Medchem Exam 3
1/118
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
119 Terms
What type of molecule are the majority of drugs?
Small molecule
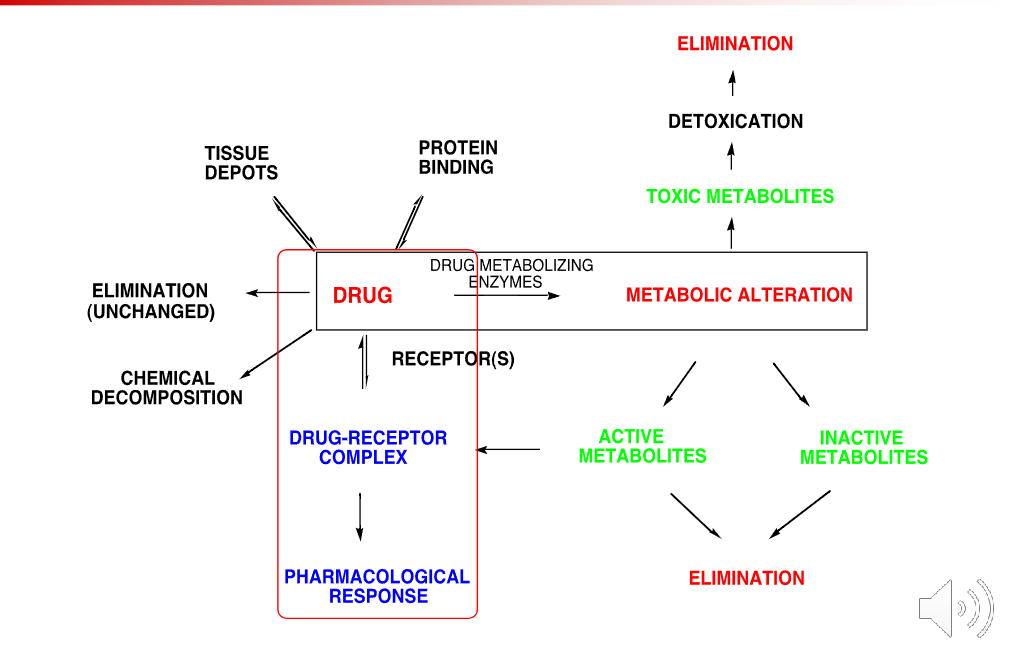
Fates of drugs in the body
Protein binding
Tissue depots
Elimination (unchanged)
Chemical decomposition
Drug receptor complex —> Pharmacoloigcal response
Metabolic alteration via Drug metabolizing enzymes
Major sources of drugs old and new
Natural extracts
Natural products —> pure molecules isolated from nature (Antibiotics and cancer agents)
Synthetic drugs (we make them)
New drugs and existing drugs (Slight modification of existing drug —> New function)
Screening synthetic Chemical and Natural product libraries —> Put a collection together and screen for drug
Rational Drug Design —> We know enough info that we can make a molecule that works
The role of serendipidy—> Drugs discovered by accident
Natual extracts and products as source of drugs
Sole source of new drugs for thousands of years
Natural drugs are usually more complicated and contain more rings than synthetic drugs
Approved drugs are more natural product like → reflects the success of natural product based drugs
Ex: Epibatidine
Took sample of skin secretion from posion tree frog and tested it for biological activity
200 times more potent than morphine and was not an opiate and was a nicotinic antagonist
Had SE in humans but was solved by synthesizing many variants
Currently an important research tool
Syntheic drugs as source
The carbon source of synthetic drugs is petroleum
2% of petroleum is used for petrochemical precursors for pharmaceuticals, plastics, pesticides, perfumes, dyestuffs, food additives, etc.
Synthetic libraries are often too conservative in their structural properties
EX:
Aspirin —> One of the first semi-synthetic drugs prepared by acetylation of salicylic acid
Chloral hydrate → Synthesized by chlorination of ethanol and used as a sedative
Chloroform and ether → synthesized from ethanol and used anesthetics
*Considering nature has been making drugs for billions of years there have been many drugs that have came and gone, and these molecules often fail in trials because of the absence of a
thorough phenotypic selection process
Making new drugs from existing drugs
Small modifications in existing drugs leads to discovery of new functions of a drug
Ex:
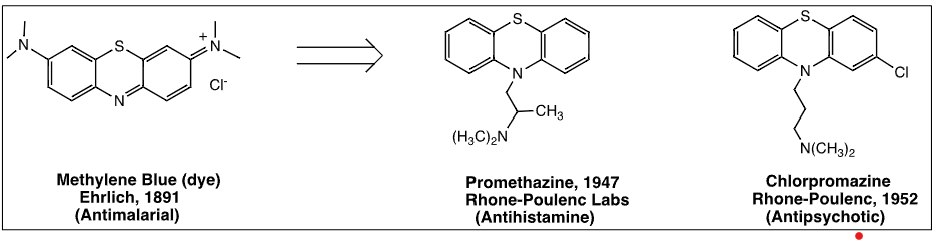
Promethazine which was made from antimalarial phenothiazine dyes has antihistamine effects
Chlorpromazine was further discovered serendipitiously by altering promethazine
Further modification of chlorpromazine via bioisosteric replacement lead to discovery of imipramine
Screening chemical libraries and natural product libraries
Researches can screen chemical libraries (such as chemical dyes) or plant extracts for drug applicability and if a match is found it can be formulated into a drug
Ex:
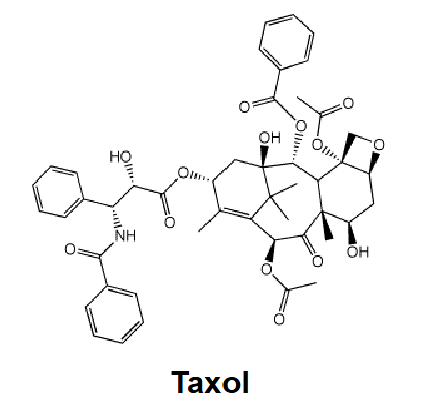
Paclitaxel (Taxol) → Anticancer drug
Plant extracts from around the world were tested for anticancer activity
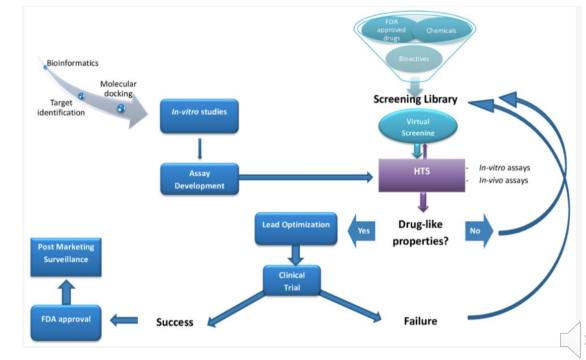
High Throughput Screening
Test each molecules or mixture of molecules against a validated drug target or organism
What is required for a good high throughput screen (HTS)?
• Robust assay that assess a validated drug target
• Assay must be statistically sound
• Assay should be rapid and inexpensive to assess many molecules
• The library of molecules should be novel and contain a variety of potentially active substructures (potential pharmacophores)
• The molecules should be at least slightly water soluble and not prone to aggregation
*Recently, molecular docking algorithms have progressed to allow high throughput in silico screening of large molecular databases to find molecules that bind to a given protein drug target
Rational drug design
uses knowledge of drug target structure or enzyme mechanism (or both) to discover molecules that bind and modulate the activity of the target
Ex: HIV protease inhibitors
• Serendipity played a role – sequencing of HIV showed it contained a protease related to pepsin and thiapepsin, already well-studied aspartic proteases
• It was immediately hypothesized that pepsin inhibitors already developed could be an excellent starting point for an anti-HIV protease based drug
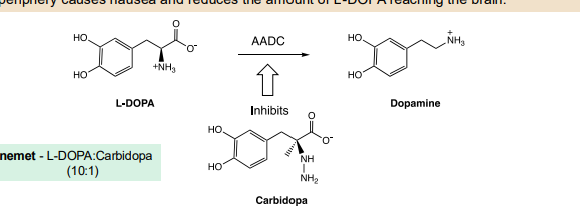
Ex: Carbidopa adjunct
Carbidopa is an inhibitor of Amino Acid Decarboxylase, the enzyme that converts L-DOPA to dopamine. Crucially, carbidopa, unlike L-DOPA, does not cross the Blood Brain Barrier
Serependipity in Drug Discovery
Finding a new drug by accident
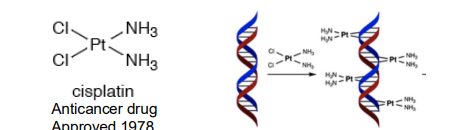
Ex: Cisplatin
Saw a similarity between mitotic spindle and magnetic force lines
Subjected E. Coli to electric current in aqueous NH4Cl and “inert” PT electrodes
The drug discovery and development process
Takes a lot of time (>10 years) and money ($2 billion) to approve a drug
Lead Optimization
The initially discovered or designed lead molecules for a given drug target are almost always not the final drug structure
This is because the structure needs to be optimized for a number of important attributes that make a small molecule a good drug
Some important characteristics of a good drug:
• Good oral bioavailability – water soluble but also small and lipophilic
• Chemically stable – acid stable for oral activity, hydrolytically stable
• Chemically unreactive – low or no reactivity with nucleic acids or proteins to avoid mutagenicity or immune response
• Metabolically stable – resistant to enzymatic breakdown in the body
• Pharmacologically specific – no off-target binding
• Potent but not too potent – potency is usually necessary for specificity
• Good toxicity profile – wide therapeutic index to avoid toxic side effects but the activity usually needs to be intermittent, especially in the CNS
• Inexpensive to manufacture – fairly simple structure for chemical synthesis or able to be prepared by fermentation (not Taxol, for example...)
Why do drugs fail in devlopment?
Toxicity is the overwhelming issue in preclinical drug development
For clinical studies, it is a combination of safety, efficacy and commercial
Issues with efficacy is that the wrong drug target is selected
But we’ve made significant progress improving PK and bioavailability
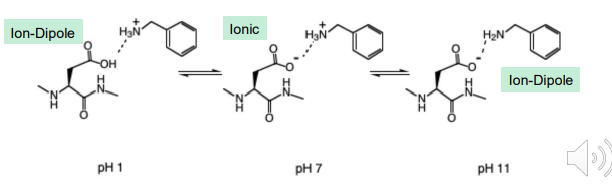
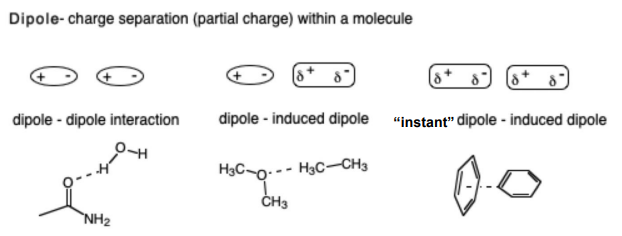
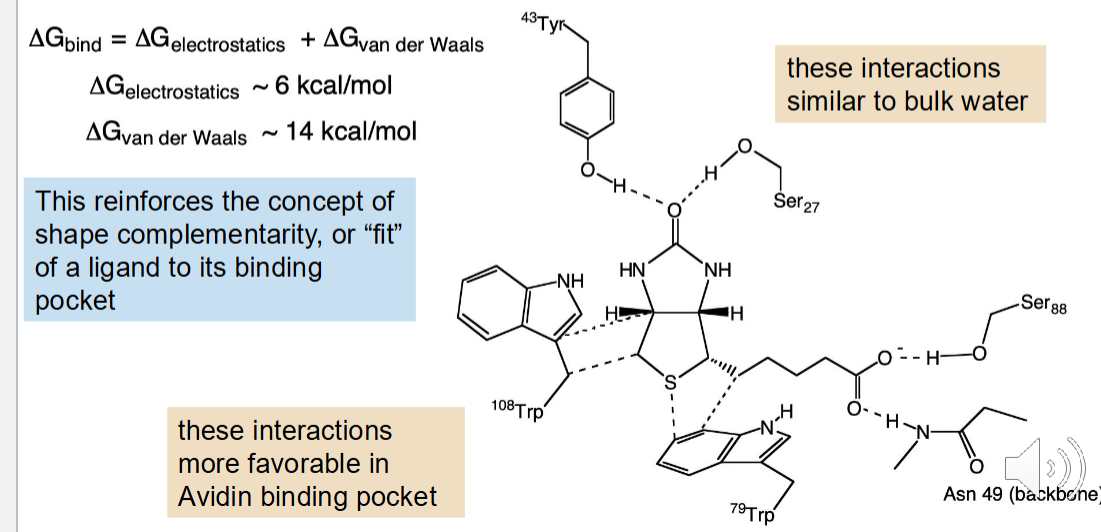
Noncovalent Interactions
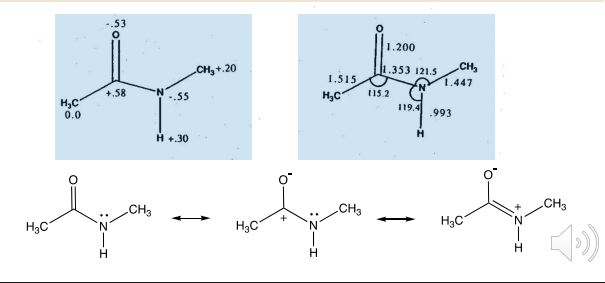
The unequal distribution of electron density in molecules results in electrostatic interactions between and within molecules
Electron density is not uniformly distributed in most molecules
The carbonyl nitrogen bond has significant double bond character
The amide bond has a large partial negative charge but so does the nitrogen
The crystal structure of this drug shows that the N group has a short bond character which is evidence for the third resonance structure
Theoretical calculations can now provide a better idea of how a molecule can interact with other molecules as well as its reactivity
Dipole bond moments and electronegativity
Bond moments are proportional to the difference in EN of two atoms
The more EN an atom is the larger the bond moment
A larger number indicates more polar covalent bonds that a more e- withdrawing
Distance dependence of Noncovalent interactions
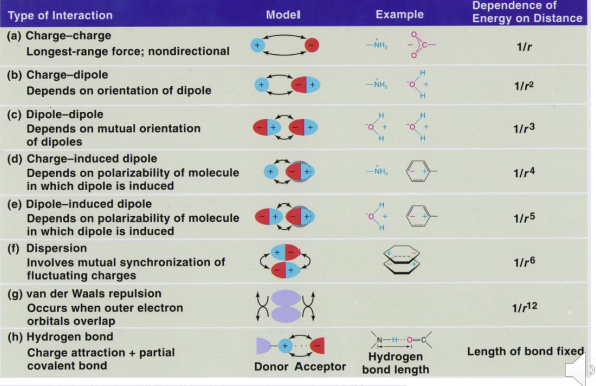
Ionic Interactions (Charge-Charge)
Ions are solvated by water which competes with this interaction
However, in a protein binding pocket water may be excluded, which enhances the energy of the interaction
Energy is proportional to 1/Dr where D = dielectric constant and r = distance
Often PH dependent
Dipole-induced dipole interactions
Dipoles can be permanent or temporarily induced by a nearby charge or occur by random fluctuation of electron density in a nonpolar group
Charged ions can also interact with permanent dipoles and can induce dipoles in other molecules or within the same molecule
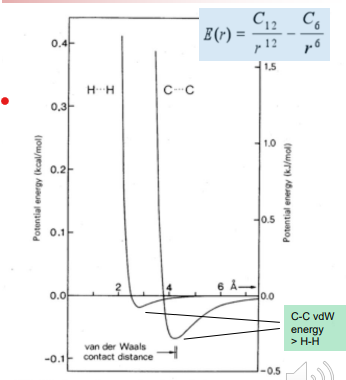
London Dispersion / Van der Waals interactions
There is an optimal distance between any two atoms that is energetically favorable
The attractive dispersion force is typically induced dipole interactions and are proportional to 1/r6 , while the repulsive force is prop. to 1/r12
This means too far away no benefit and too close together huge energetic penalty
The Dispersion induced dipole – induced dipole interaction is related to the “polarizability of the atoms involved. This is proportional to the distance of the valence electrons to the nucleus. Sulfur and bromine, iodine are examples
The large number (n) of van der Waals interactions (n x 0.5 kcal/mol) can add up to a significant amount of binding energy, but only if the atoms are close → This explains the importance of “fit” between a drug and it’s protein target binding site
Hydrogen bonding
An electrostatic interaction between heteroatoms (N,O) and hydrogen atoms bound to heteroatoms → Can be considered a special case of a dipole-dipole interaction
Strongest when linear (nonlinearity does not greatly decrease energy of interaction)
All atoms remain neutral
Ex: Water can solvate polar groups
Ex: Proteins use hydrogen bonding to interact with polar groups in small molecules and use intramolecular H-bonding to fold into secondary and tertiary structures
Clinical effect of a hydrogen bond
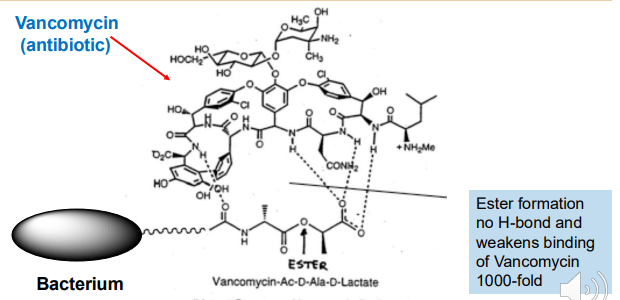
The antibiotic Vancomycin kills gram positive bacteria by clamping down on the D-Ala-D-Ala terminus of its peptidoglycan.
A hydrogen bond from the Vancomycin to the amide of the D-Ala-D-Ala is key to binding
Basically, strains were isolated in which the peptidoglycan had an ester in the place of the amide which decreased the binding affinity by 1000 fold rendering the antibiotic useless
Cation - Pi interaction
A special Ion - Dipole Interaction
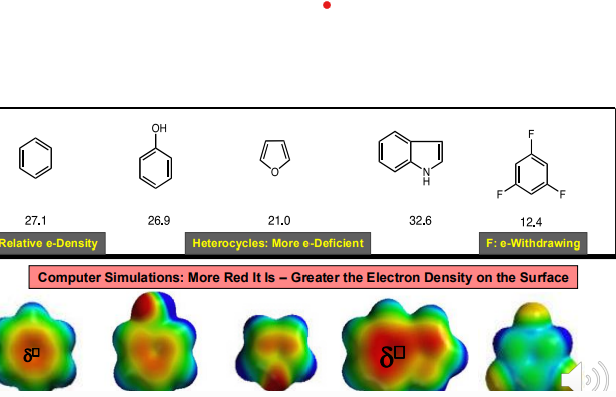
Cation - PI interaction: A non-covalent interaction between the face of an electron-rich system (i.e., benzene) and a nearby hard metal cation (i.e., Li+, Na+, K+), or a softer, more diffuse ammonium cation (i.e., NR4+).
An Electrostatic-Like Interaction: Positive charged species being attracted by the negative PI-electron cloud (or PI-cloud)
The more negative the ring surface is, the stronger the cation-π interaction
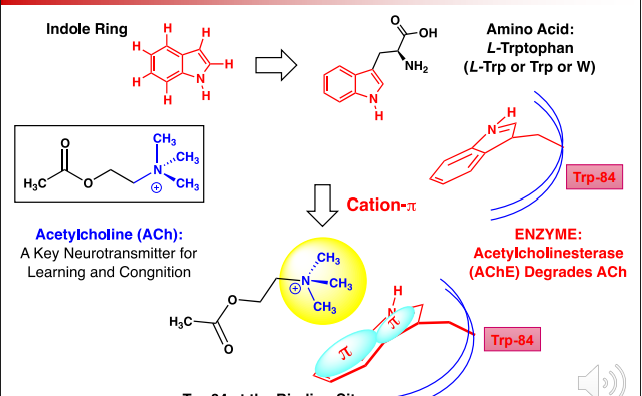
Cation PI AcHE Example
In AcHE, Trp-84 forms a cation-pi interaction with the N-(Ch3)3 group
This binding allows for AcHE to degrade AcH which is important for regulation
Entropy
A measure of uncertainty or probability —> specifically related to the number of microstates available to a system for a given macrostate
If a system has a greater number of possible microstates or greater uncertainty—> greater entropy
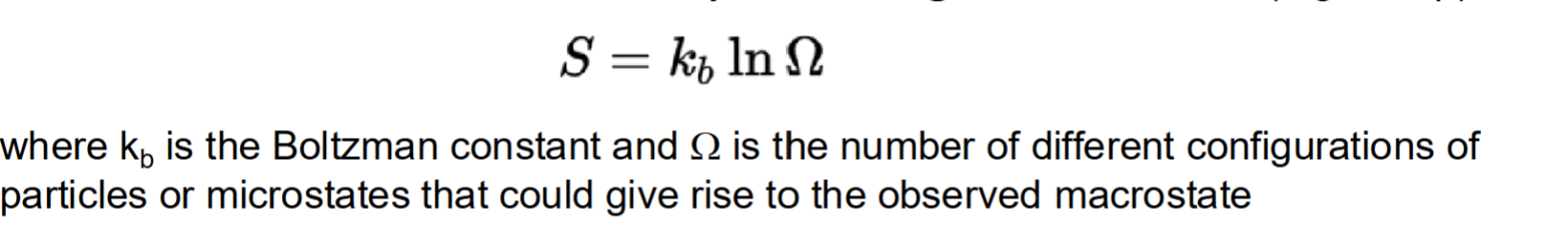
Second law of Thermodynamics
The entropy in the universe always increases for spontaneous processes.
