Pharm Unit 1 Exam
1/304
Earn XP
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
305 Terms
what’s the scientific definition of a drug?
mc’s that BIOLOGICALLY + PHYSIOLOGICALLY change an org
what’s the GOVERNMENT definition of a drug?
something (NOT food) that treats / prevents DISEASE
changes STRUCTURE/FUNCTION of animal

what’s the dif btw pharmacoDYNAMICS + KINETICS
dyn: drug ON body
kin: body ON drug (ie metabolism)

what’s the dif btw a physiologic + generalized receptor?
physio: NORMAL body substance acts on NORMAL receptor normally (ie epinephrine on beta 2)
general: drug ACTS on body receptor to alter function

what types of mc’s can be drugs?
any chemical that causes physiologic affect

what is lipinski’s rule of 5?
< 5 H bond donors
>10 H bond acceptors
< 500g/ mol
partition <5

goal of drug abs due to lipinski’s rule?
sticky to bind to intended receptor but NOT others
small enough to be abs acr gut

what are up-and-coming drugs?
peptides / proteins / antibodies
but they’re so big they have to be administered via INJECTION

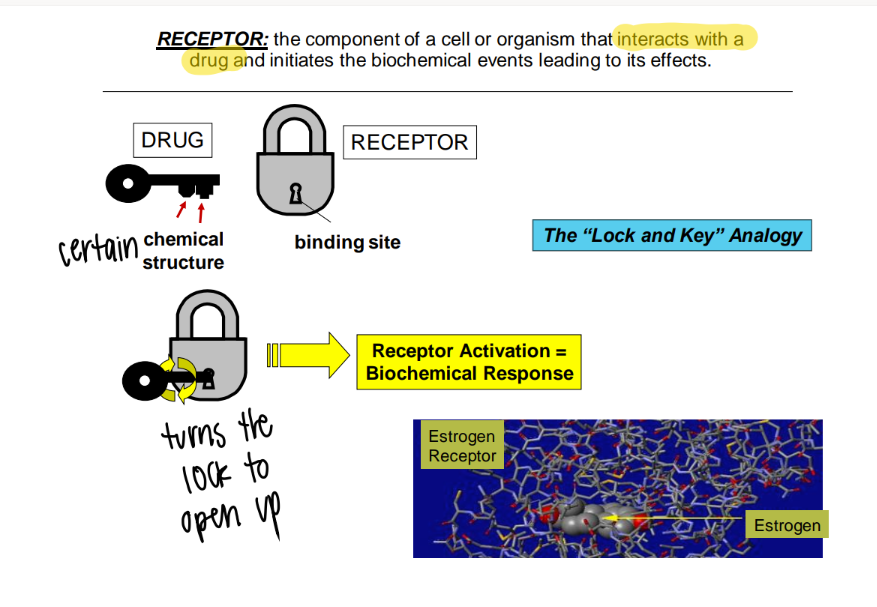
lock and key analogy
BIND: structure
ACTIVATE: pathway
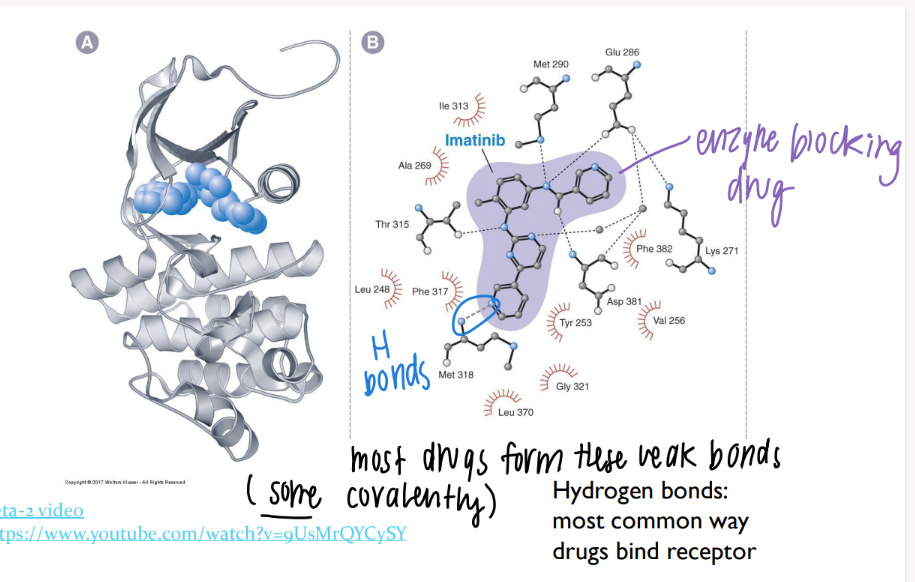
what types of bonds do drugs form?
