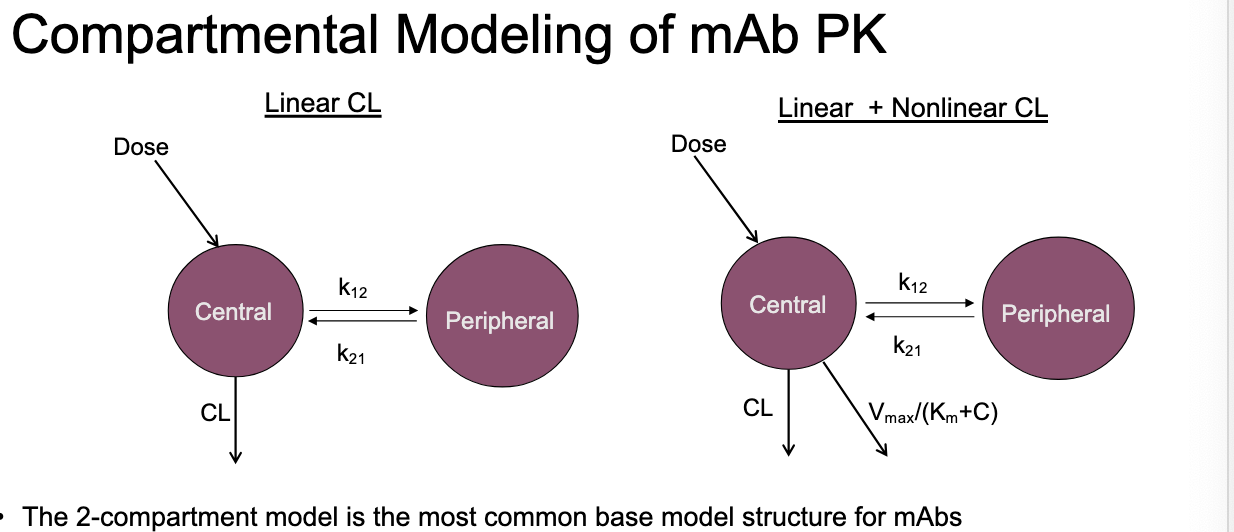
compartmental modeling of mAb PK
2 compartment model = most common base model structure for mAbs
clearance pathways are described using linear, non-linear or combo of terms
with extravascular admin, model is sometimes compressed to 1-CM structrue
biphasic PK profile is NOT always apparent for SC dosing
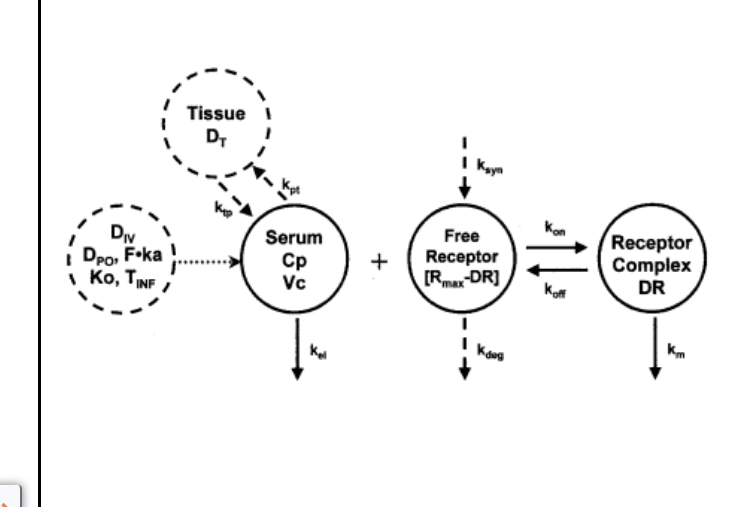
target-mediated drug disposition (TMDD)
mechanism: high affinity binding of a large fraction of the dose to its pharmacologic target will significantly impact PK
hypothesis made based on the increasing # of drugs in development with high affinity for targets
general expectations
NCA-derived Vss and CL will decrease with increasing dose to a limiting value
greater than dose proportional changes in AUC at low doses
AUC = total drug exposure
@ low doses → most of the drug gets cleared b/c targets are NOT saturated (CL is high) but as dose increases the targets start to fill up and less of the drug is cleared via TMDD so more remains in circulation → hence, small increase in dose results in a larger than expected change in AUC