Always released in the form of heat
Molecular interactions and entropy
Entropy is increased when the system has more possible configurations or microstates
Examples:
Molecules with translational freedom have greater entropy than those that are ordered or in bound arrangements where movement is restricted (Molecules in crystal vs solution)
Molecules with greater freedom of rotation have greater entropy than those that are rigidified in rings
For a spontaneous process, change in G < 0
According to the second law of thermodynamics the total entropy is always increasing

Hydrophobic effect
Placing nonpolar molecules in water (polar solvent) is energetically unfavorable
Due to dipole-induced dipole noncovalent interactions
The restriction of motion reduces the entropy of the system → Entropic cost
Desolvation and the hydrophobic effect
Oil molecules interact with each other to create one giant oil droplet which has less SA to cover with ordered water molecules —> Entropically favorable because less ordered water molecules
Why does this matter?
Most small molecule drugs are hydrophobic
Binding of a drug to a receptor is high entropy because there is less ordered molecules in the receptor pocket
The binding of hydrophilic groups is not always better in a protein binding site because polar AAs that interact with these groups may not be oriented to interact any stronger than with solvent water
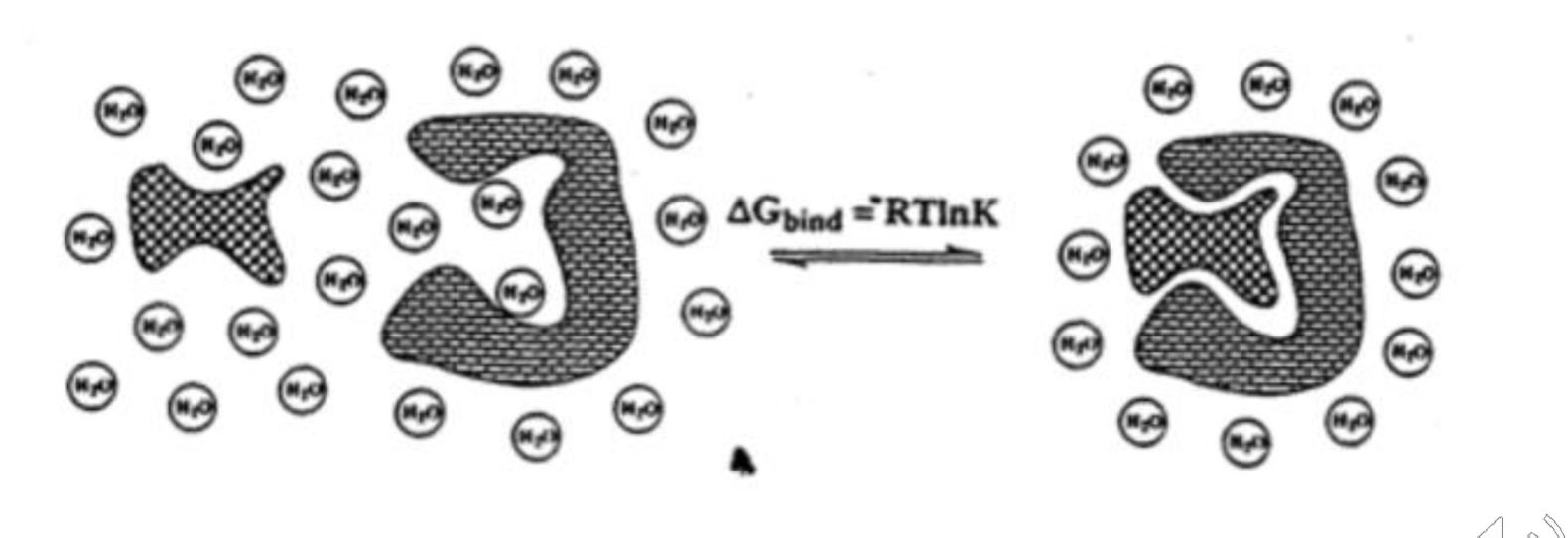
Measuring hydrophobicity of Molecules
Can be measured experimentally by the use of partition ratios
Add a test molecule into a mixture of water and an immiscible organic solvent (usually octanol) and measure the concentration of the molecule in the organic vs aqueous phase
The ratio of the concentrations are reported as the Log P
Higher Log P = more nonpolar “more greasy” —> less soluble
Lower Log P = more polar “less greasy” —> more soluble

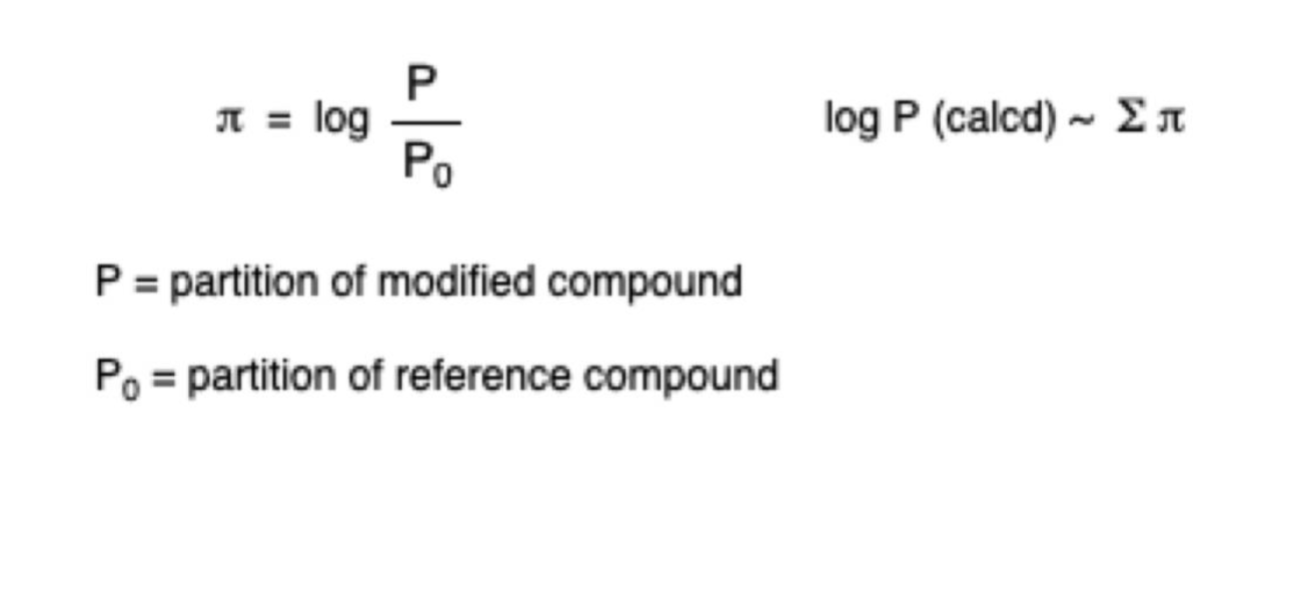
Pi values
Estimate Log P using data from a variety of reference molecules that provide (group) hydrophobicity values
An intramolecular hydrogen bond has a positive value
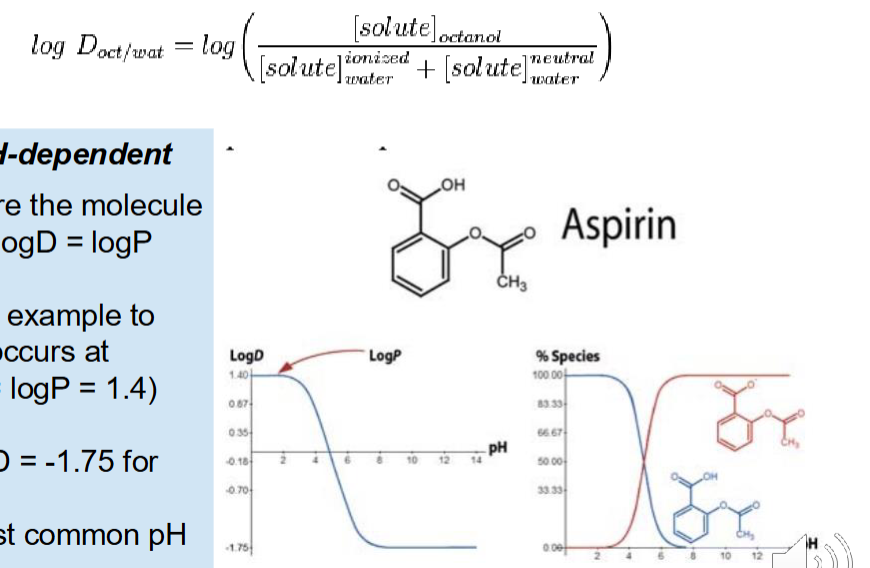
LogD
pH dependent
tells us how much of a drug is ionized at a certain pH
When negative —> Ionized
When neutral —> Log P
When positive —> Unionized
Estimating Total (intrinsic) binding energy
A summation of the independent binding interactions
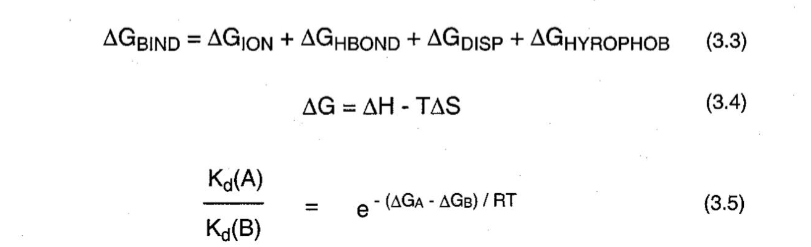
Relationship between binding energy of a small molecule and equilibrium
Small changes in binding energy can result in large changes in Kd
Large +Kd means it takes a lot of energy for the molecule to dissociate —> Large -Ka
Large +Ka means it takes a lot of energy for the molecule to come together —> Large - Kd
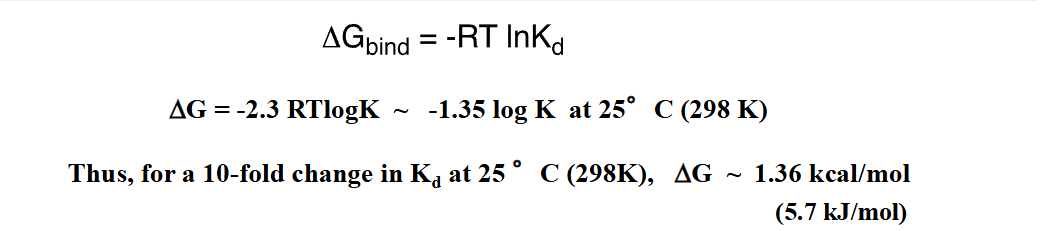
Van Der Waals forces vs H-Bond
Having multiple Van der Waals forces add up in energy and makes the molecule harder to dissociate vs one H-Bond
Drug Targets
Most small molecule drugs target protein receptors
G-Protein Coupled Receptors (GPCRs) and nuclear receptors are most common
Ion channels (voltage and ligand gated) make up largest class of drug targets
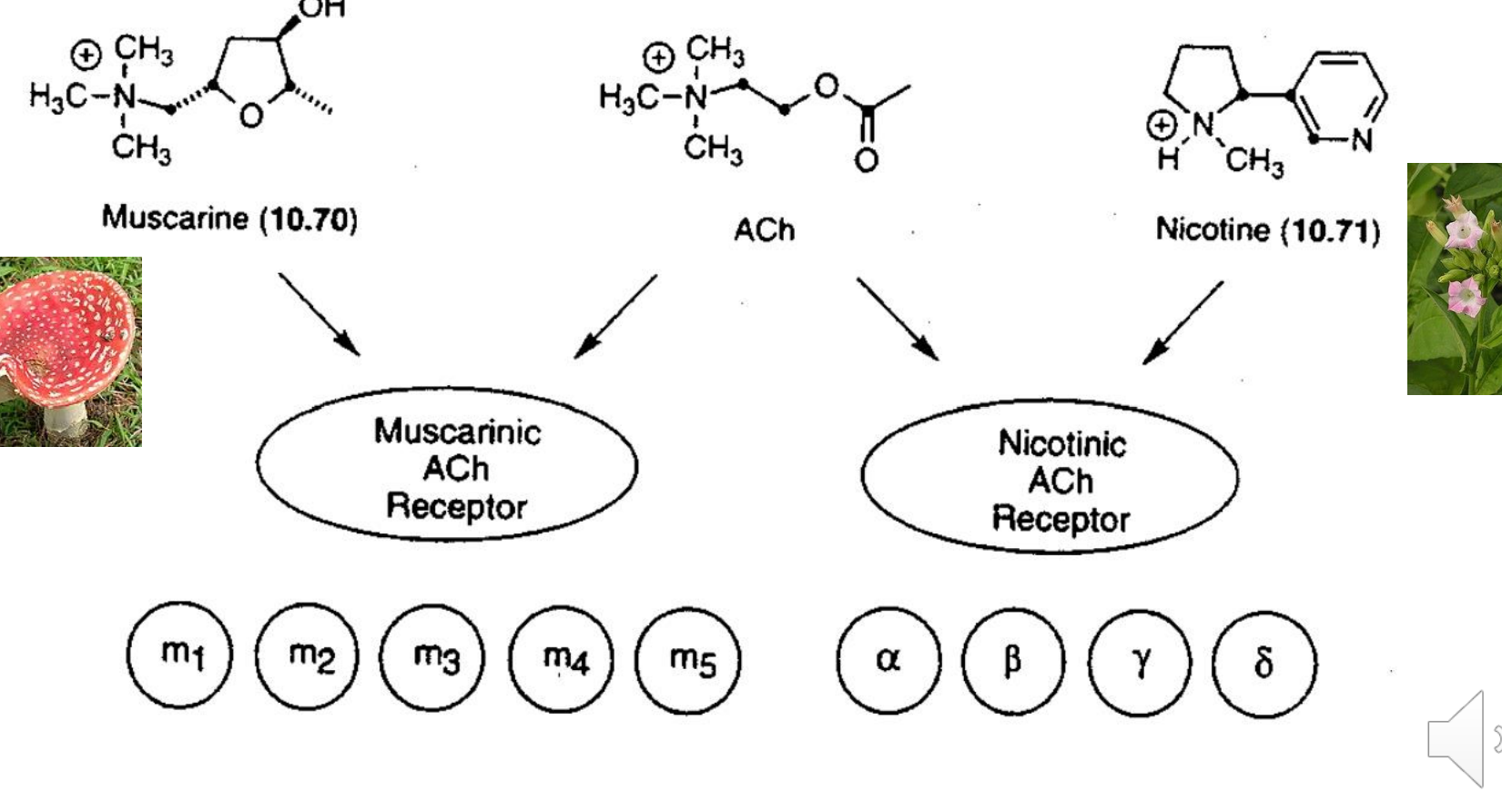
ach drug targets
Can bind to Muscarinic (G protein) → 5 subtypes (M1-M5)
Makes pi-cage for cation pi interactions which then makes an acetylcholine salt bridge with the Ach
Produce second messenger molecules leading to a signaling cascade
Can bind to Nicotinic (Ion receptor) → 4 subtypes
ligand gated ion channels that increase intracellular Ca2+
Alzheimer’s and PD
Tourette’s Syndrome, ADHD
Schizophrenia, Pain, Nicotine addiction
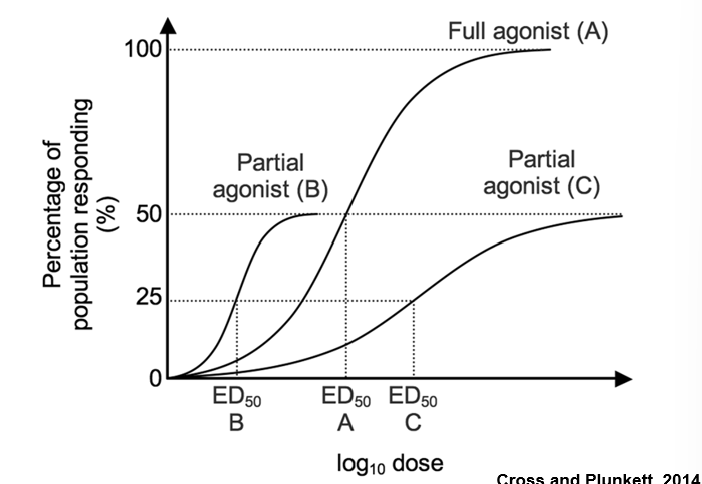
Agonists
Active Ligands are able to induce the proper conformational change in their target receptor that triggers the downstream signaling events
mimics the full activity of the endogenous ligand
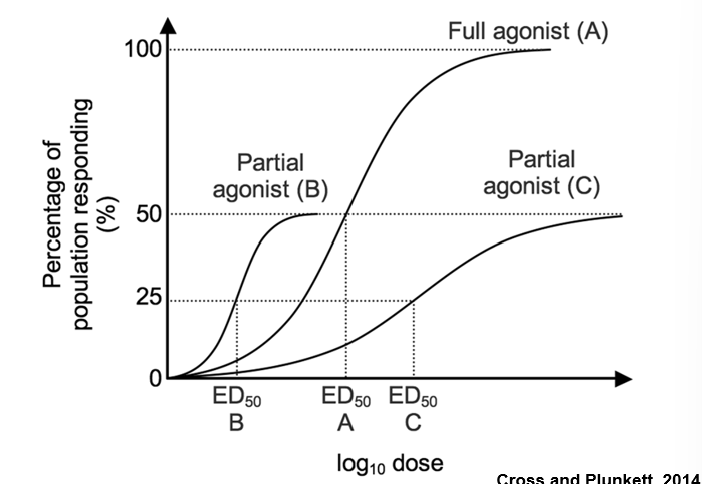
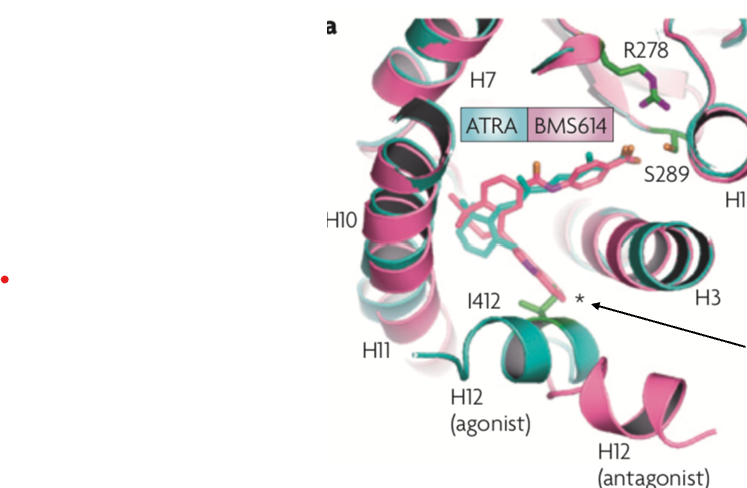
Antagonists
inhibit this change in conformation due to a steric clash
blocks the effect (binding) of the endogenous ligand
Kon
The rate constant for the association of a ligand with its receptor, representing the speed at which a drug binds to its target
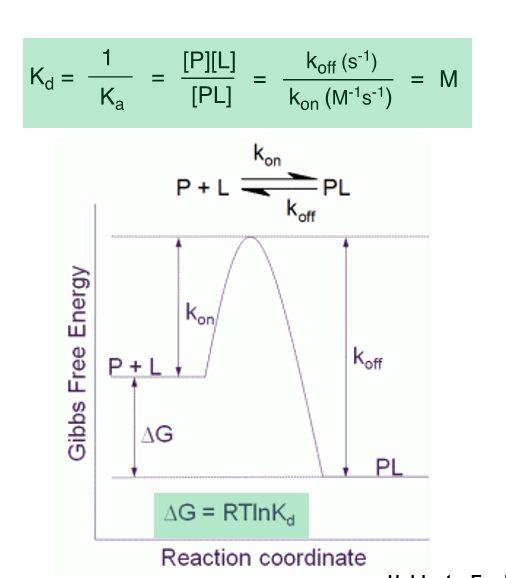
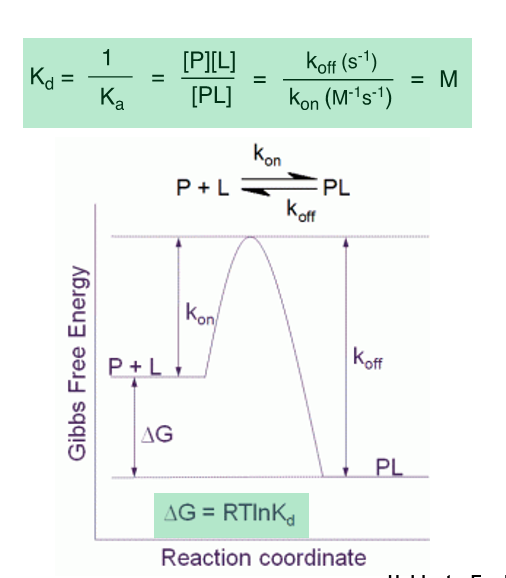
Koff
Varies more widely than Kd
A small koff value is desirable as it will increase a drug’s residence time, which has been correlated with efficacy
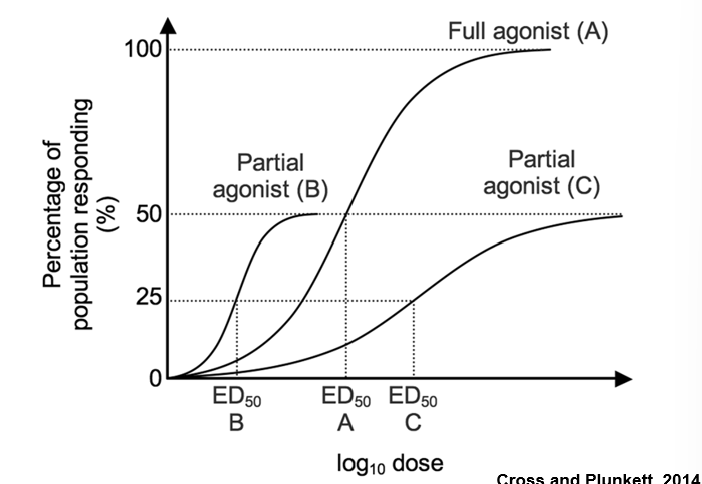
Partial Agonist
mimics the activity but plateaus at a lower level
Inverse Agonist
has the opposite activity of the agonist
Effect
Measure of biological output
It depends on binding but is a measurable action after binding takes place
Potency
is measured along the x-axis by the ED50 – the concentration of the ligand that gives 50% of its maximum effect
ED50 is the same as an EC50 except it is specific for compounds used in human or animal studies
The EC50 is often lower than the Kd due to the presence of “spare receptors”
—> Often the reason is related to the biochemical amplification in a ligand-receptor signaling pathway
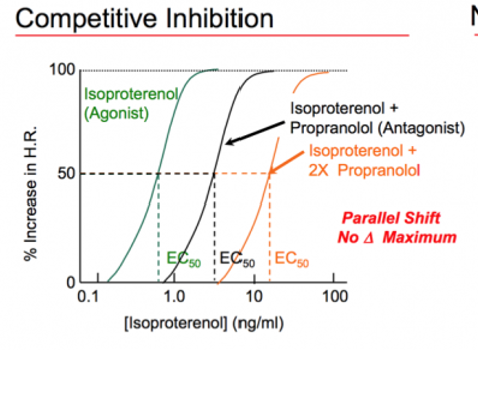
Competitive inhibition
•simply shift the activity curve of an agonist to the right.