most are WEAK hydrogen bonds
(some STRONG covalent bonds)
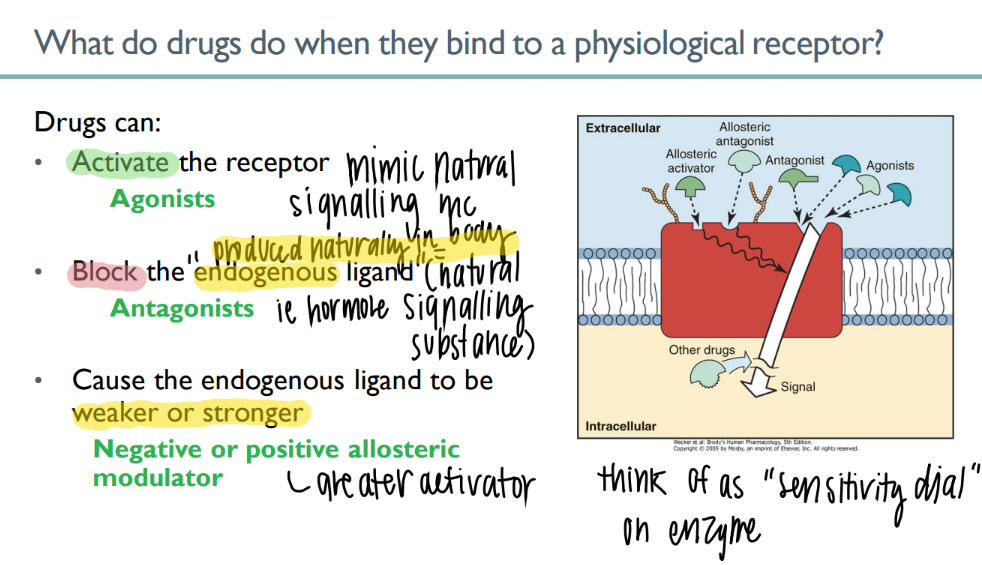
what’s an antagonist?
