Biotransformation/Drug metabolism (Pharmacokinetics)
1/26
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
27 Terms
Define Biotransformation
Chem alteration of drug in body converting lipophilic drug —> hydrophilic drug
to be easily excreted by kidney
Occurs in liver through Phase I and Phase II.
Example of Active drug —> inactive drug (bio activity of drug change)
Most drugs
Example of active drug —> active drug (bio activity of drug change)
Codeine —(oxidation)—> Morphine
Diazepam —(oxidation)—> Oxazepam
Phenacetin —(oxidation)—> Acetaminophen
Inactive drug —> active /prodrug (bio activity of drug change)
Enalapril —> Enalaprilat
Active —> More active/ toxic (bio activity of drug change)
Chloral hydrate —> Trichloroethanol
Too much paracetetamol
Sites of metabolism
1st pass metabolism (1st site of drug administration)
Liver (major metabolism site)
Lungs, Kidneys, adrenals
Drug after phase 1 and phase 2
Following Phase I = Drug may be activated, unchanged, inactivated (most often)—> Oxidation (most imp), Reduction, Hydrolysis
some drugs go directly enter phase II metabolism
Phase 2 product = conjugation product (conjugated drug usually inactive)
Describe phase I of metabolism
Phase I =
Convert lipophilic -—> more polar molec
May ↑ (active metabolites) . ↓( inactive metabolites) or unaltered drugs pharmacological activity
Drug metabolism most freq reaction involved catalysed by cytochrome P450 syst (AKA microsomal mixed funct oxidases)
What do enzyme inducers do and how does that affect biotransformation
Enzyme inducers induce selected CYP isozymes —> ↑ drug biotransformation—>
(significantly)↓plasma conc of drugs that are metabolised by CYP isozymes
Enzyme inducers
Phenobarbital (convulsion)
Rifampin (TB)
Carbamazepine
CYP2D6 vs CYP3A4/5
CYP3A4 = Most imp for metabolism 50-60% drugs
CYP2D6 has no enzyme inducers
What do enzyme inhibitors do and what are the consequences?
Def: Inhibit CYP isozyme activity
Consequence = Higher blood levels and potential ↑ drug therapeutic and/or toxic effects
(imp source of drug interactions but can lead to serious adverse events)
Examples of enzyme inhibitors
Ketoconazole (anti fungal)
Omeprazole
Cimetidine
Erythromycin
Ritonavir
Define Conjugation (Phase II reaction)
Def: Coupling of drug or its metabolite with endogenous substrate
like glucuronide, sulfate, gluathione
Others = methyl group, acetic acid, amino acid, carb
Enzymes catalysing phase II biotransformation
Glucuronyl transferase (glucuronide conjugation)
Sulfotransferase (sulfate conjugation)
Transacylase (amino acid conjugation)
Other = acetylase , ethylases, methylases , gluathione transferase
Where are the enzymes for phase II biotransformation
Numerous tissues , plasma, (sub cellular incl) cytosol mitochondria and
endoplasmic reticulum
Glucorinide conjugation vs non glucorinide conjugation
Glucuronide =
Most common
Drug/ metabolite coupled glucuronic acid
Catalysed by microsomal enzymes
Occurs with phenols, alcohol, carboxylic acids
non glucorinide =
Less common
Catalysed by non - microsomal enzymes
Occurs with Sulfate, O,S and N - methylation, N-acetylation, Glycine and glutamine, Gluathione and carbs.
Note:
Sulfate (eg:steroid).
O,S and N - methylation (eg:norepinephrine),
N-acetylation (eg:salicylic acid),
Glycine and glutamine amino acids (eg:salicylic acid),
Gluathione (eg: etharynic acid).
Phase I vs Phase II reaction (Def, Reactions, Catalysed, Results)
Phase I
a. Converts lipophilic drug (non polar) —> Hydrophilic compound (polar)
b. Oxidation, reduction and hydrolysis reaction
c. Most reactions catalysed by cytochromeP-450
d. Results:
Active drug —> inactive metabolites , toxic metabolites, more active
Inactive drug —> prodrugs (active)
Phase II
a. Converts drug/metabolites into highly polar compound
b. Conjugation (glucuronic acid or other)
c. Most reactions catalysed by glucuronic acid
d. Results = Inactivate drug and make polar so easily excreted
Phase II can be done without going through phase I
Explain 1st pass metabolism
Drug absorbed GIT first enters portal circulation then systemic. If drug metabolised in liver or gut wall during initial passage ↓ amount of uncharged drug in systemic circulation.
Consideration of 1st pass hepatic metabolism and an example
First pass metabolism by intestine or liver limits drug efficacy when taken orally
Drugs that exhibit high 1st pass metabolism should be given in sufficient dose or change route of administration
Eg: >90% nitroglycerin cleared during single passage through liver so administered sublingual route.