It takes more of the agonist to give the full effect.
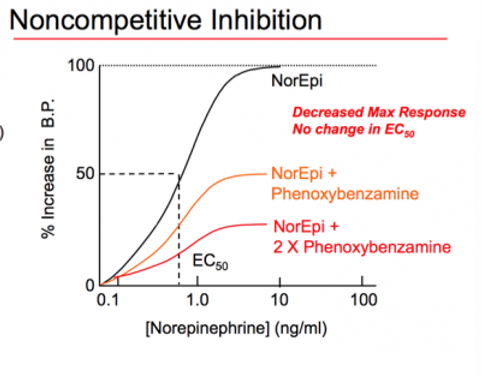
Noncompetitive inhibition
affect both binding and activity and prevent agonist from achieving the full effect by binding at a different site on protein
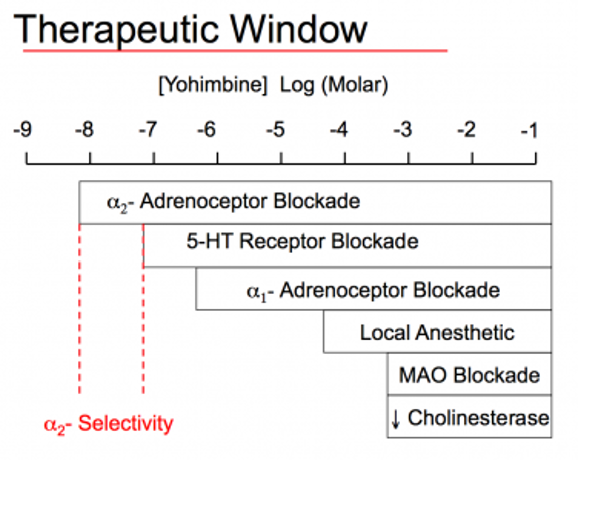
Therapeutic Index
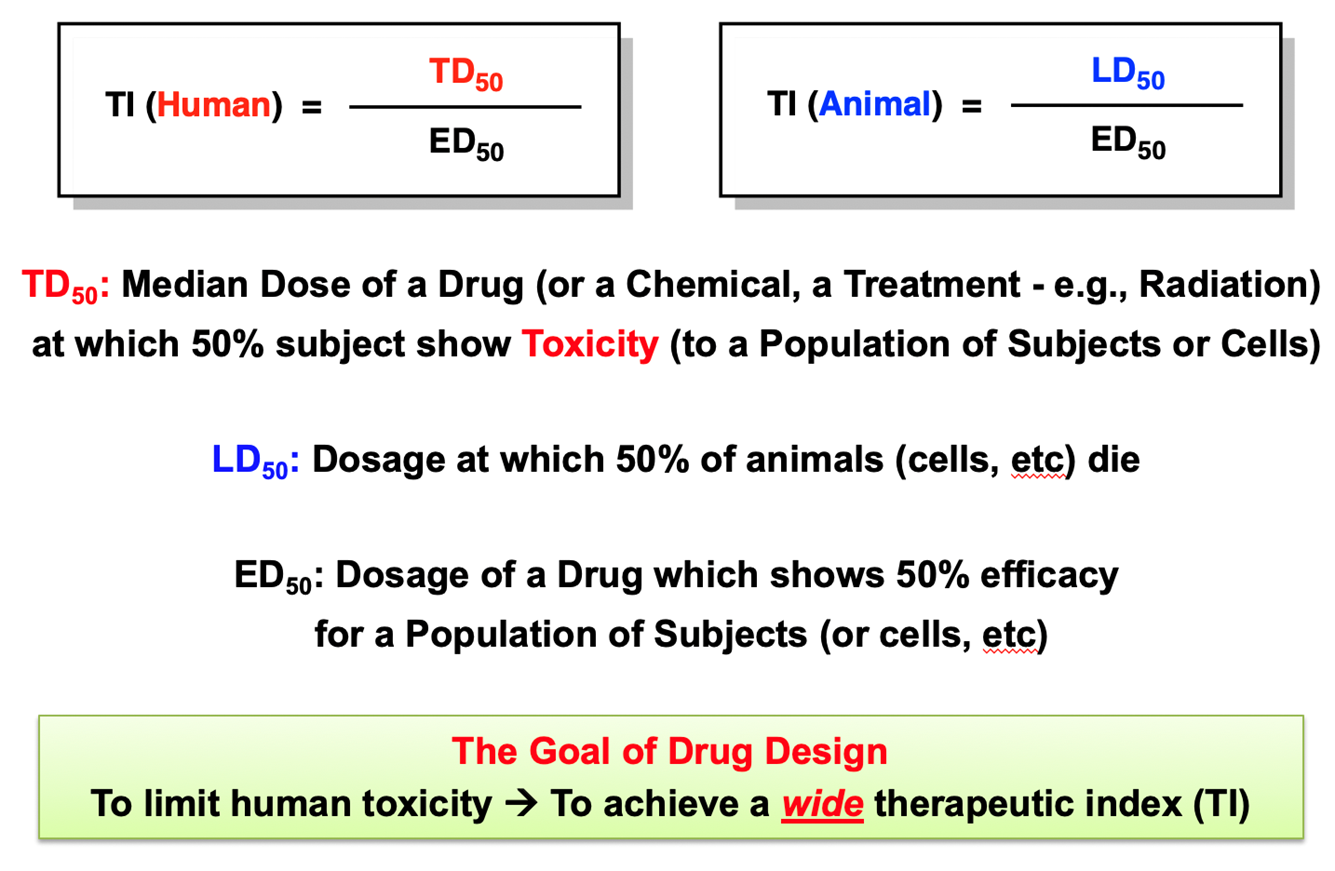
Toxic when higher concentrations are given because the drug can occupy multiple receptors which leads to side effects
Racemic vs Single Enantiomer Drugs
Most approved drugs that contain chiral centers are now marketed as a single enantiomer
However, many older racemic drugs are still on the market
In many cases, one of the enantiomers dominates the desired activity
Example: Warfarin – S-enantiomer is 5x more potent than R enantiomer at inhibiting coagulation. There are also substantial differences in the serum
half-life for each enantiomer because each is metabolized by different enzymes in the body
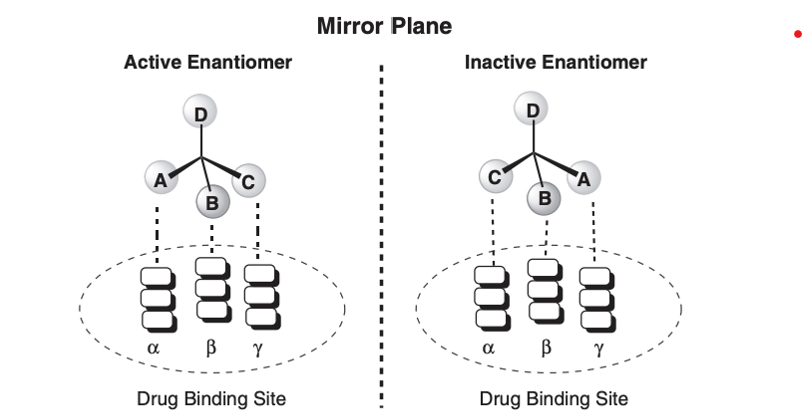
Protein binding sites are spatially specific and made up of chiral amino acids
Thus, two enantiomers do not bind in the same orientation to a given binding site and have different pharmacological activity
Enantiomers
Two molecules with non-superimposable Mirroring Molecules
Possess molecular chirality
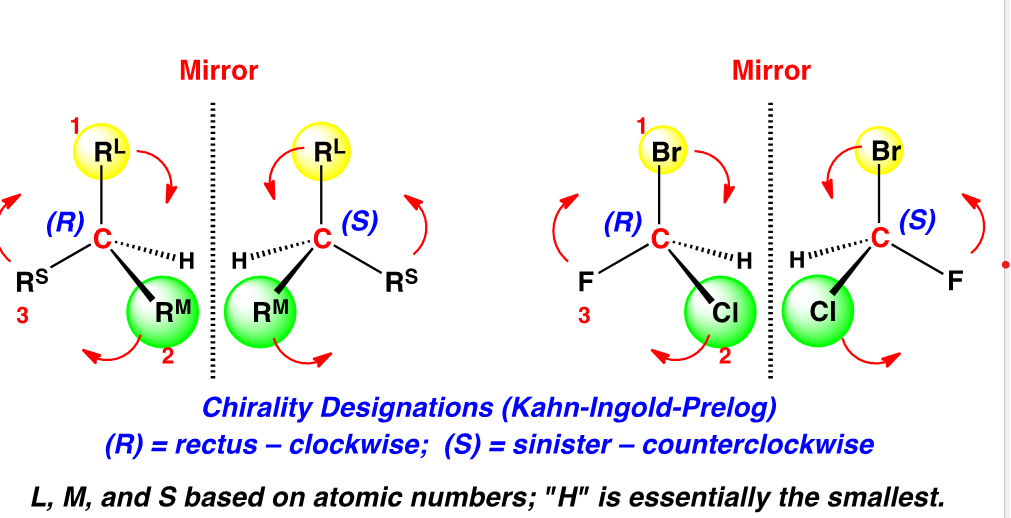
R = clockwise
S = counter-clockwise
Put thumb in direction of lowest group (typically H → D) and curve hands in the direction that goes in order (1→ 2 → 3 → 4)
dash = down wedge = up
Eutomer → active enantiomer
Distomer → less potent enantiomer
Problems posed by the distomer of a drug lead to an imperative to develop the drug as a single stereoisomer
When the distomer causes adverse effects the drug is a candidate for developing the other enantiomer as a new drug with a better safety profile
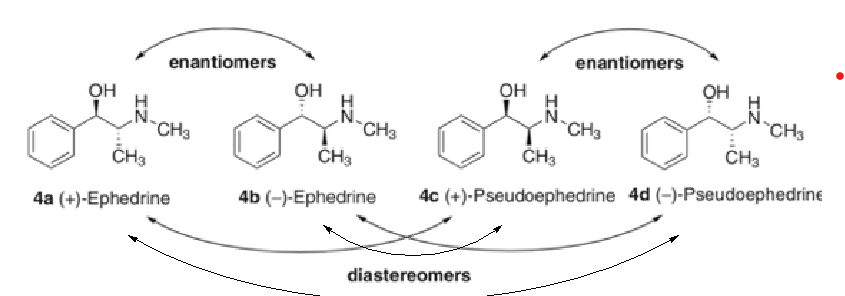
Diastereomers
Any stereoisomers that are not the enantiomer
Racemic vs scalemic
•A racemic mixture is a mixture of each enantiomer at a ratio of 1:1
•Any other ratio is termed a scalemic mixture
•The total number of stereoisomers of a chiral molecule = 2N,
where N = number of chiral centers
Issues with companies preparing a single enantiomer of a new drug
Most companies prepare the single enantiomer of a new drug candidate to avoid potential pitfalls in the approval process due to problems of the other stereoisomers
Problem: Preparing enantiomerically pure drug molecules is more difficult and more expensive than preparing racemic mixtures
Challenge: How are single enantiomers formed in molecules?
1.Use enantiomerically pure starting materials (sugars, amino acids, natural products) – inefficient, used before chiral technology
2.If the molecule has a carboxylic acid or basic amine can separate enantiomers at the end by making diastereomeric salts
3.Use an enzyme to selective modify one of the enantiomers of the racemate to facilitate separation or prepare a pure enantiomer from an achiral starting material
4.Use synthetic chiral catalyst to prepare one enantiomer at a key step in the synthesis from an achiral starting material
5.Separate enantiomers by chiral chromatography
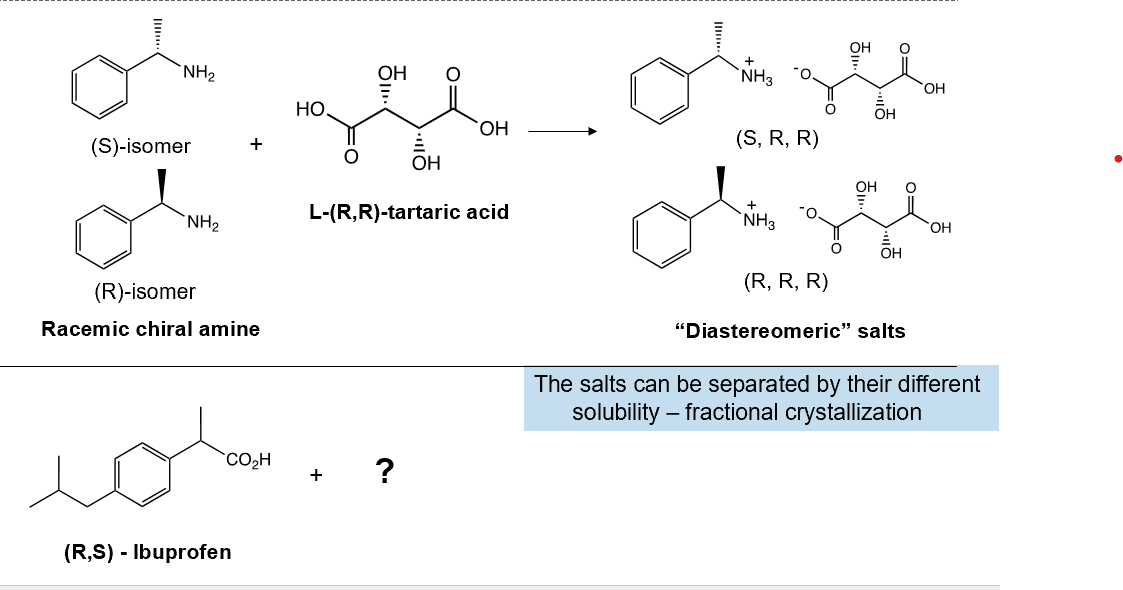
Chiral Resolution in diastereomic salts
add a single enantiomer of a chiral acid to resolve mixture of amine bases, or visa versa
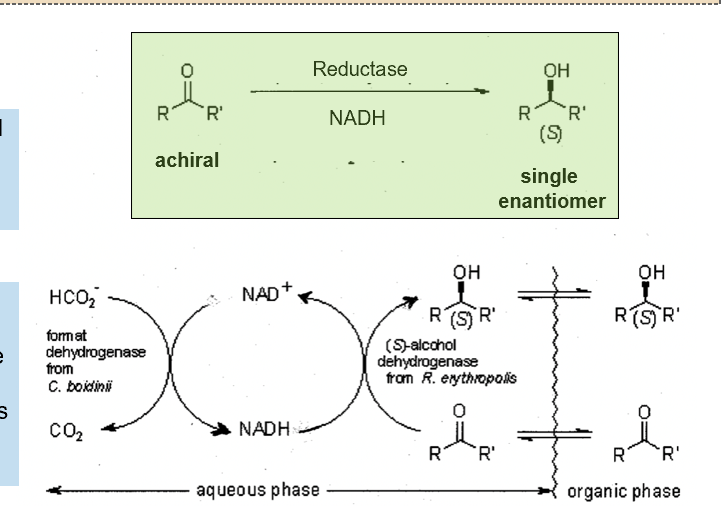
Enzyme catalysts
Esterases, reductases and other enzymes are often able to distinguish between enantiomers and can create single enantiomers. This allows:
•Separation of racemic mixture by modifying only one enantiomer
•Creating a single enantiomer from an achiral starting molecule
Synthetic Chiral Catalysts
These catalysts act like enzymes in that they bind the starting molecule and modify it to produce mostly one enantiomer of a chiral product
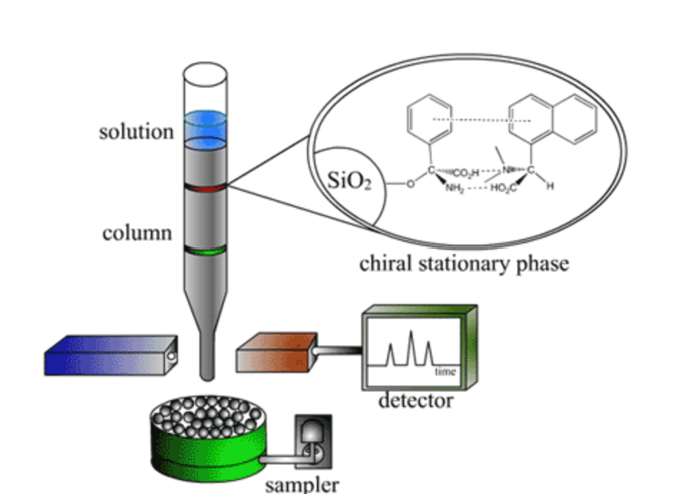
Chiral Chromatography
Chromatographic supports can be prepared that have a single enantiomer attached to polymer or silica beads. These columns are extremely expensive and are usually not practical on large scale. Notice how they use the same noncovalent interactions that we have been discussing, this time used for an entirely different application.