BLOCKS naturally-occuring body-made ENDOGENOUS ligand
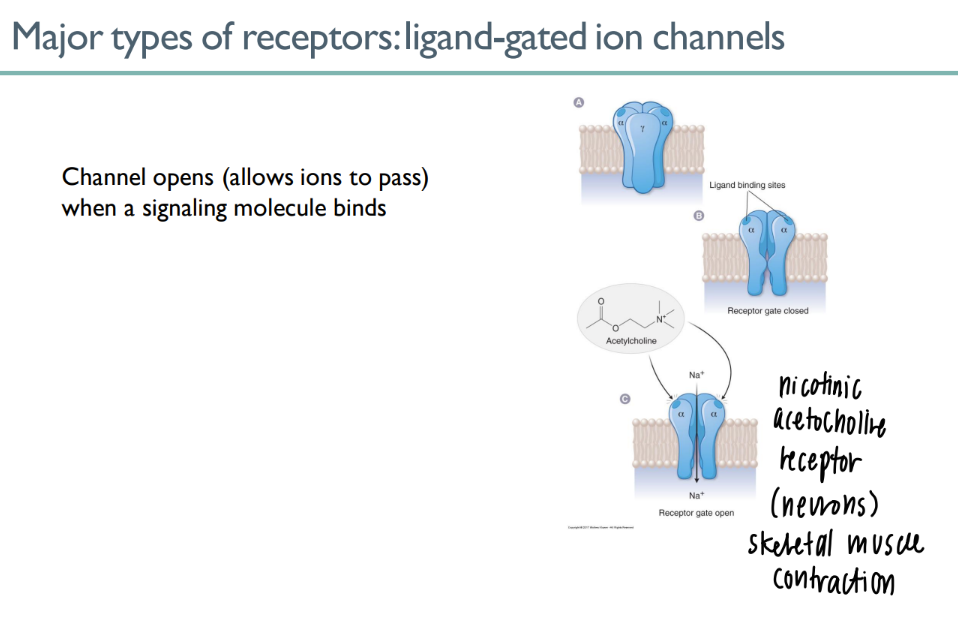
how do ligand gated ion channels work
ligand binds to site on receptor → channel opens!
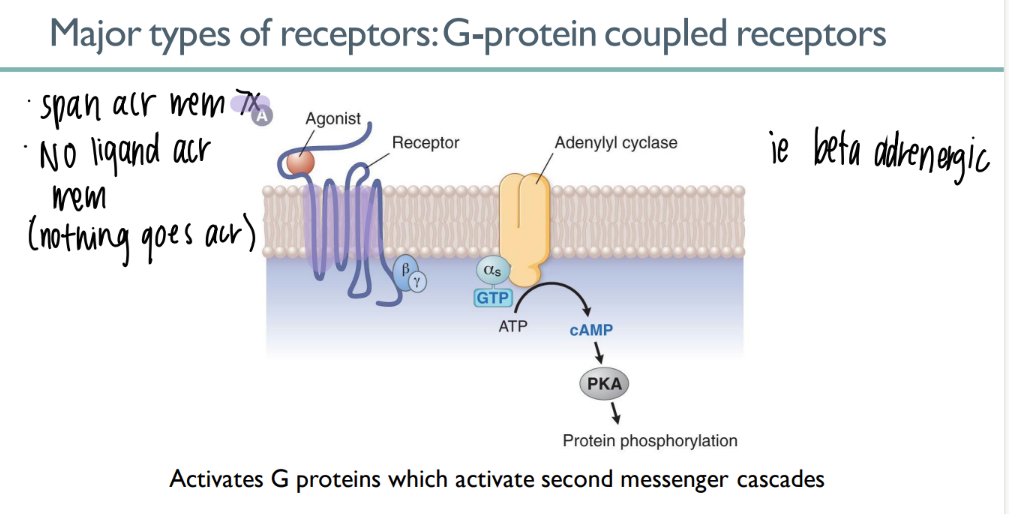
how tf does a GPCR work?