at higher doses this is not the case b/c targets are saturated so at that point nothing is getting cleared (CL rate stays the same) there is more of a difference at low doses b/c CL is a factor
mathematical modeling of TMDD
accounts for:
kinetics of drug-receptor association and dissociation
receptor turnover
kinetics of drug-receptor complex elimination
in many cases, the kinetics of binding occurs much more quickly than other PK processes
binding parameters might NOT be identifiable when fitting the model
published approximations that assume equilibrium binding
characteristics of TMDD for mAbs
key features
non-linear PK
dose-dependent decreases in clearance
typically observed for mAbs against cell surface targets
TMDD may occur with soluble targets, but less frequent observation
TMDD is saturable in nature
dependent on target expression, accessibility, turnover
PK studies at a range of doses are required to characterize TMDD
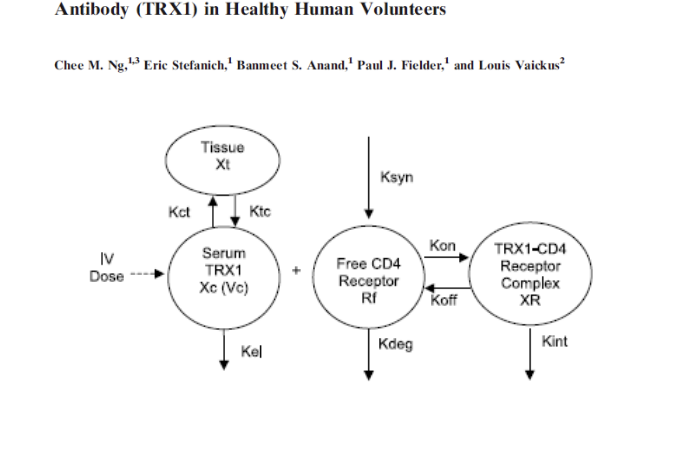
application of TMDD models - TRX1
TMDD model is able to well characterize
plasma TRX1 PK
free and total CD4 concentrations (PD)