Ex:
In this example, a chiral single enantiomer of an amino acid is bound to silica and packed into a column
The racemic mixture is passed through the column and one enantiomer interacts more tightly than the other by noncovalent interactions which causes a separation in the elution time
Synthetic Biology
Use of recombinant DNA technology to create genetically modified organisms for the synthesis of novel products
For drug discovery, this is typically either for more efficient synthesis of bioactive molecules or their precursors
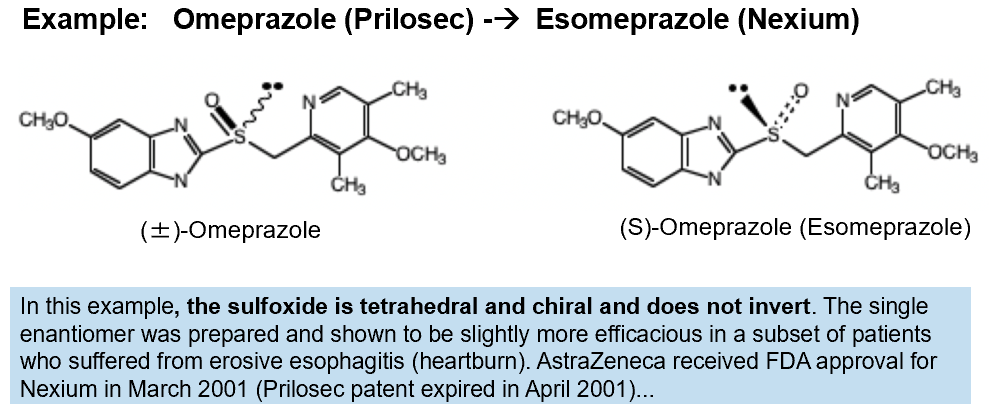
Chiral Switch
the development of a single enantiomer version of a racemic drug that has already been approved.
Since the efficacy of some racemic drugs is dominated by one of the enantiomers (eutomer), the other enantiomer (distomer) is unwanted and may contribute to side effects
If the drug company can prove that the single enantiomer drug has better efficacy or fewer side effects they can get a new patent and market it as a new improved version of the drug
Exclusivity period of chiral switch drugs varies widely and is assessed on a case by case basis
Thalidomide
Thalidomide would be a perfect candidate for a chiral switch drug
however, it was soon found that the chiral center of the molecule is unstable in vivo, due to the relative acidity of the C-H proton at the chiral center:
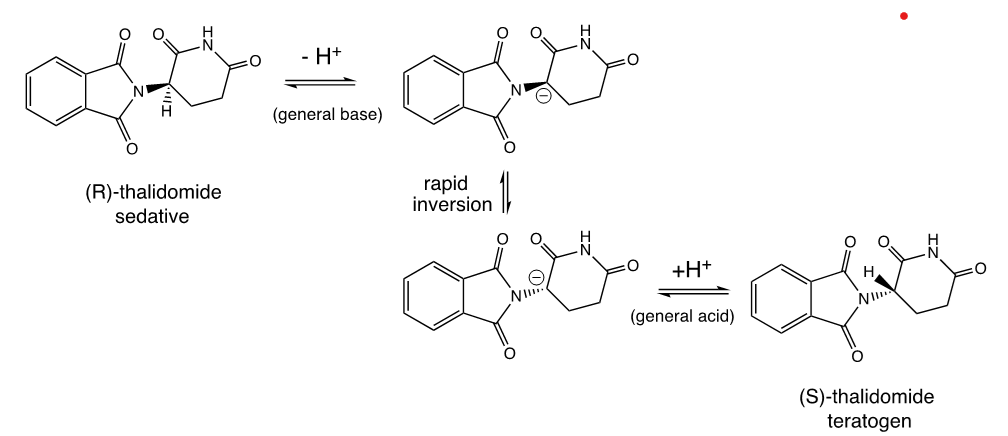
Reasons why chiral switch drugs fail
Some chiral switch attempts failed spectacularly – Lily with fluoxetine (Prosac)
Was assessed in clinical trials at too high of a dose which revealed a statistically significant level of cardiac problems in the treated patients. The cost to repeat the trial was deemed too high so it was abandoned.
Some chiral switches are approved but then fail in the market due to lack of advantage over the racemate
The single enantiomer of albuterol, levalbuterol (Xopenex) has not been adopted at the expected rate due to lack of increased efficacy over the parent racemic drug.
The question will always be whether the pure enantiomer gives a better clinical outcome that justifies the large increase in cost for the new drug → The public backlash over high drug prices doesn’t help to produce a favorable environment for these endeavors
Conformational Isomers
Conformational isomers have inherently different energies
For all Acyclic systems look through the C-C axis
Staggered > Eclipsed because it avoids torsional strain caused from steric repulsion
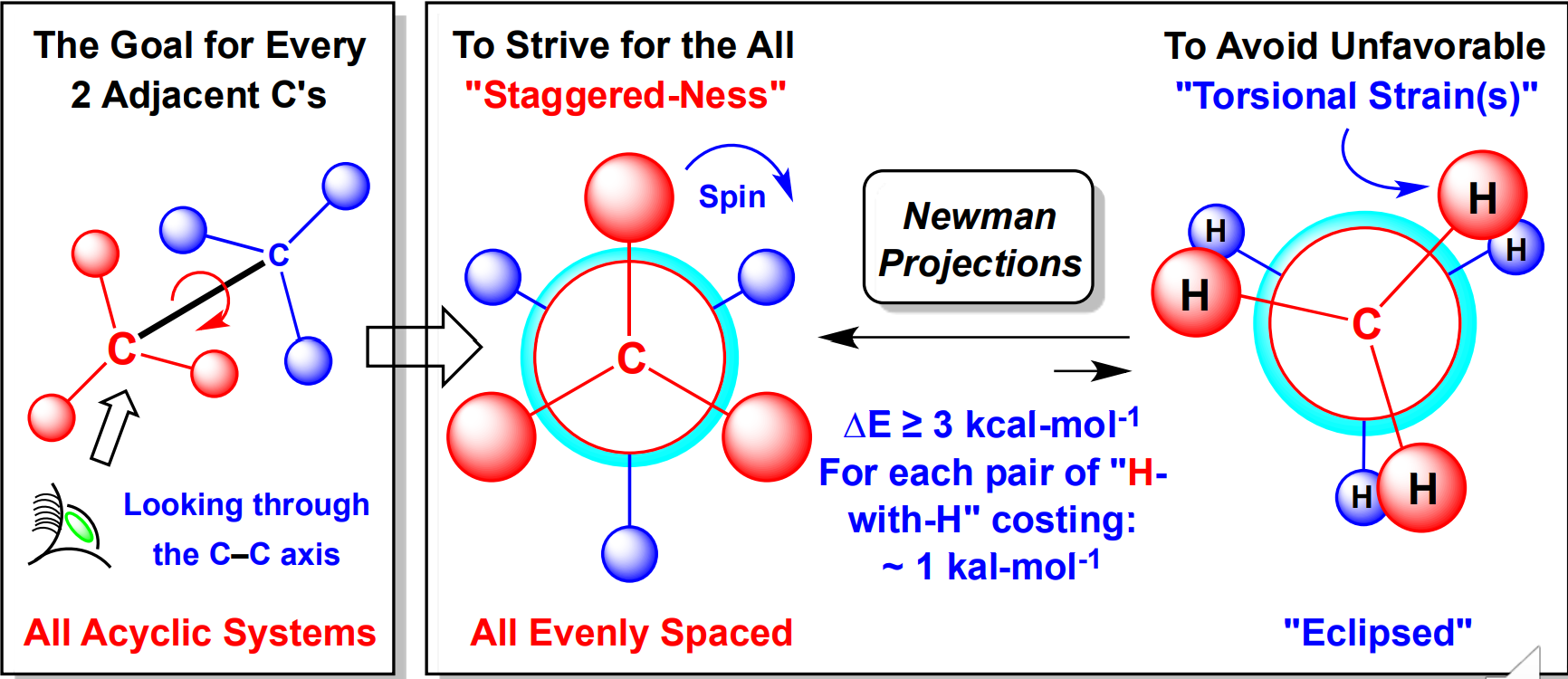
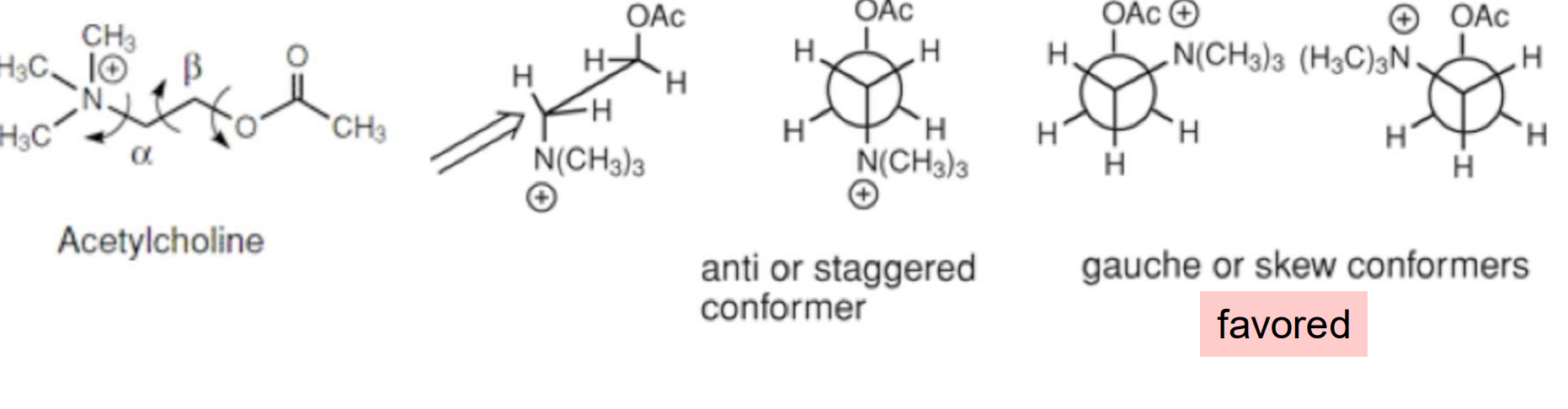
Gauche vs Anti
Gauche → 30 degrees
Anti → 180 degrees
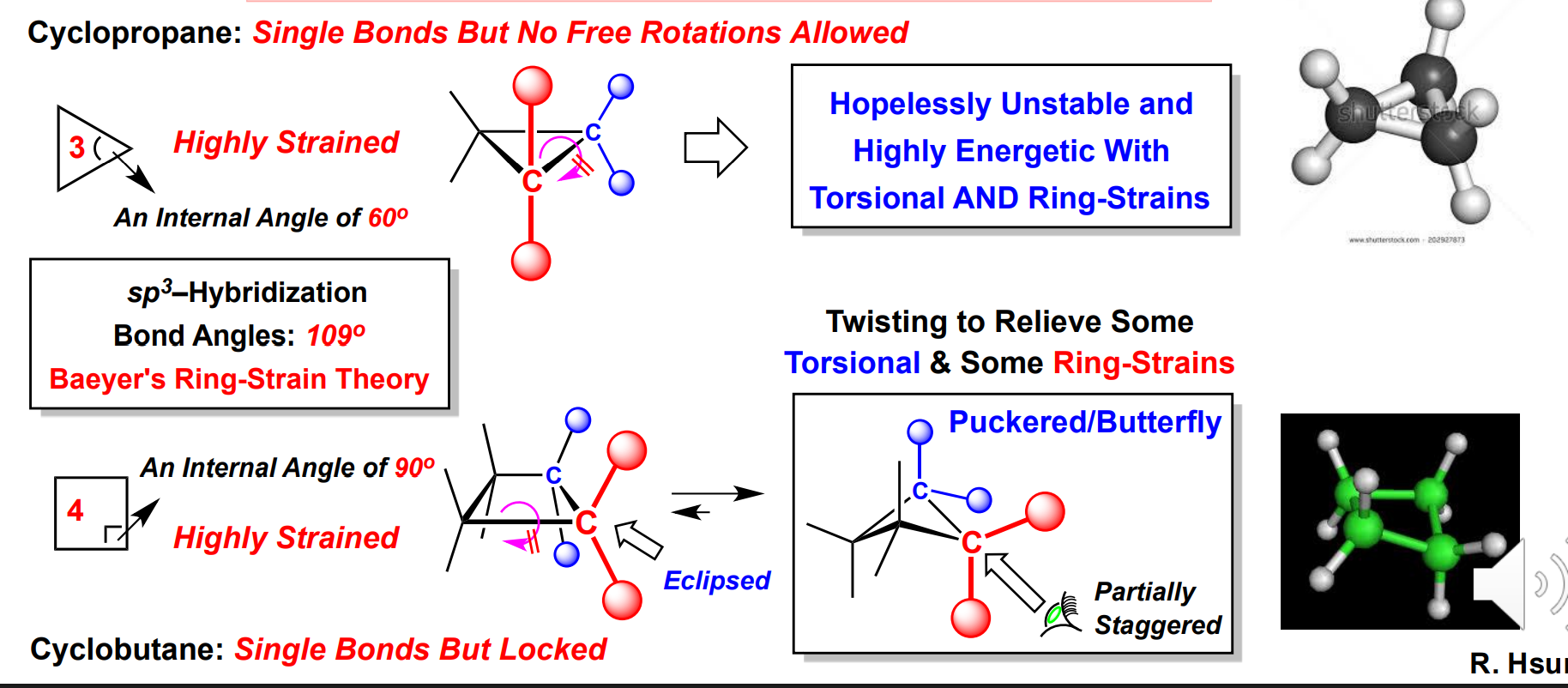
Baeyer’s ring strain
Acyclic single bonds rotate to a more stable conformation
Ex: Cyclopropane undergoes conformational change to Cyclobutane because of it being unstable
Ex: Cyclopentane undergoes conformational change to an envelope
Ex: Cylohexane undergoes chair flip to relieve torsional strain → Best of all rings → most stable
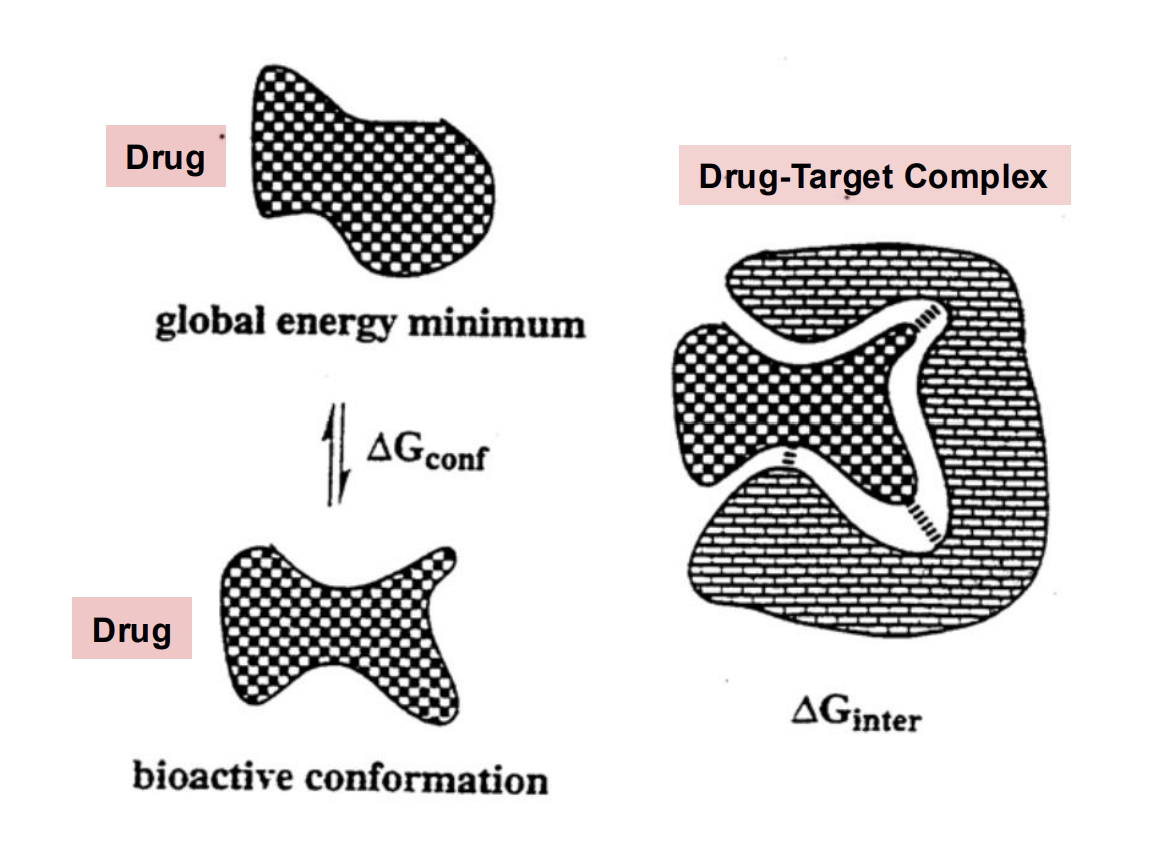
Bioactive conformation
The target/protein-bound conformation
Drug molecules that have flexibility in their conformation due to multiple rotatable bonds can have a bound conformation that is different from the solution conformation
Change in Gconf is the free energy change required to reach the bioactive conformation
There is an “entropic cost” associated with freezing bond rotations and upon binding.