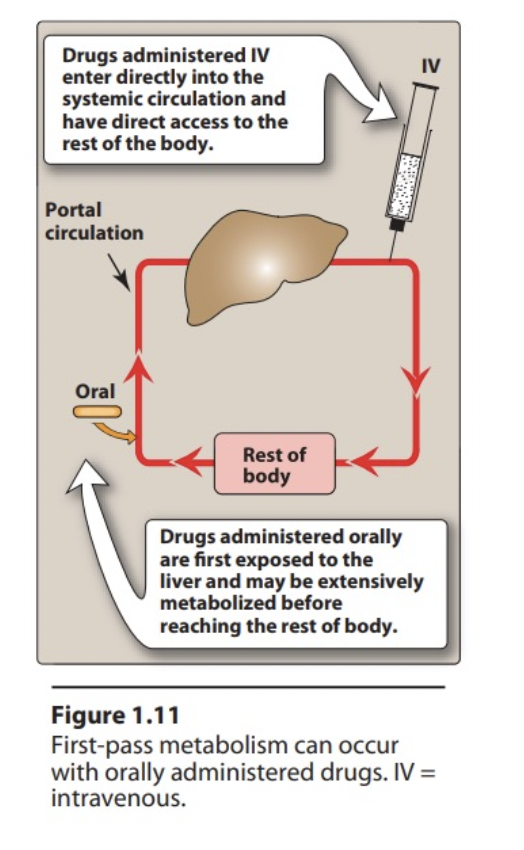
1st pass metabolism vs metabolism
First pass metabolism
Metabolism of drug before reach systemic circulation
Inactivates drug
Decreases bioavailability
Occurs in Liver, intestine, lung, and skin
Metabolism
Drug metabolism after reach systemic circulation
May activate drug
Terminates action of most drugs
Occurs in Liver (microsomal enzymes) & non microsomal (liver, GIT and plasma)
Define therapeutic blood range and define toxic blood range
Therapeutic blood range: conc of drug which majority of treated population can receive therapeutic benefits
Toxic blood range: conc majority of treated population can have toxic manifestations
Factors affecting biotransformation related to the patient
Genetics (**)
Diet (starvation deplete glycine conjugation)
Age (liver cant detoxify chloramphenicol in neonates = Gray baby syndrome)
Gender ( young males > females sedation from barbiturate)
Disease ( liver disease ↓ its ability to metabolize drugs)
** Acetylation of isoniazid (rapid or slow) , succinylcholine hydrolysis by pseudocholinesterase enzyme
Factors affecting biotransformation related to the drug
Chem properties: (drugs may stimulate/inhibit other drug metabolism like enzyme inducers or inhibitors)
Route of administration: oral can result in extensive hepatic metabolism of drugs (high 1st pass metabolism eg:propranolol or morphine)
Dosage : Toxic doses can deplete enzymes needed for detox reactions
A 30-year-old female patient is taking ketoconazole for a severe fungal infection. She undergoes routine blood tests and her physician notices elevated liver enzymes. Considering the pharmacological profile of ketoconazole, what should the physician be most concerned about regarding this patient's condition?
(Possible options at the back if want MCQ style)
A) Potential renal toxicity due to prolonged use of ketoconazole.
B) Risk of drug-induced liver injury due to inhibition of CYP450 enzymes.
C) Enhanced activity of anticoagulants due to enzyme inhibition.
D) Accumulation of ketoconazole due to its renal clearance.
Ans = Risk of drug-induced liver injury due to inhibition of CYP450 enzymes. (B)
A 24-year-old woman with a history of epilepsy is being treated with phenobarbital, which she has been taking for the past year. She presents to the clinic for a routine check-up. The physician considers adding a new medication for her anxiety and prescribes diazepam. What effect does phenobarbital have on the metabolism of diazepam, and what should the physician monitor for as a potential consequence of this interaction?
(Possible options at the back if want MCQ style)
A) Increased metabolism of diazepam leading to decreased therapeutic effects
B) Decreased metabolism of diazepam leading to increased therapeutic effects
C) No effect on the metabolism of diazepam
D) Phenobarbital will cause diazepam to be entirely ineffective
Ans = Increased metabolism of diazepam leading to decreased therapeutic effects (A)
A 45-year-old male with a history of chronic fungal infections is prescribed ketoconazole to treat his condition. Alongside ketoconazole, he is also on a regimen of atorvastatin for hyperlipidemia. Which mechanism explains the potential drug-drug interaction between ketoconazole and atorvastatin?
(Possible options at the back if want MCQ style)
A) Ketoconazole acts as a CYP450 inducer, increasing atorvastatin metabolism.
B) Ketoconazole inhibits CYP450 enzymes, decreasing atorvastatin metabolism.
C) Ketoconazole enhances the renal excretion of atorvastatin.
D) Ketoconazole increases the absorption of atorvastatin in the gastrointestinal tract.
Ans = Ketoconazole inhibits CYP450 enzymes, decreasing atorvastatin metabolism (B)