model-estimated affinity does NOT agree with in-vitro measurements
KD (model) = 19.4 nM
KD (measured) = 0.6 nM
TMDD models are useful tools to characterize blood PK data
use caution with extrapolation of results beyond the range of data used for fitting

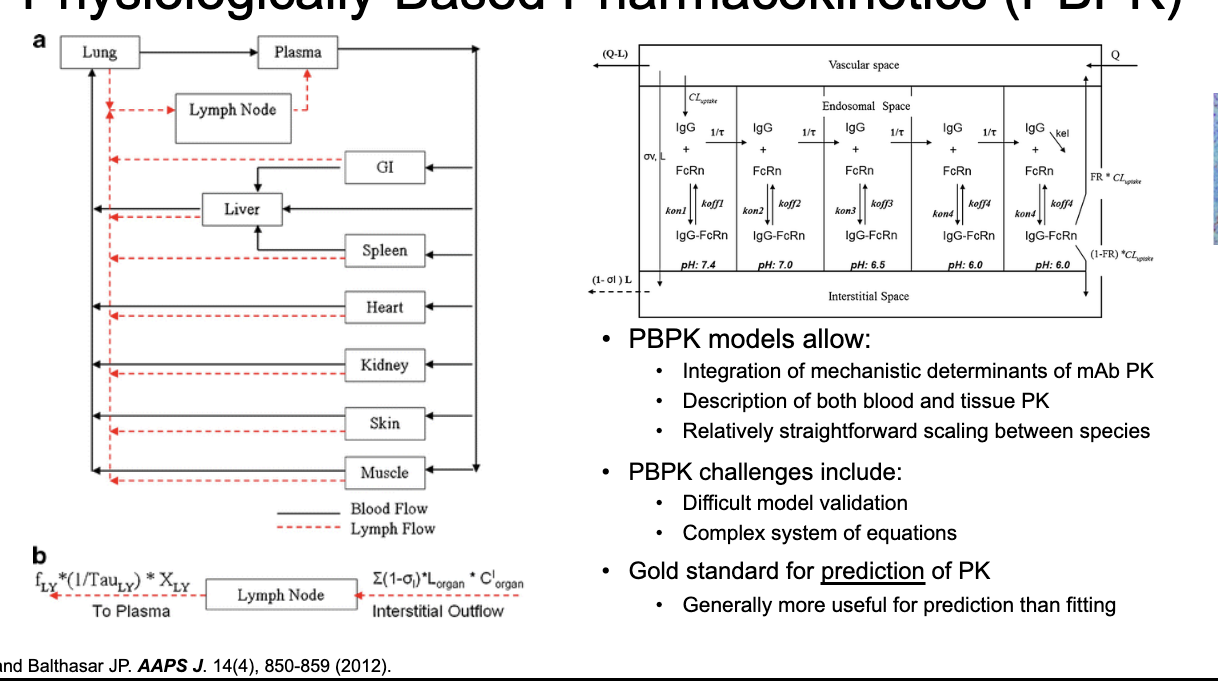
physiologically-based PK (PBPK)
PBPK models allow:
integration of mechanistic determinants of mAb PK
description of both blood and tissue PK
relatively straightforward scaling between species
PBPK challenges:
difficult model validation
complex system of equations
gold standard for prediction of PK
generally more useful for prediction than fitting
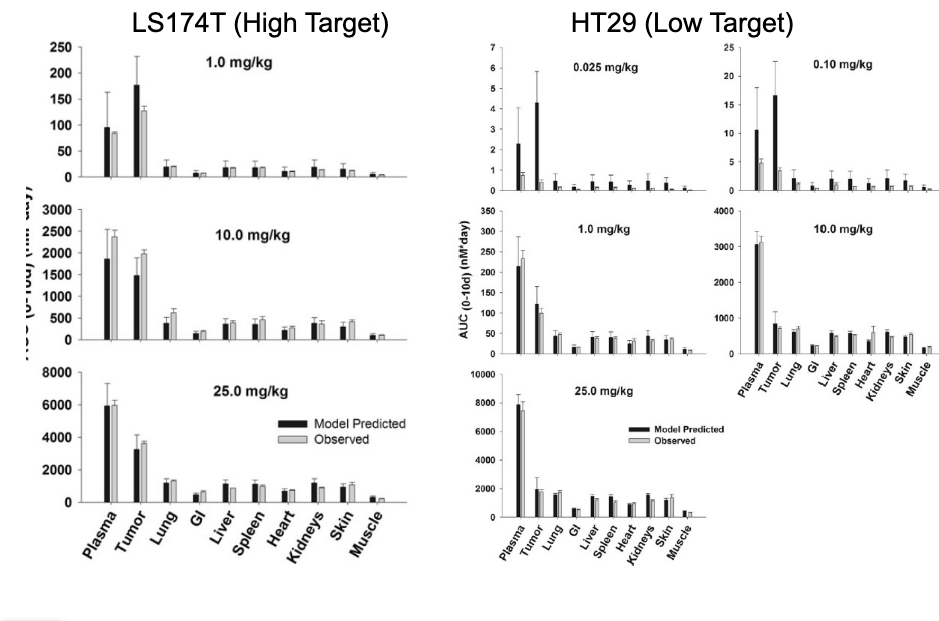
PBPK as predictive tool
PBPK model was used to make a priori predictions of the PK of an anti-CEA mAb
2 tumor models (high/low target)
NO target expression
WITHOUT fitting any parameters, the model was able to make relatively good predictions of both blood and tissue PK across a range of doses
PBPK = excellent predictive tool
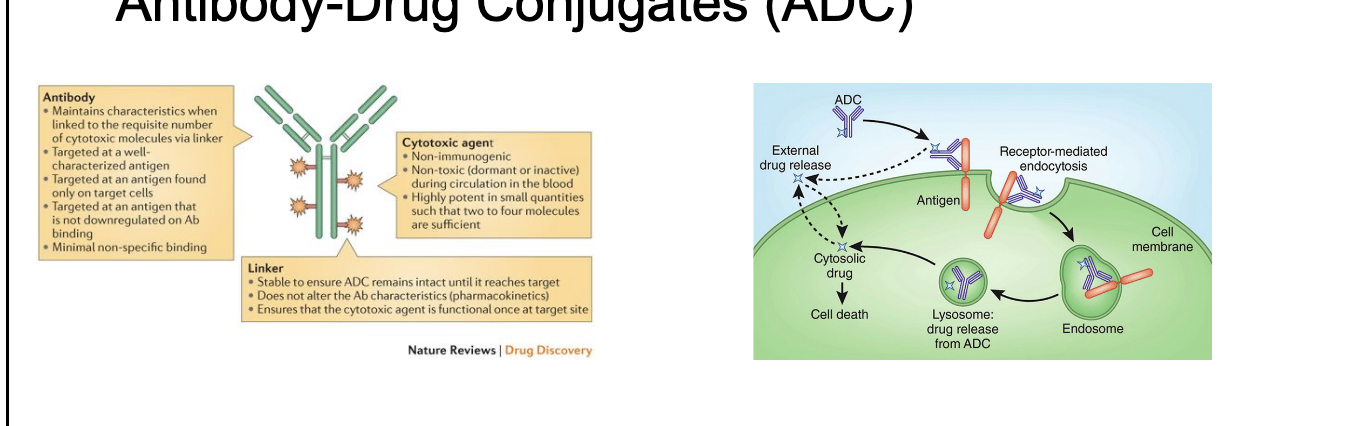
antibody-drug conjugates (ADC)