Times when the intramolecular interactions override the conformational energies
Sometimes the most stable conformation is based on an intramolecular interaction that can override steric considerations →Intramolecular ion-dipole interaction favors the gauche conformer
Example: Acetylcholine, a highly flexible molecule, is surprisingly found in both solution and crystals in the gauche conformation
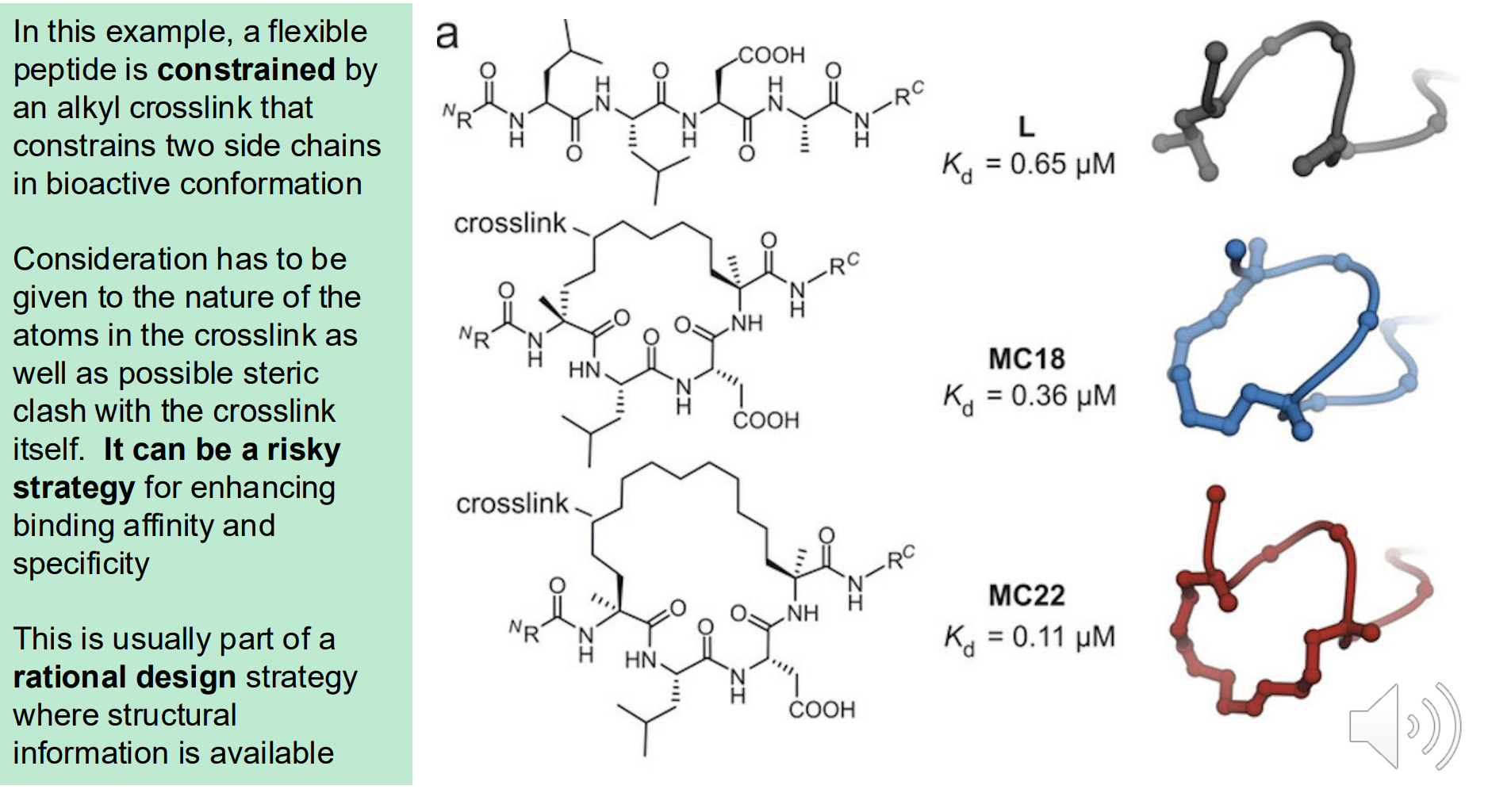
Crosslinkers
Atoms that link two areas of a molecule to constrict its conformation
This “pre-pays” the entropic cost before the binding step (Gconf is lowered)
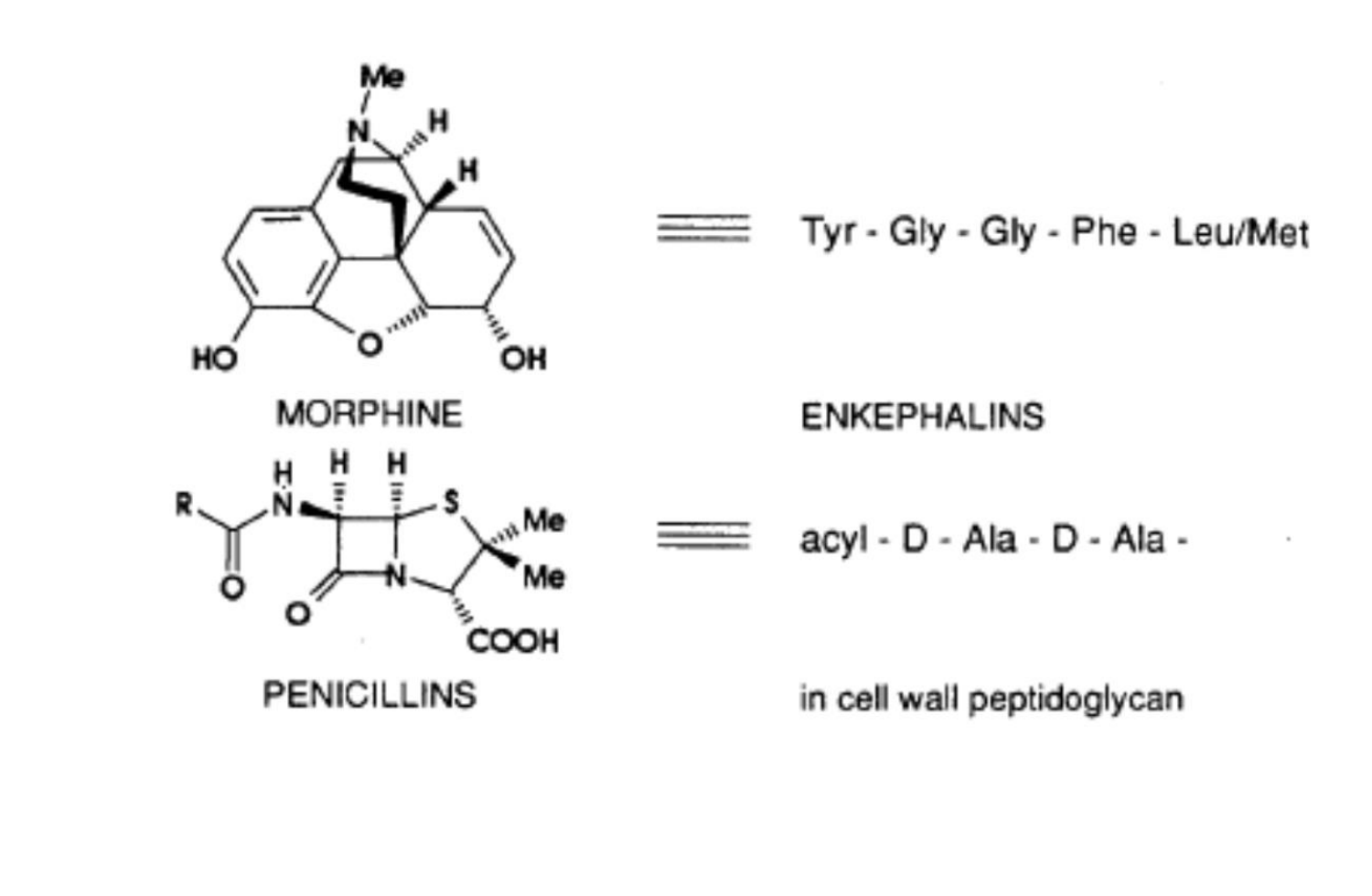
Conformational Restriction in nature
Most bioactive natural products contain rings in their structure
Rings constrain atoms into a limited number of possible conformations and improve binding affinity and specificity to the target
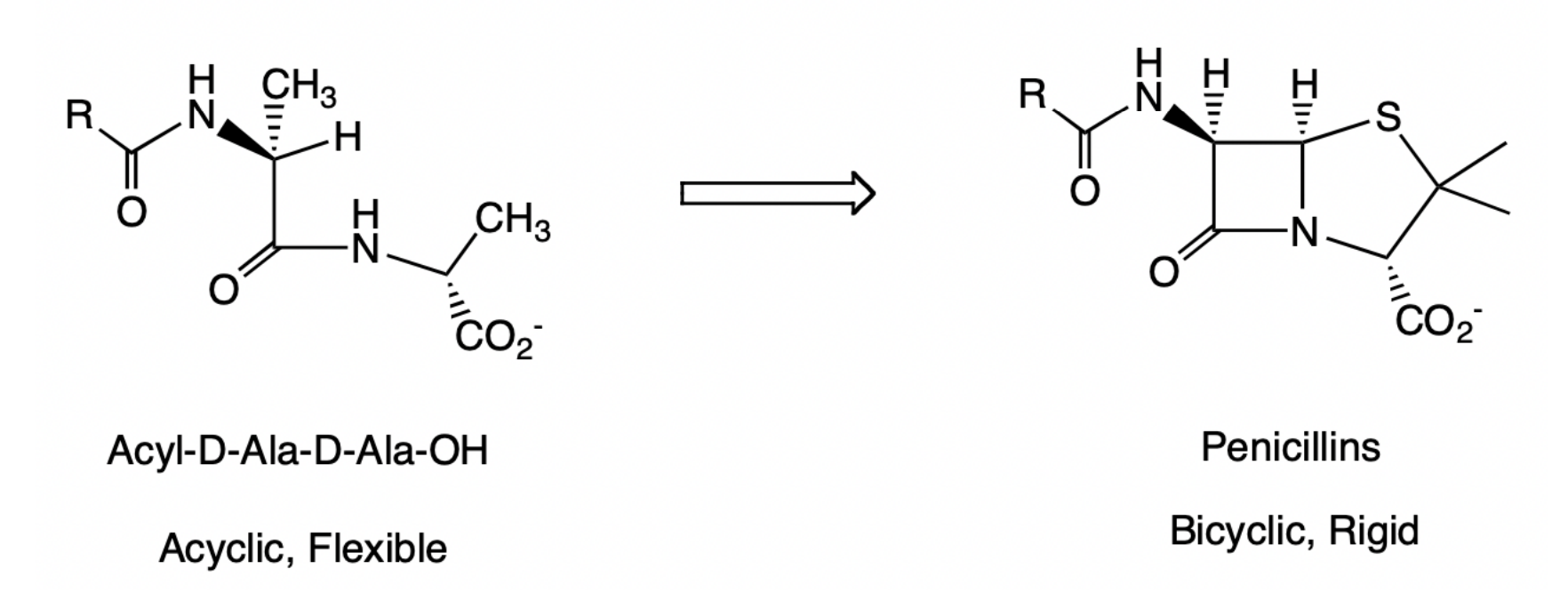
Conformational restriction by ring formation
1) Can increase affinity to target receptor or enzyme by reducing entropy loss upon binding. Atoms in the ring have already been fixed before binding to protein.
2) Can increase specificity because rigid molecule cannot access alternative bioactive conformations via bond rotations
Binding specificity in conformational changes
Acetylcholine has the flexibility to assume the bioactive conformation to activate both muscarince and nicotinic receptors
Plants have found a way to selectively modulate these receptors as a defense mechanism by the production of conformationally restricted selective ligands
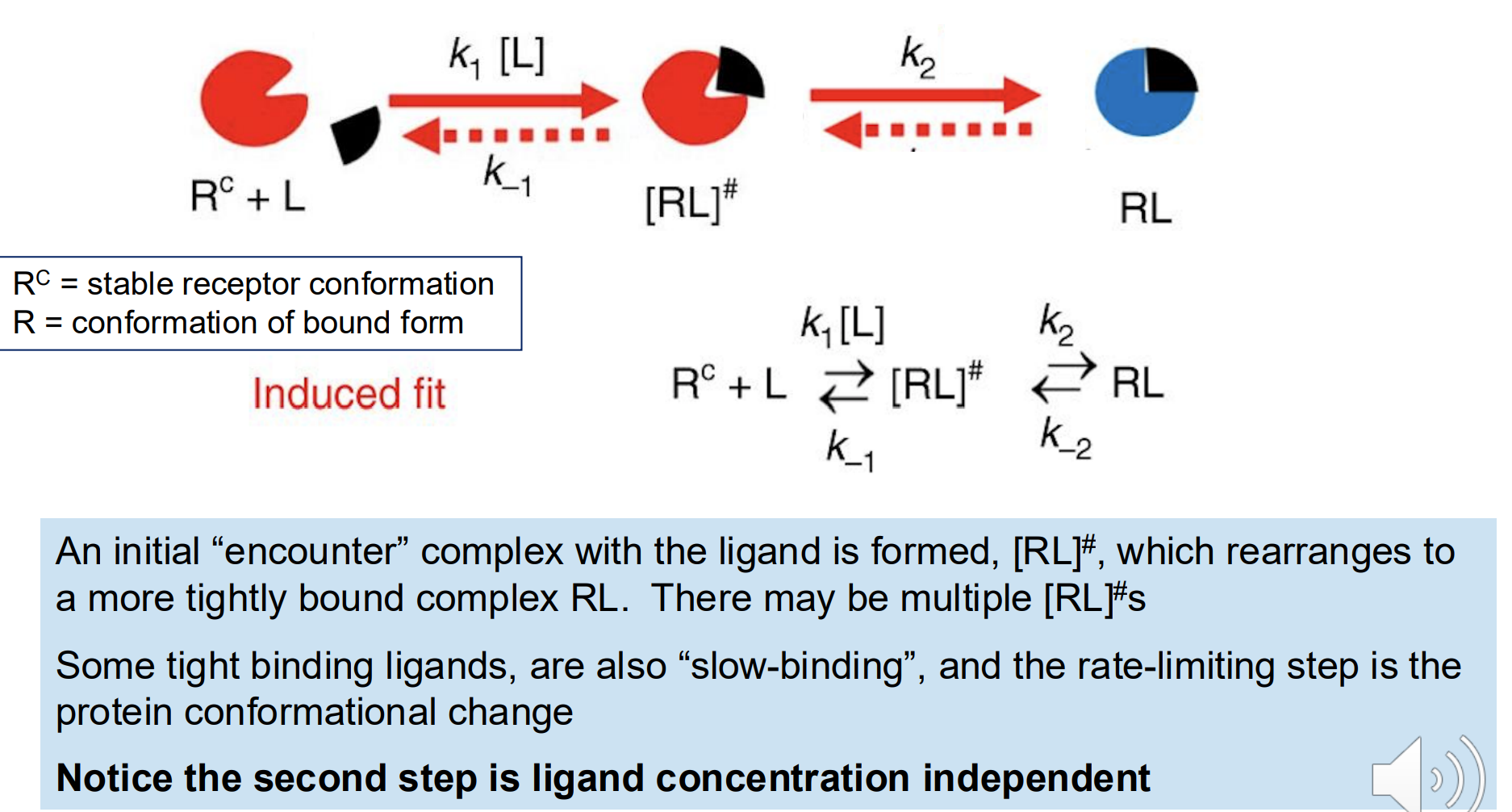
Conformational flexibility in proteins
Classical approach is that the protein is rigid
This is now clearly found not to be the case
Proteins have conformational flexibility in the presence and absence of ligand
• Previous work suggested that the main way to increase drug efficacy with a protein target is to minimize Kd (maximize binding energy)
• New evidence points to focus on increasing the drug residence time, (T) (tau), that it is bound to its target
• A binding model in which a protein conformational change is initiated by ligand binding is termed induced fit because the ligand is inducing the change
• A model in which a rare protein conformation binds the ligand directly is termed conformational selection
• The true mechanisms can be a combination of these two extremes
Induced fit
A binding model in which a protein conformational change is initiated by ligand binding
a classical mechanism that allosteric enzymes employ to regulate catalysis
Nonintuitive Scenarios with Entropy
One emerging view is that in some cases, the binding of a ligand/drug to the target receptor/protein will unlock conformational freedom in another part of the protein that makes the binding event more favorable entropically.
It is not clear how prevalent this is because we have only recently been able to measure protein conformational dynamics experimentally and it is still difficult
Further progress is required and important because we still have a poor understanding of whether a molecule is going to be a fast or slow binding drug and if it will be an agonist, antagonist or inverse agonist, etc
Tau
involves both a thermodynamic and kinetic understanding of the individual steps in complex formation
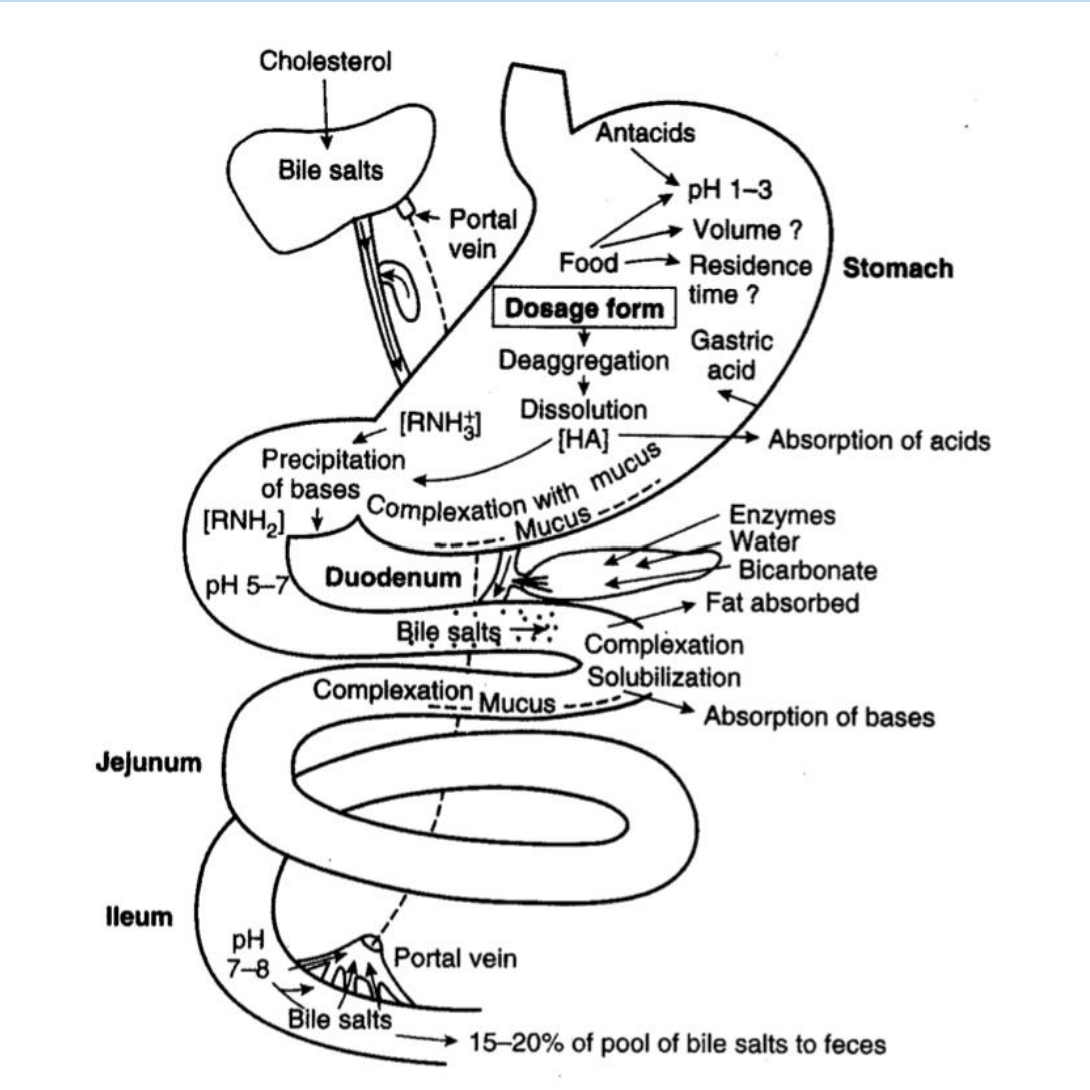
What is required for an oral drug to be absorped?