ligand binds to 7-spanning GPCR → signal CASCADE
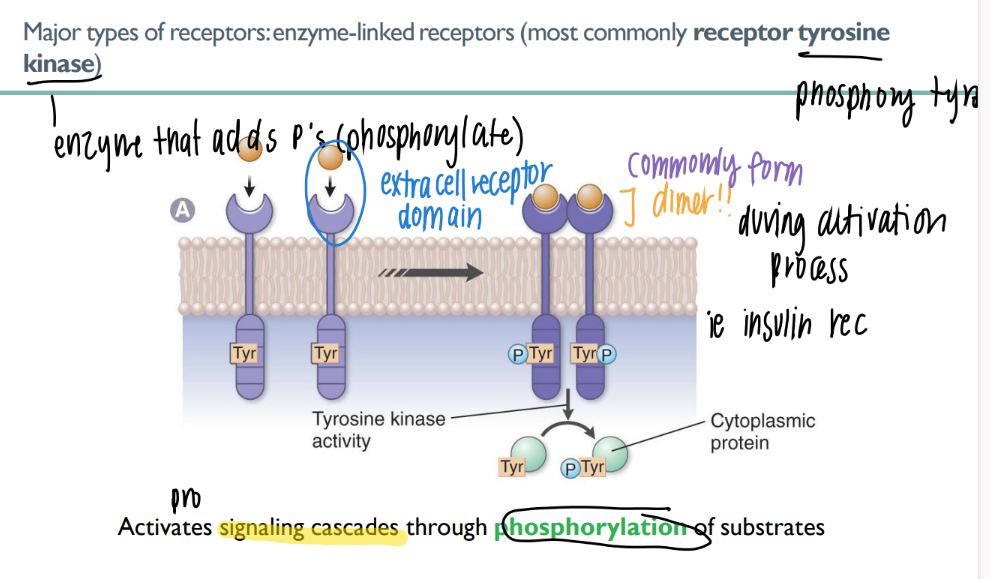
how does an enzyme-linked receptor work?
what’s an example of one?
ligand binds extracellular
receptor phosphorylates (adds P) to smth
cascade!
ex: RTK (receptor tyrosine kinase)
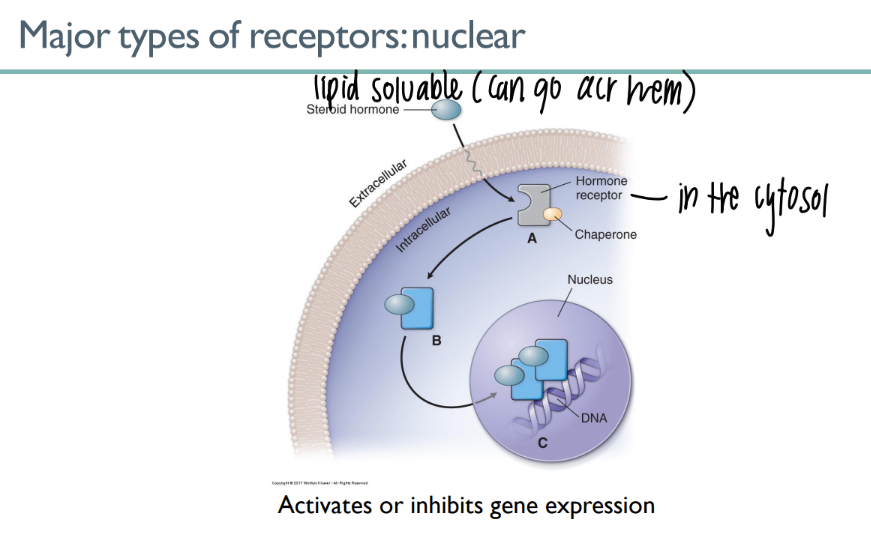
how do nuclear receptors work?
lipid-soluable ligand in cytosol
CROSSES the membrane n goes straight to the nucleus
gene expression act/inhib
what do GPCR’s, RTK’s, and LGIC’s have in common?