ADCs combine the selectivity of mAbs with the potency of chemotherapeutic drugs
careful selection of target antigen is critical to avoid severe toxicities
primary application: oncology
some investigational compounds in the area of infectious disease
more than 10 FDA-approved ADCs
ADC PK
ADC PK measurements require assays for several analytes
conjugated mAb (ADC)
free/total mAb
free/total drug
drug to antibody ratio (DAR) affects PK/PD
increased DAR → faster clearance
more drug attached → more hydrophobic and unstable ADC may be which can increase uptake by liver or spleen
some degree of deconjugation can be expected with time
understanding PK of all analytes = critical to proper characterization
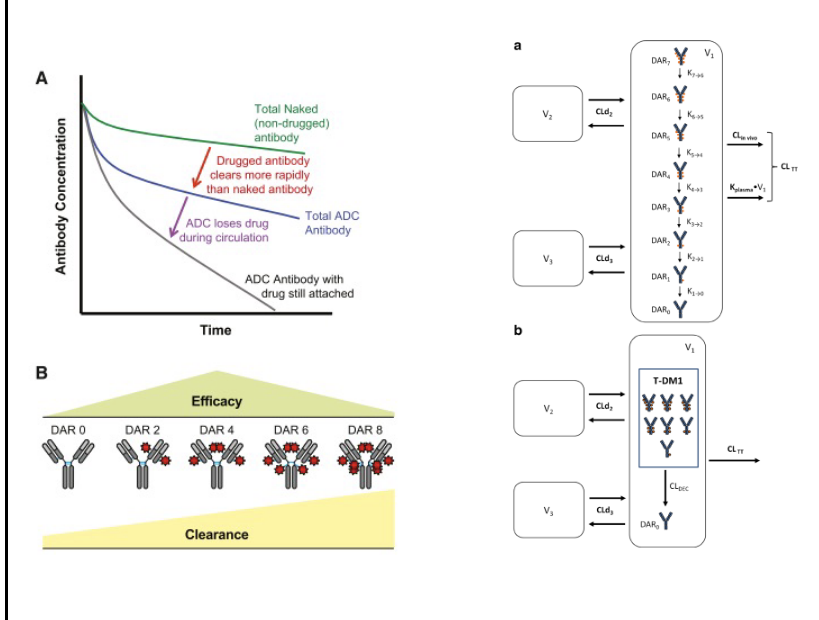
bispecific antibodies (bsAb)
bsAb have affinity for 2 distinct targets
several molecular formats can be produced
generally include fragments from 2 different mAbs
examples of approved products
catumaxomab
maligant ascites in patients with epCAM-positive cancer
binds to epCAM and CD3
blinatumomab
philadelphia chromosome-negative relapsed ALL
binds to CD19 and CD3
emicizumab
hemophilia A
binds to factor IX and X