pH varies from 1-8
Must be stable to acid
Ionizable groups must be neutral for best absorption
Volume and surface area differences affect absorption rate
Lipophilic molecules can be better absorbed by complexation with bile salts
Microbes in gut can alter structure of some drugs
Small intestine is rich in metabolic enzyme and transporters
Lead optimization
The process of improving the drug for use in humans
Initially discovered bioactive compounds for a given disease state (”lead compounds”) are often not the final structural versions of the marketed drug

Lipinskis rule of 5
Approved oral drugs are getting a little larger and complex
Mean values and trends of Lipinski-type parameters for marketed small molecule drugs means that most drugs follow this rule
Exceptions:
Some surprises are due to intramolecular interactions
Ex:
The immunosuppressive cyclic peptide cyclosporine can shield polar groups by forming 3 intramolecular hydrogen bonds

Active vs passive transport
Unlike passive diffusion, active transport is directional, specific and shows saturation kinetics due to requirement for a protein binding step
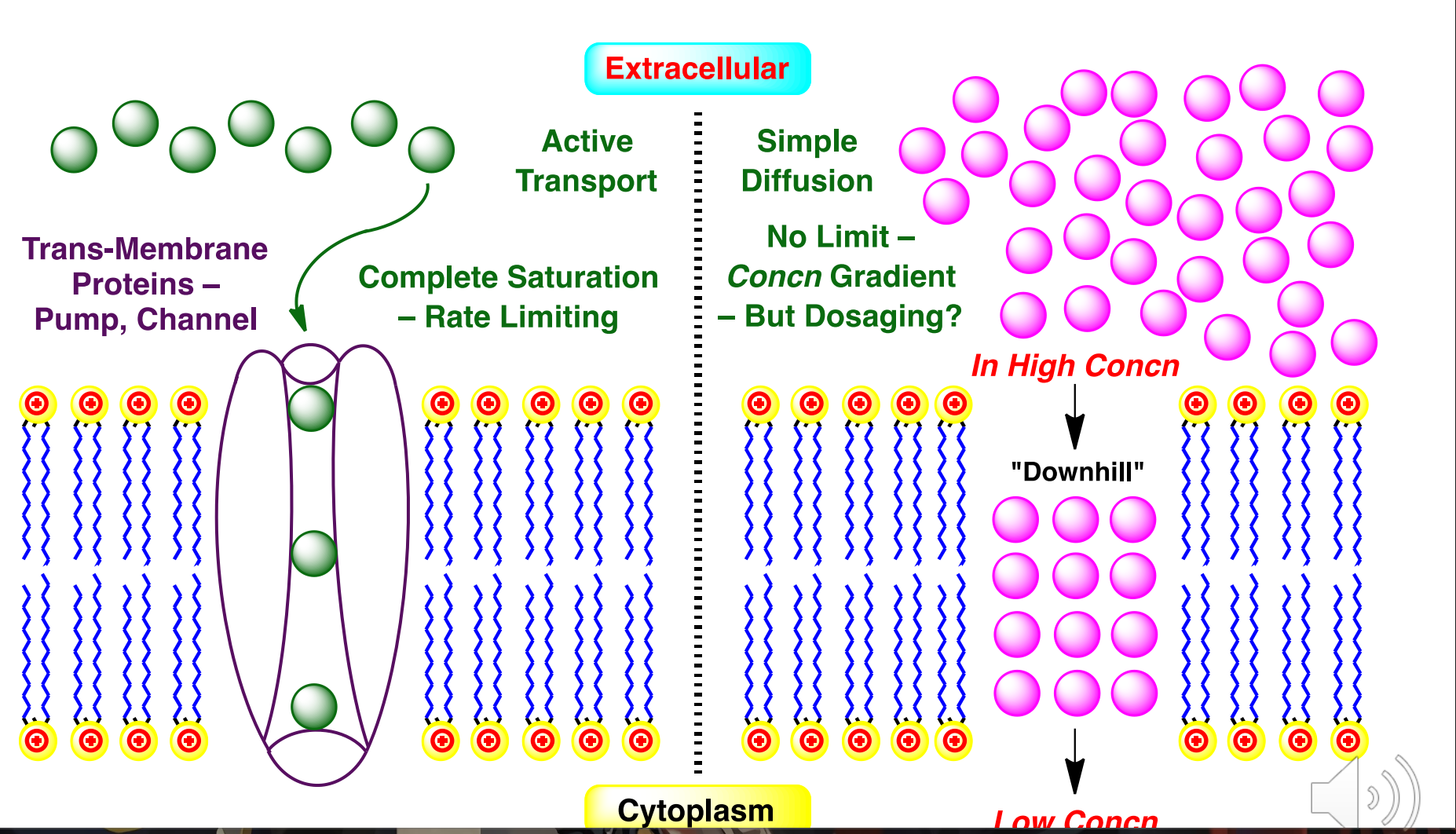
Importance of transport proteins
Human cells contain an enormous variety of membrane bound transport proteins which have overlapping substrate specificity and are crucial for moving molecules in and out of cells
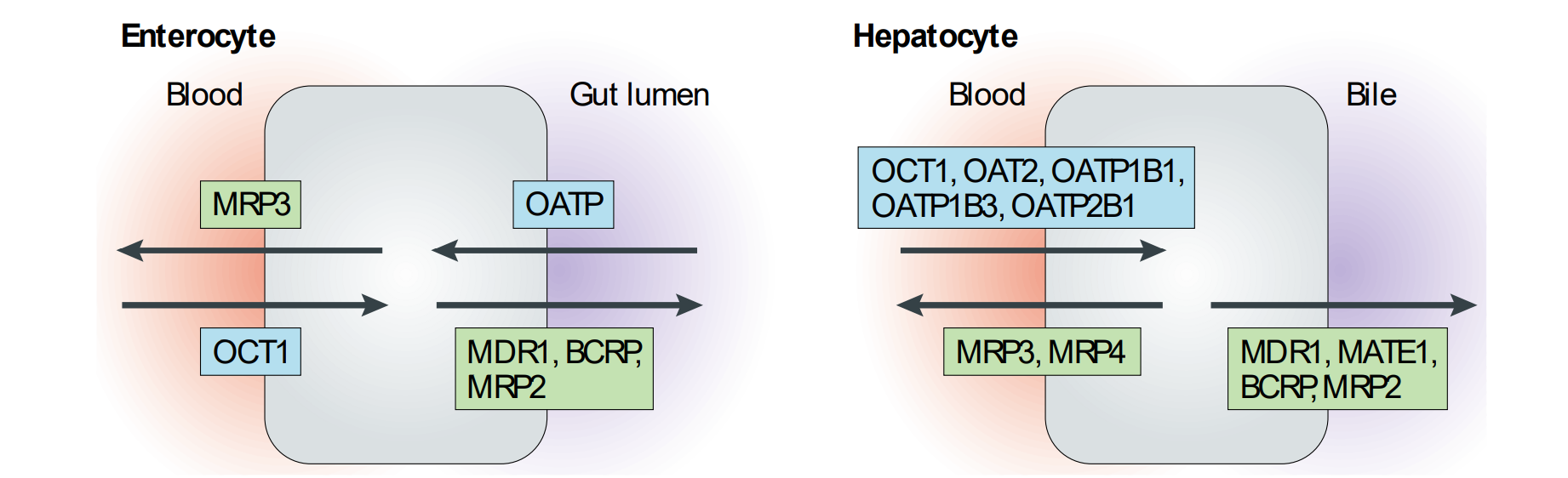
Drugs taken orally may be substrates for the many transporters that are present in both enterocytes (intestine) and in hepatocytes (liver), as well as cells lining the BBB and target tissue
Two superfamilies - Solute carrier proteins (SLCs) that import molecules into cells OCT = Organic Cation Transporter; OATP = Organic Anion Transport Protein ATP-binding cassette proteins (ABCs) that export molecules out (efflux)
Drugs that are both extensively metabolized and are substrates for ABC/MDR/MRP efflux pumps have poor oral bioavailability. Inhibitors of efflux proteins can increase bioavailability.
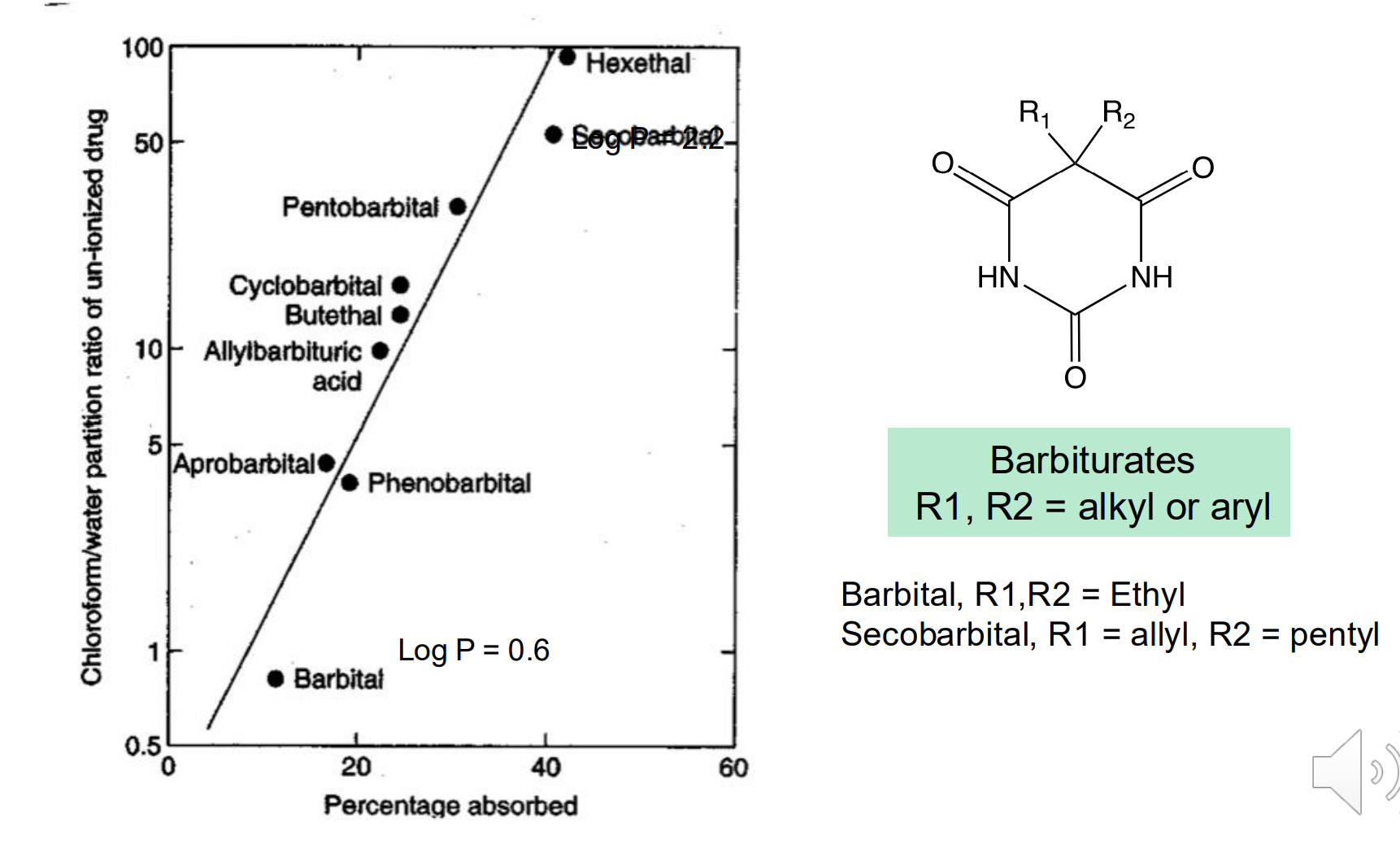
Relationship between LogP and Permeability
There is a positive correlation between lipophilicity and passive diffusion across membranes
Relationship between Log P and BBB passage
CNS bioavailability requires neutral species for passive diffusion Molecules above the trend line likely undergo active transport into the brain Molecules below the trend line higher MW or may be substrates for efflux pumps such as Pglycoprotein In this data we don’t see an optimum logP for permeability however a molecule like cholesterol (logP = 8.7) will mostly stick in the membrane of cells or form micelles
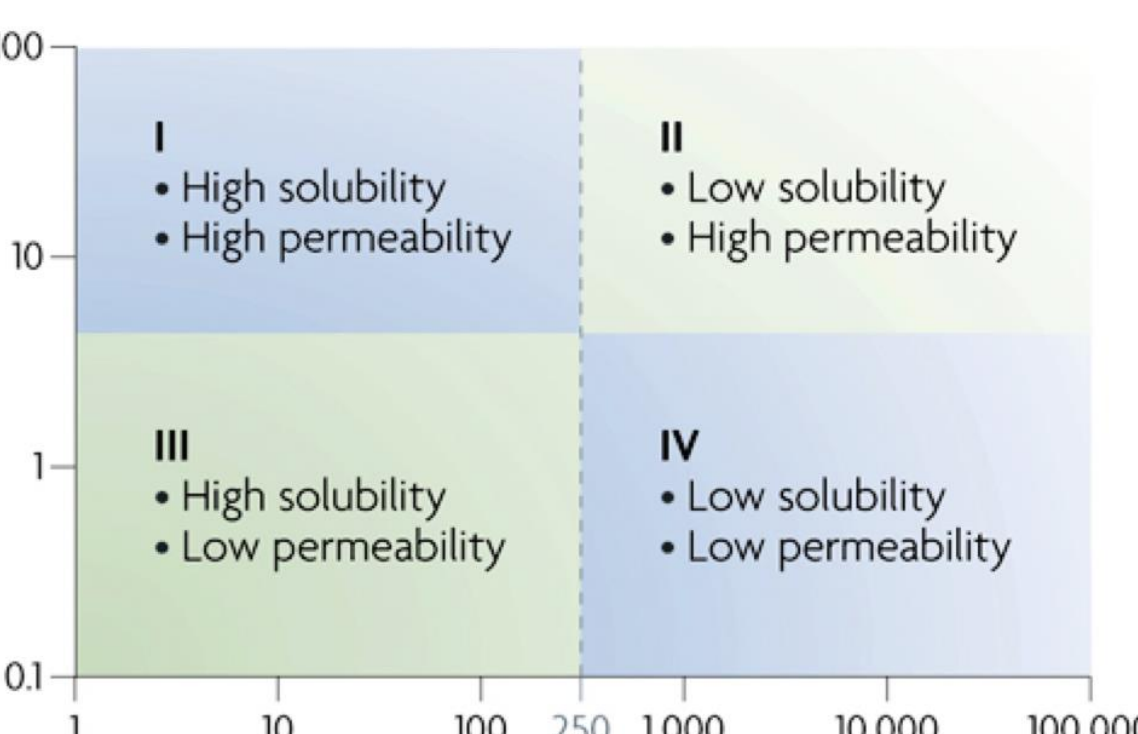
BCS classes
Group I – generally good oral activity
Group II – solubility is rate limiting step for absorption
Group III – permeability is rate limiting
Group IV – usually low oral bioavailability. Often substrates for Pglycoprotein
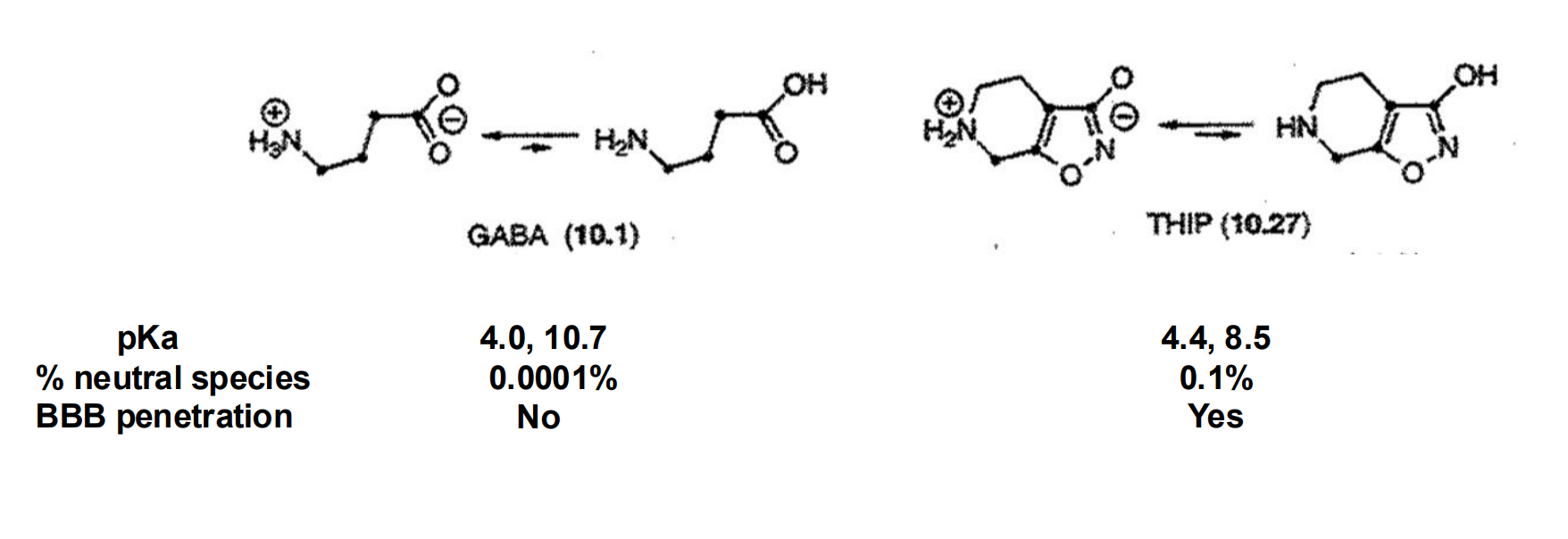
pKA considerations for BBB passage
NS bioavailability requires neutral species for passive diffusion in addition to low MW
EX: THIP is a partial agonist at GABAA receptor and a conformationally restricted GABA mimic which, unlike GABA, has the ability to penetrate the blood brain barrier (BBB)
Notice the difference in pKa values for the anionic and cationic functional groups between GABA and THIP
EX: amines
For non-quaternary amines, there is a fraction of neutral species that can pass through the BBB via passive diffusion
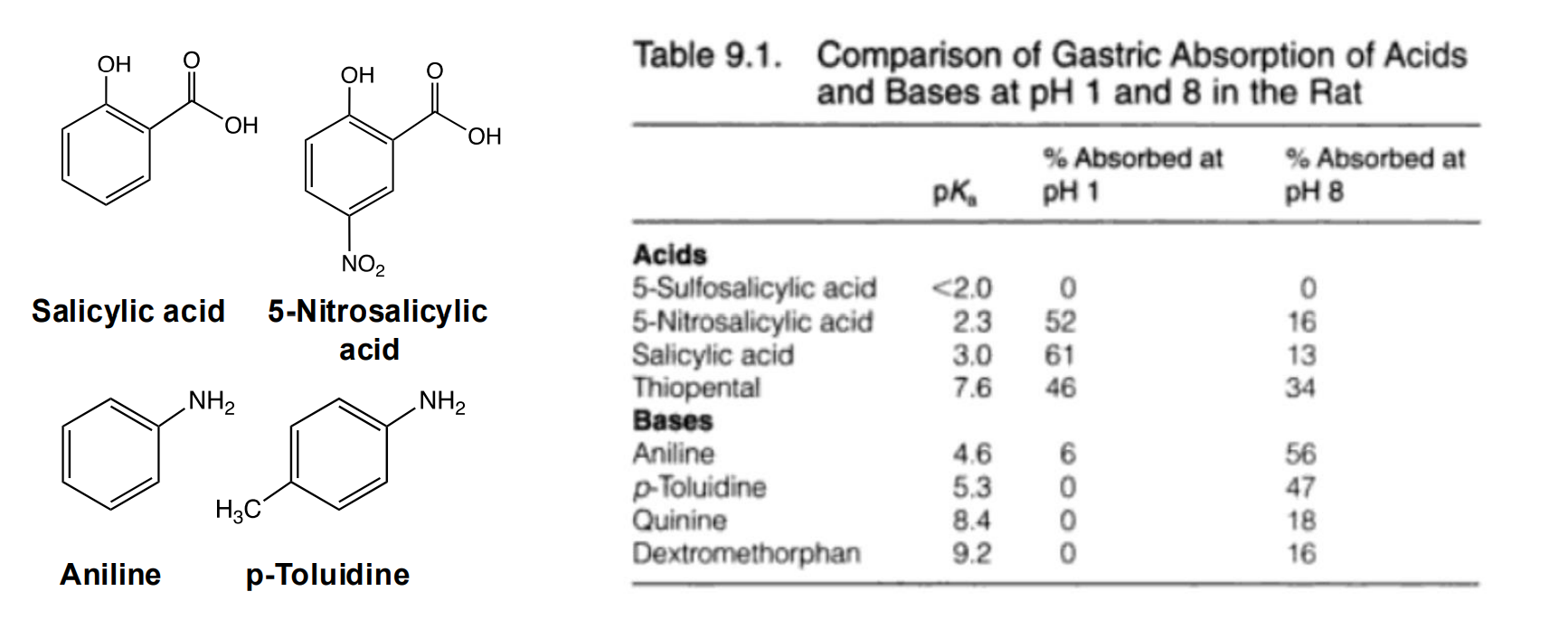
Absorption of Acids vs Bases
Absorbance of ionizable groups is predictable based on % ionized
Acids are absorbed fastest in the stomach because a larger fraction of the molecule is neutral
In the intestine:
the correlation is not as clear, but the trend across pH values is consistent with a correlation to the neutral form having increased absorption The physiology of the intestinal barriers is more complex and less well mixed which may explain local pH aberrations near the absorption sites Due to the larger volume and surface area, even acidic drugs are mostly absorbed here
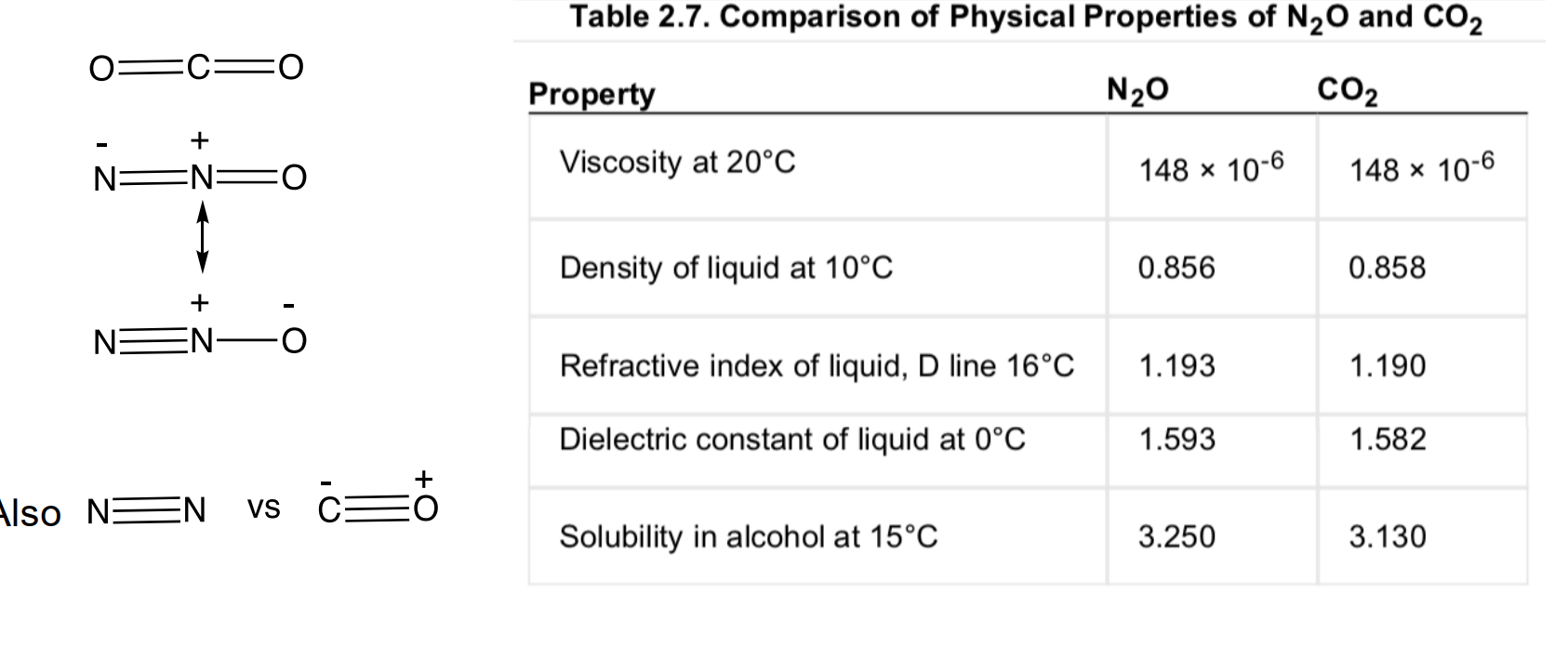
Isoteric Substitution
Langmuir (1919) observed similar physicochemical properties were displayed by simple isosteric substitutions in molecules.