ON cell mem
cascades + cell mem potential changes
INDIRECTLY act/inhib gene exp

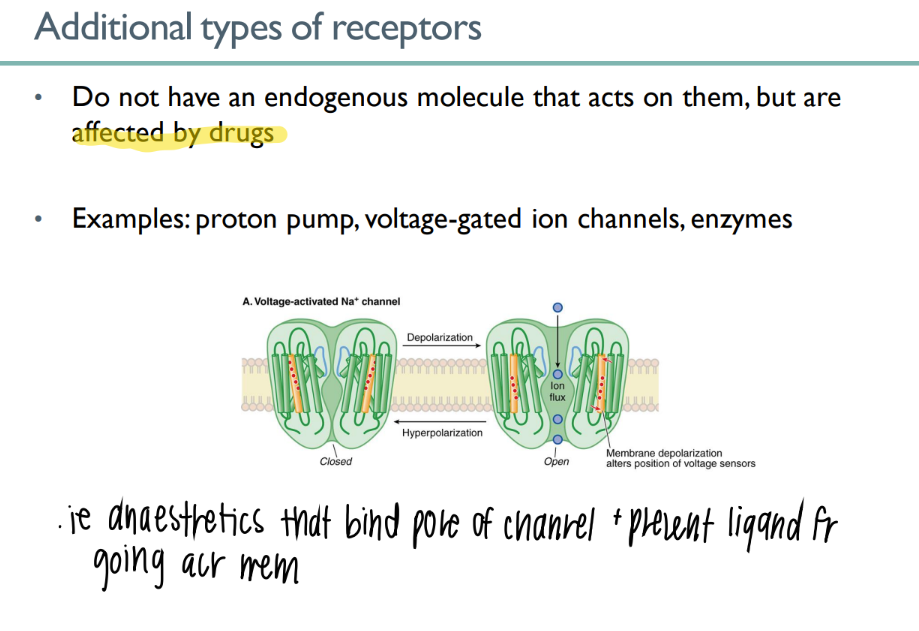
some receptors are not ACTIVATED via endogenous mc’s. how r they activated? what’s an example of this?
act by DRUGS (ie Na we eat)
ex: voltage-activated Na channel
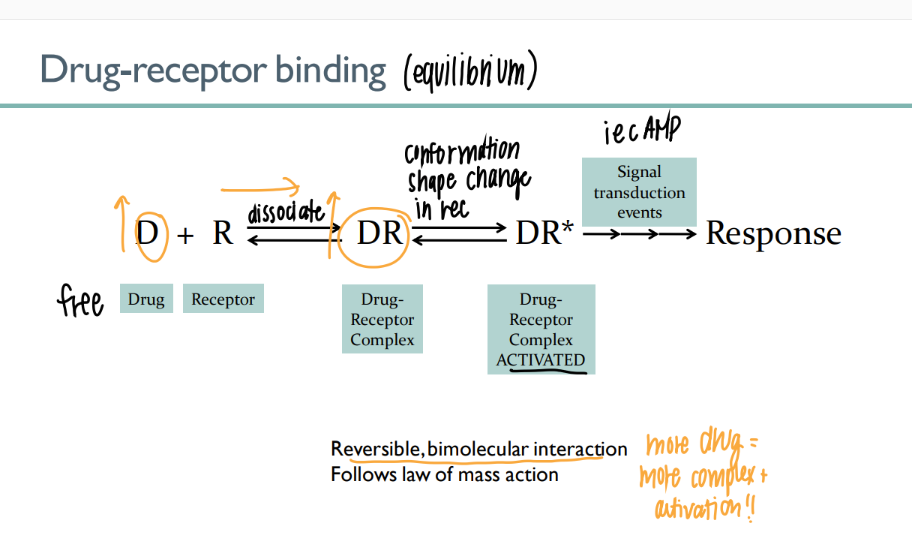
the drug-receptor complex is what type of interaction? how tf is this relevant?