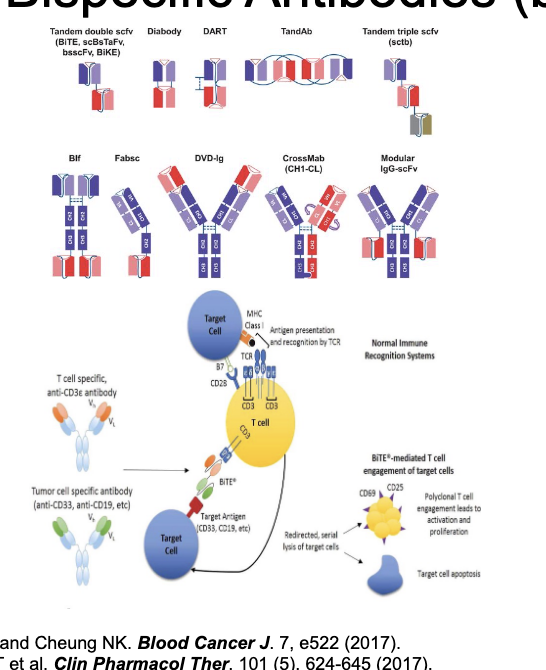
bsAb PK
PK of bsAb is complicated by affinity for 2 targets
generally described using multiple TMDD models
target molecules are often moving throughout the body - have to consider target kinetics/cycling
few good examples of bsAb PK modeling
bsAb = often eliminated very quickly
affinity for 2 targets
many are antibody fragments that CAN be renally filtered
blinatumomab = dosed as 28 day IV infusion to maintain efficacious concentrations
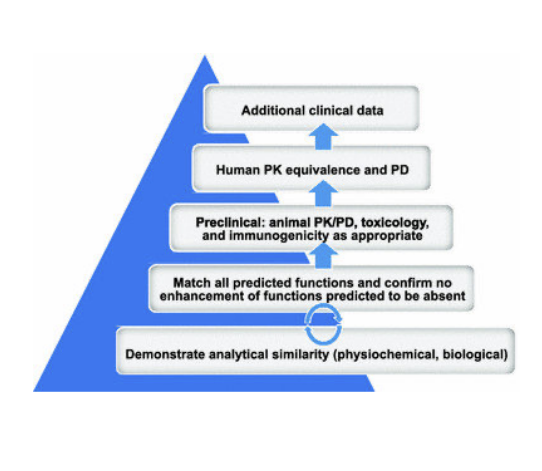
biosimilars
FDA def: a biological product that is highly similar to and has no clinically meaningful differences from an existing FDA approved reference product
similarity is assessed by comparing purity, chemical identify and bioactivity
clinically meaningful differences are assessed by comparing PK, PD, immunogenicity
biosimilars are interchangeable with reference products
may be substituted by pharmacist WITHOUT physician’s intervention
biosimilar mAbs
more than 15 approved products
remicade - inflectra, renflexis, avsola
humira - amjevita, cyltezo, hyrimoz, hadlima, abiralada