Example: nitrous oxide vs carbon dioxide
Strict isosterism principle has evolved: first to small groups of atoms to whole rings, to bioactive functional groups
RIng isosterism
the nitrogen and sulfur substitutions are conservative isosteric replacements for C-H
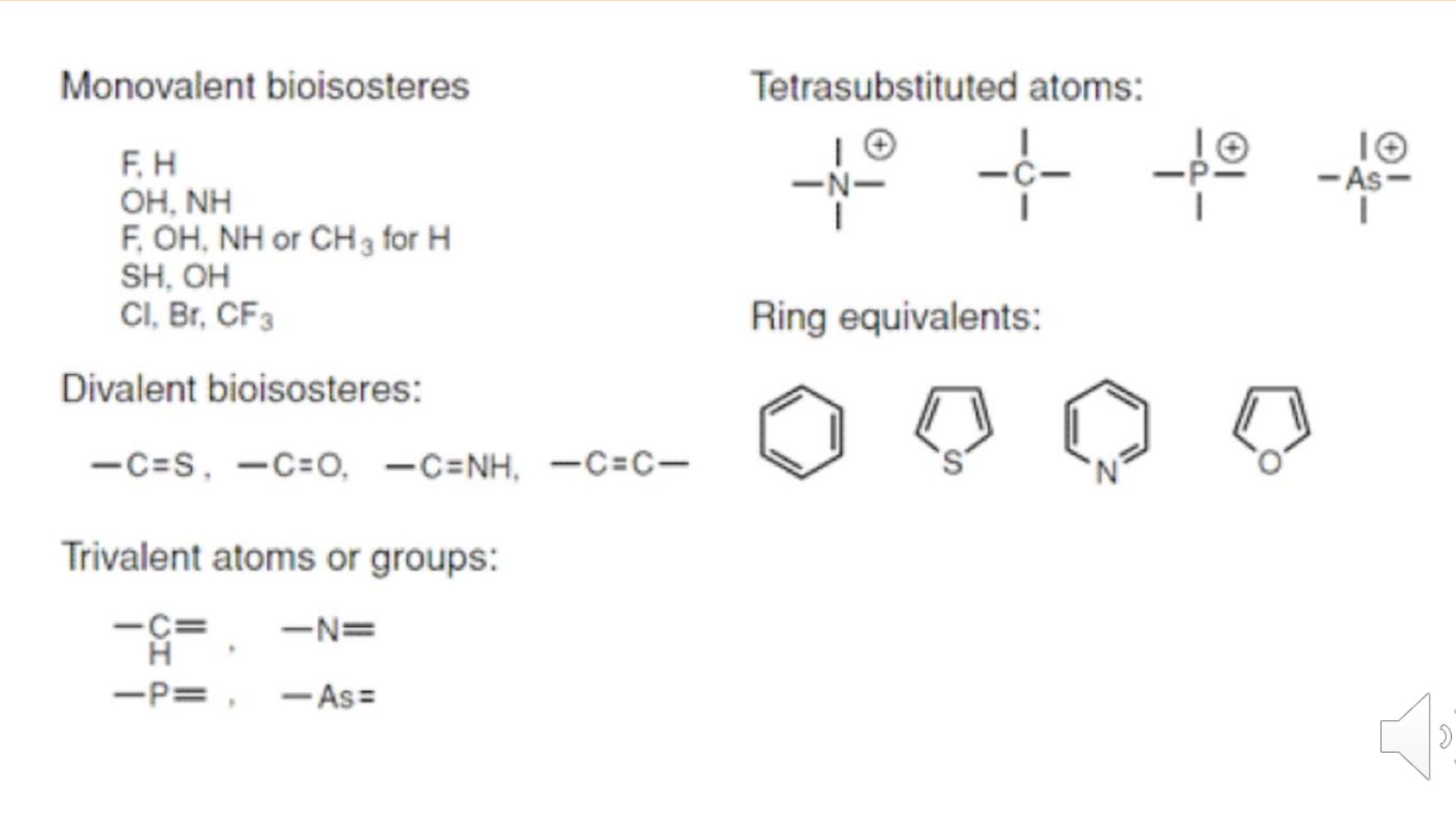
Bioisosteric substitution
Classic Bioisosteric Replacements have very similar steric properties and involve single atom substitutions and in rings accompanying contraction.
Properties however are sometimes changed significantly by these substitutions
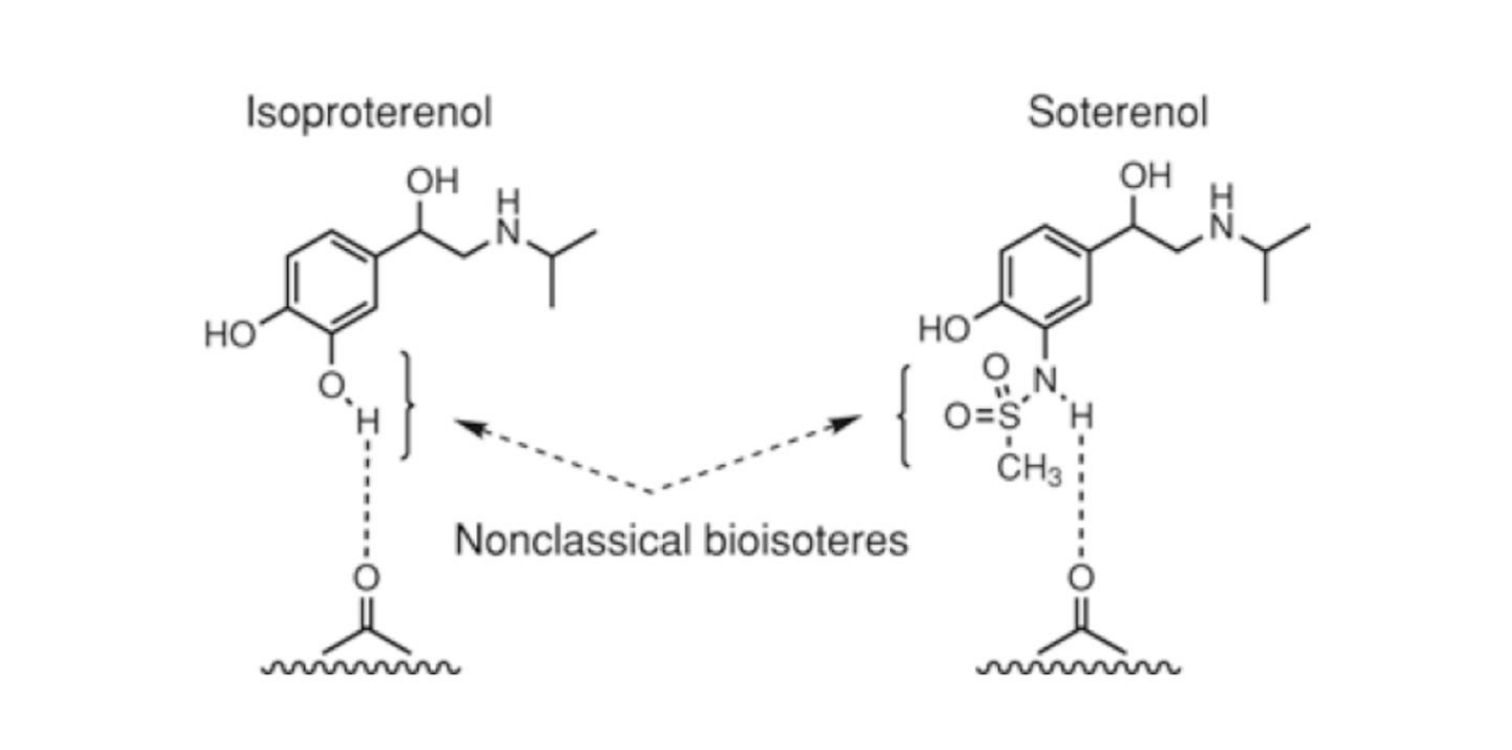
Nonclassical bioisosteric substitution
have sometimes widely different steric properties but involve group replacements to maintain properties such as electronic distribution or pKa that are important to retain bioactivity
In the above example, the phenol and sulfonamide have similar pKas and can both donate a hydrogen bond, even though the sulfonamide is significantly larger steric demand
bioisosteric rings
Cyclic bioisosteres for carbonyl containing groups can mimic the electronic distribution of the group as well as the pKa and other properties
Some of the replacements can be head scratchers... The rigidity of the ring can also introduce risk
Me-Too Drugs: H2 Antagonists
Clear SAR in the Histamine H2 Antagonist drug class led to a series of bioisosteric substitutions that resulted in the first in class drug Cimetidine followed by a series of “Me-Too” competitors
In this case, problems with metabolic inhibition, and poor solubility and bioavailability opened the door to competitors with improved ADME properties
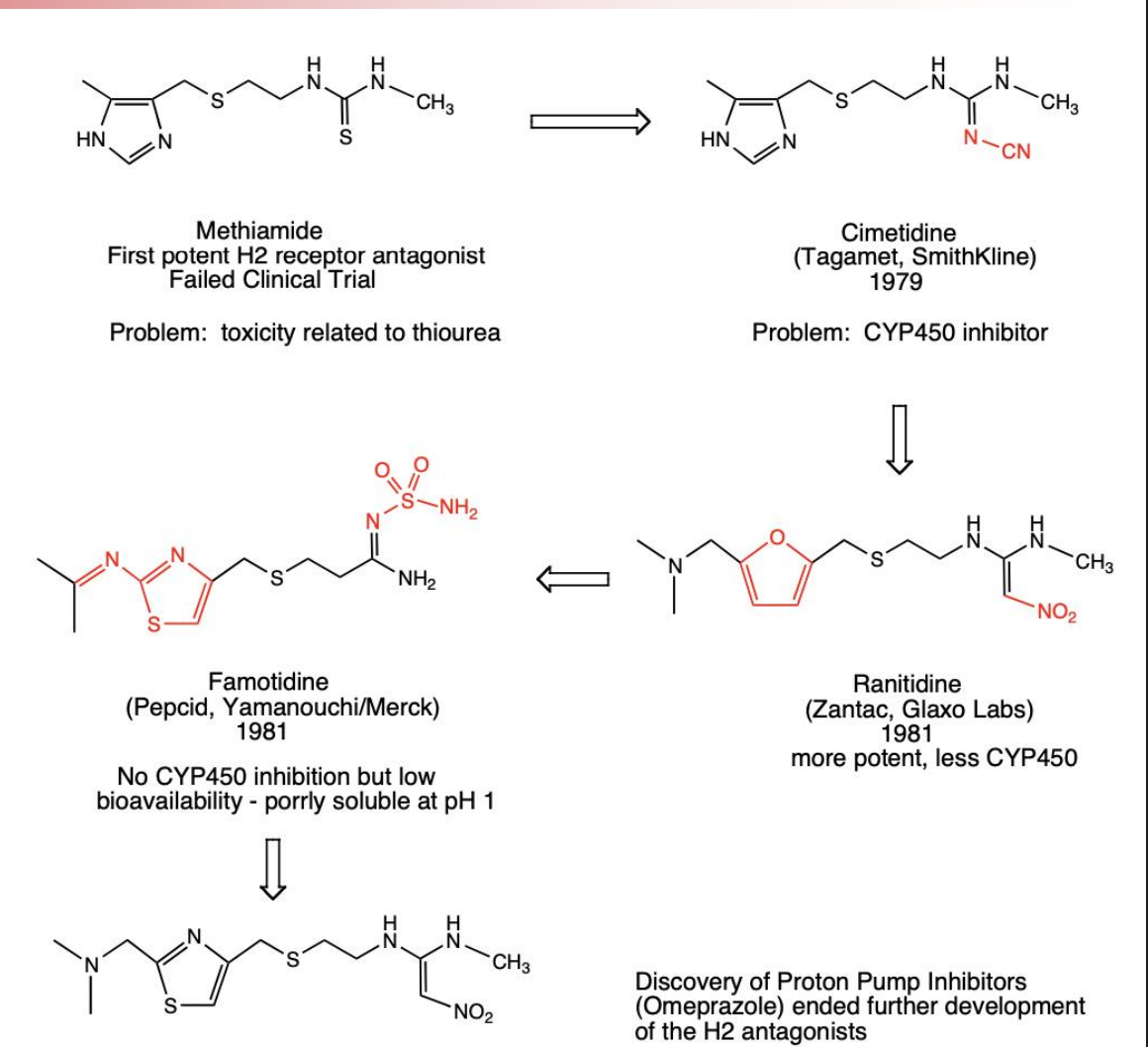
Me-Too drugs pros and cons
Positive aspects for Me-Too Drugs are:
They can improve on ADMET or specificity issues with previous drugs in the same class
They sometimes lead to entirely new activities which lead to new drugs (antihistamine to antipsychotic to antidepressant tricyclics)
They can compete with the first in class drug to drive down prices
Negative aspects are:
They can use up resources that could otherwise be spent on novel drugs to treat more pressing diseases
They can sometimes be of minimal additional benefit to patients
Marketing may exaggerate the benefit of the new drug
They can drive prices up by delaying entry of generics
Me-Too Drugs are attractive largely because there are few clinical trials that involve driect comparison between two drugs in the same class. The FDA only requires that a drug be better than placebo not the current standard of care. Then marketing can influence which drug is adopted if the two are similar in efficacy.
Deuterium substitution by hydrogen
Unique case of bioisosterism by isotopic substitution
The prediction is that binding will be identical because it is dominated by interaction of valence electrons between drug and target.
Although there is evidence this is not totally accurate
Deuterium is a heavy isotope of hydrogen which has an additional neutron in its nucleus. This gives a slightly stronger and shorter bond that is kinetically slower to break because it sits in a deeper kinetic energy well (“zero point energy”)
The slower rate of C-D bond breaking vs C-H is termed a kinetic isotope effect

Pioglitazone
Type-2 diabetes drug
Activity is due to the R-enantiomer, while S-enantiomer has shown weight gain and edema (swelling) side effects due to PPAR receptor binding
PXL065 – deuterium stabilized R-enantiomer completed Phase II successfully as treatment for NASH (Nonalcoholic steatohepatitis) – no PPAR side effects
The deuterium substitution slows the rate of racemization in vivo, which is catalyzed by base removal of the proton/deuteron.
The difference in rate, (kH/kD), can be up to 10-fold In mice, admin of racemic pioglitazone gives 3:1 (S:R) after 5 days, while admin of PXL065 reverses that ratio to 2:1 (R:S) (R) Hep
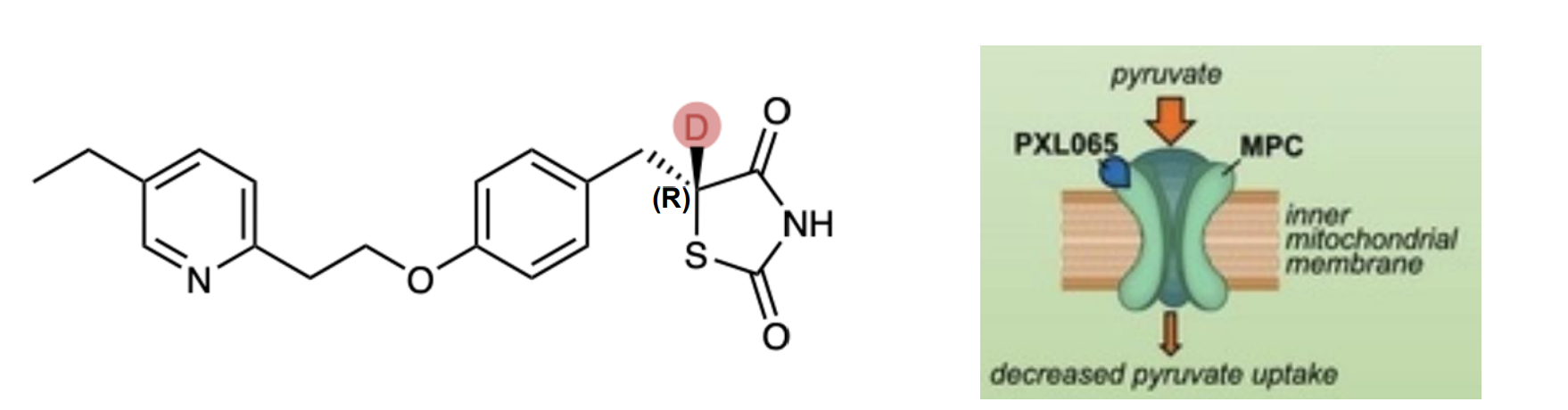
Antimetabolites
Alternative substrates that block vital metabolic pathways
Reversible inhibitors
Multisubstrate analog inhibitors → derive binding energy by tethering multiple substrates in one molecule
Transition state analogs → derive binding energy by mimicking high energy reaction intermediates and/or transition states
Irreversible inhibitors
Affinity labels → substrate analogs specific for enzyme active sites and contain a highly reactive electrophilic group
Mechanism Based Inhibitors (“suicide substrate”) → substrates that are converted to a reactive species by the action of the target enzyme
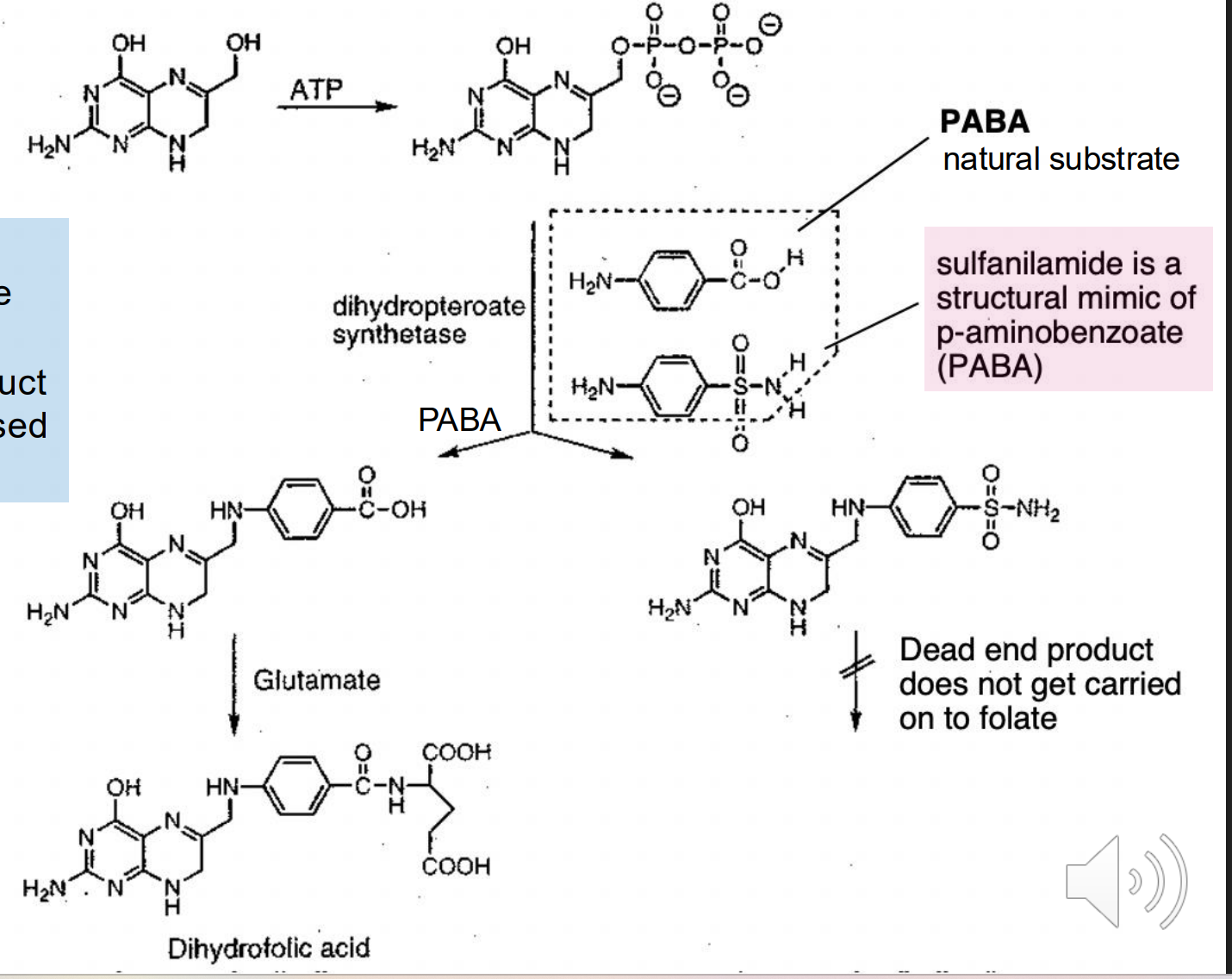
Prontosil
A red dye that was discovered that protects mice from death by steptococcus
Converted to Sulfanilamide
Would not have been discovered if screened against bacterial cultures in vitro → No mouse to convert the molecule to sulfanilamide
Sulfanilamide is an alternative substrate in folic acid biosynthesis (mimics PABA which leads to cell death)
Insoluble in water so must be injected
“Elixir of Sulfinaliamide” led to creation of more restrictive FDA
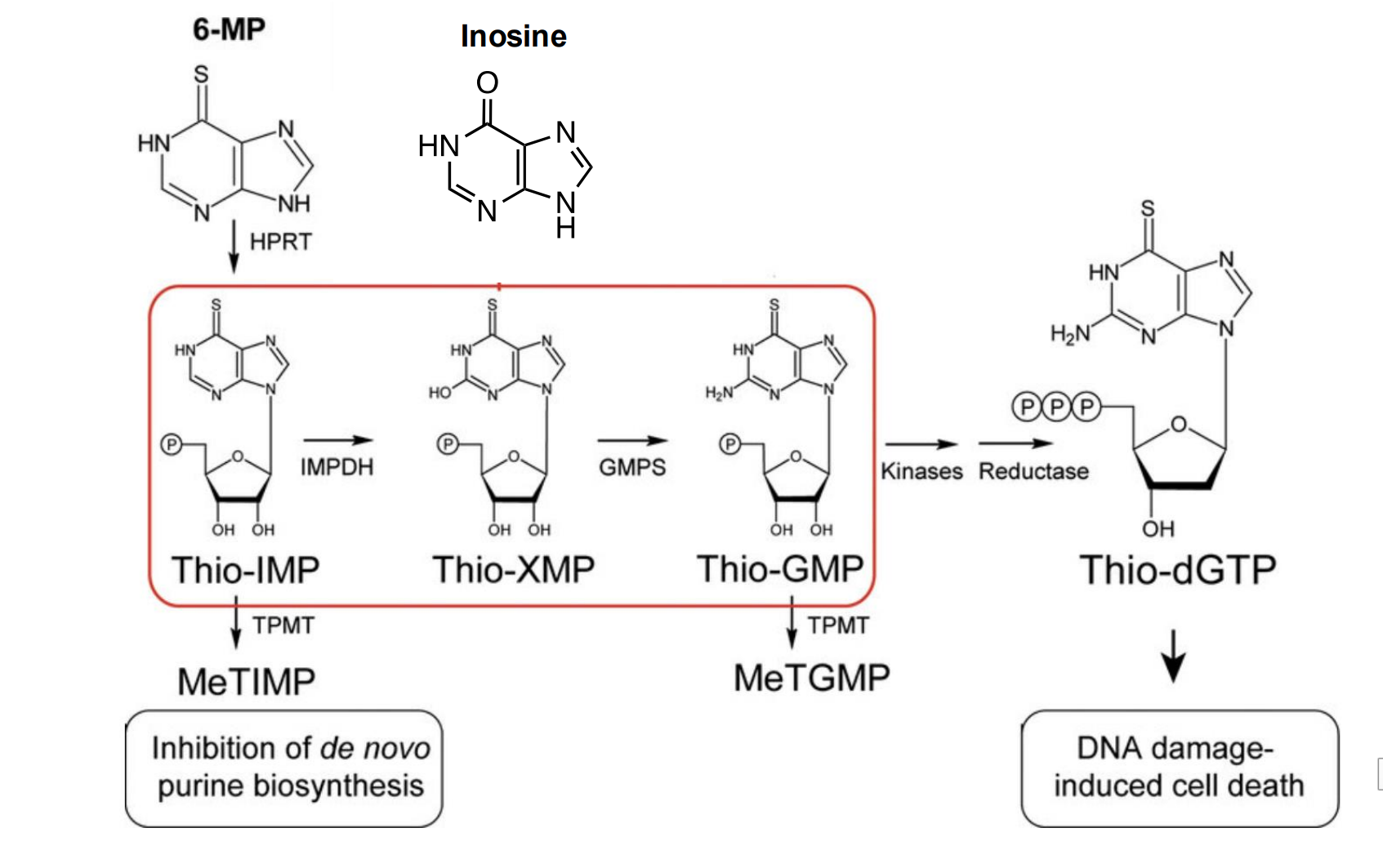
6-mp
Taken up as substrate for the purine salvage pathway
This leads to dead end pathway toxicity and cell death
Multisubstrate Analog Inhibitors
mimic two or more substrates in one molecule
They gain an entropic advantage as well as enthalpic because they “prepay” the loss of entropy by freezing the translation of one of the substrates.
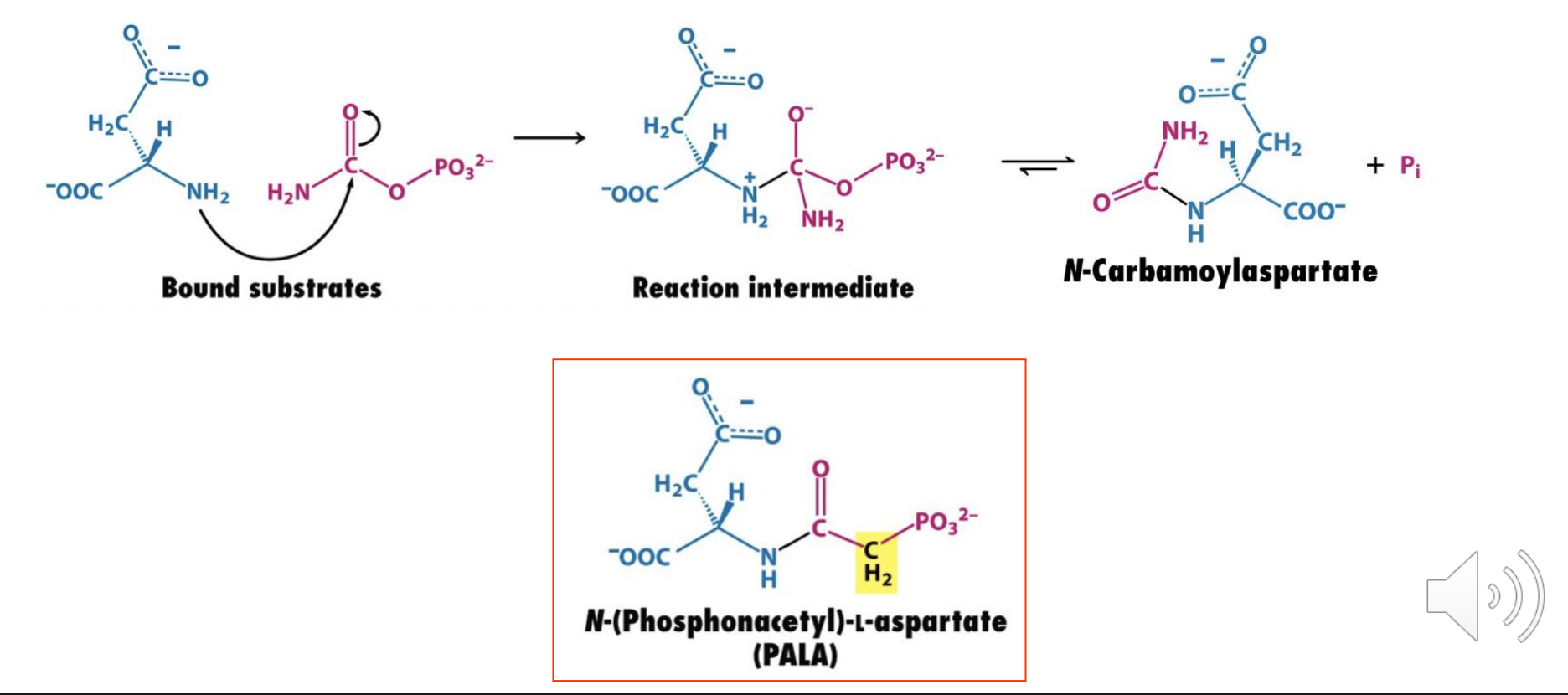
Ex: PALA has structural features of both substrates L-aspartate and carbamoyl phosphate and is a tight binding inhibitor of aspartate transcarbamoylase
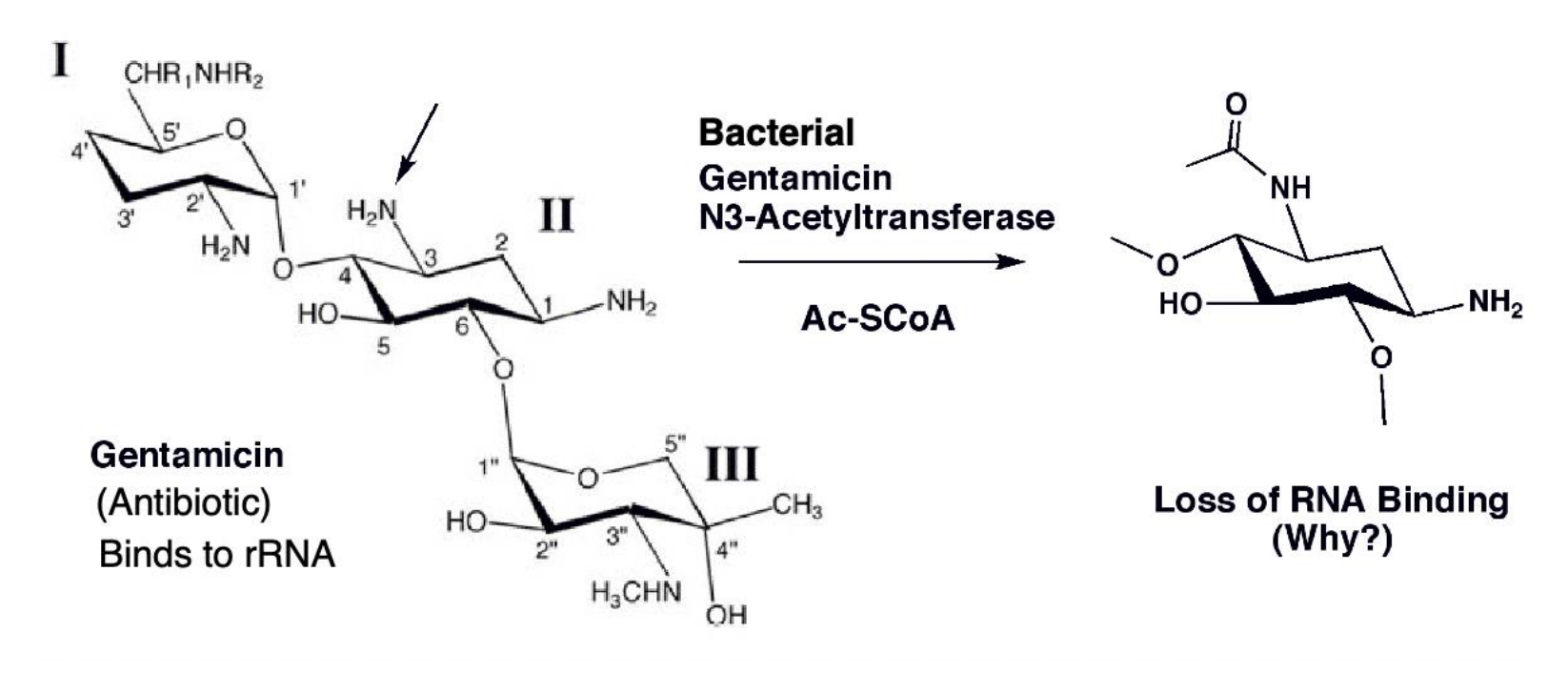
Gentamicin resistance
Bacterial evolution of gentamicin resistance via acetylation of the drug molecule
The enzyme selectively transfers an acetyl group to the 3-amino of the gentamicin which decreases the affinity of the drug for its rRNA target
The mechanism of the acetylation reaction proceeds via a tetrahedral intermediate in which both substrates are in close proximity for reaction
The enzyme accelerates the reaction in part by approximation, or the bringing together of the two substrates in the proper orientation.
Transition state analog inhibitors
Mimic the transition state of the enzymatic reaction
Enzymes catalyze reactions in part by stabilizing the transition state (reducing Change in G‡ )
They use noncovalent interactions to do this
Thus, TS mimics can have more of these interactions (and bind more tightly) than either the ground state substrates or products
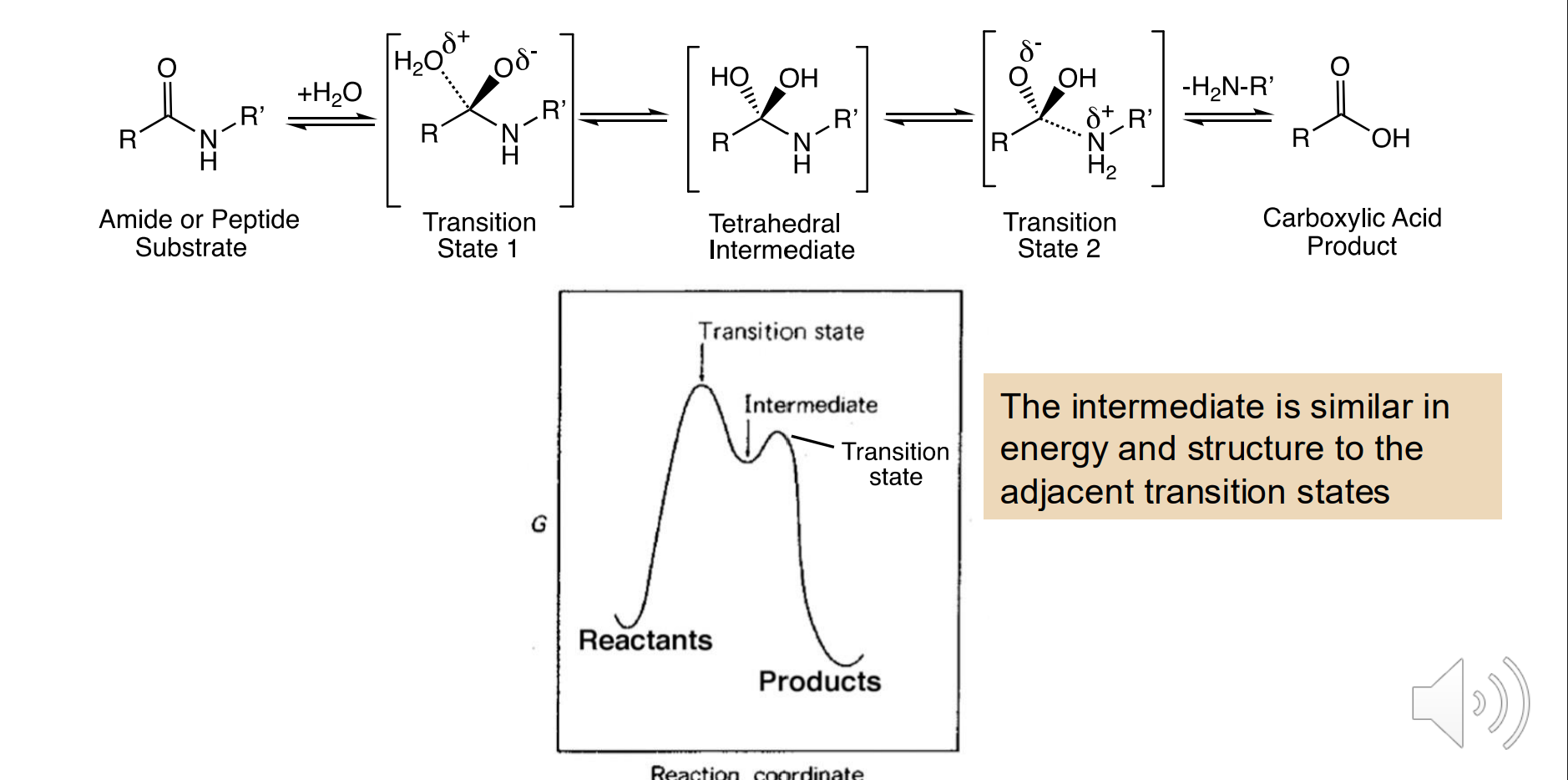
Unstable reaction intermediates are usually similar to each transition state, both in energy and usually in structure
So many are known as reaction intermediate analogs