REVERSIBLE, BI-molecular interaction → more drug = more activation
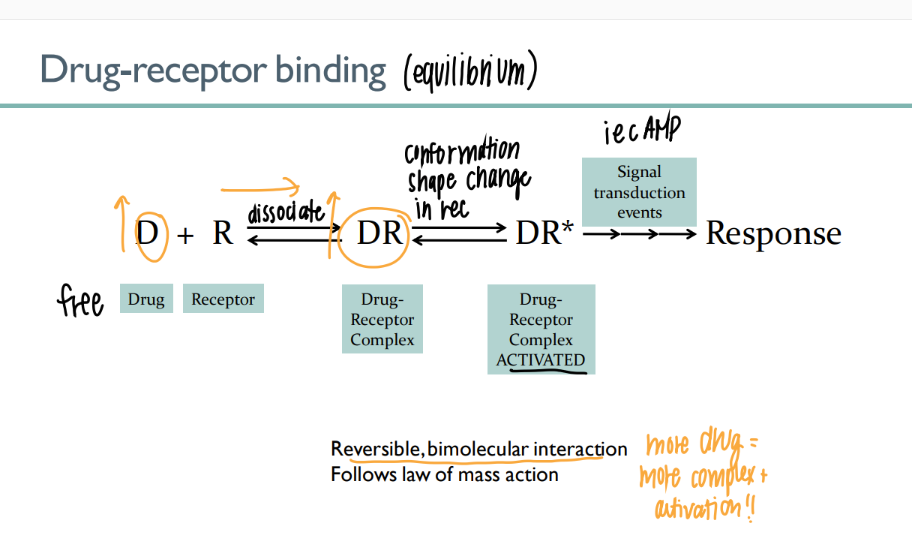
what are the 4 parts of the drug-receptor equilibrium?
drug receptor (free)
drug-receptor complex (bind)
drug-receptor ACTIVATED
signal transduction → response
drug-receptor selectivity is messed up in what 2 ways?
binds to the RIGHT receptor but on dif organs
binds to the wrong receptor. (too much drug conc)
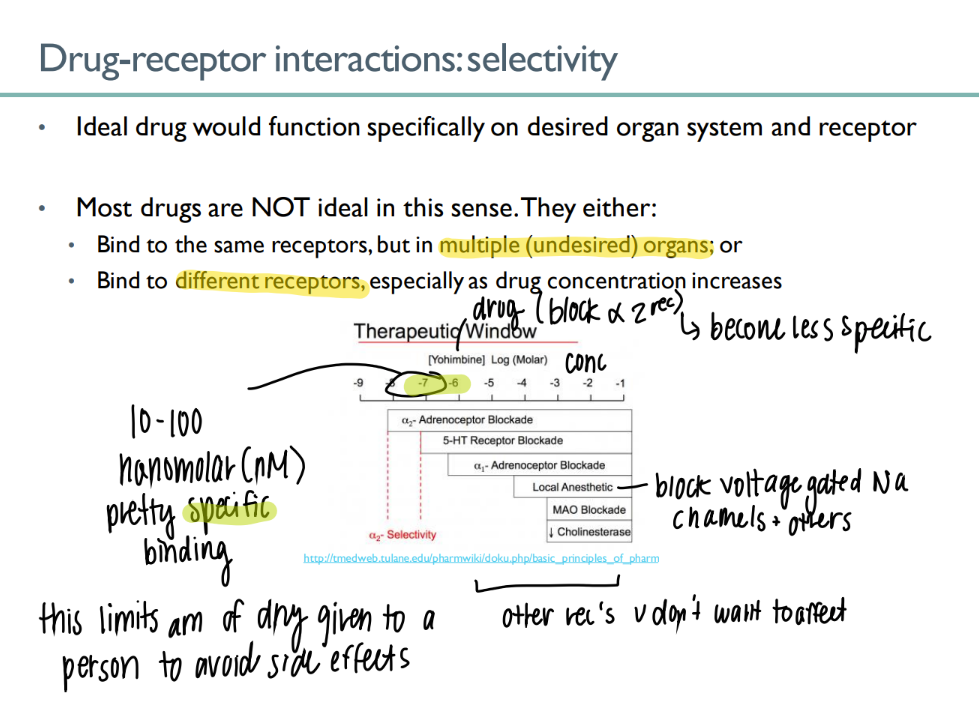
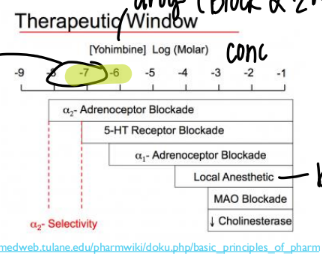
explain this window
certain RANGE of drug conc to get EXACT effect
(in this case, the small highlighted region)
else get SIDE effects
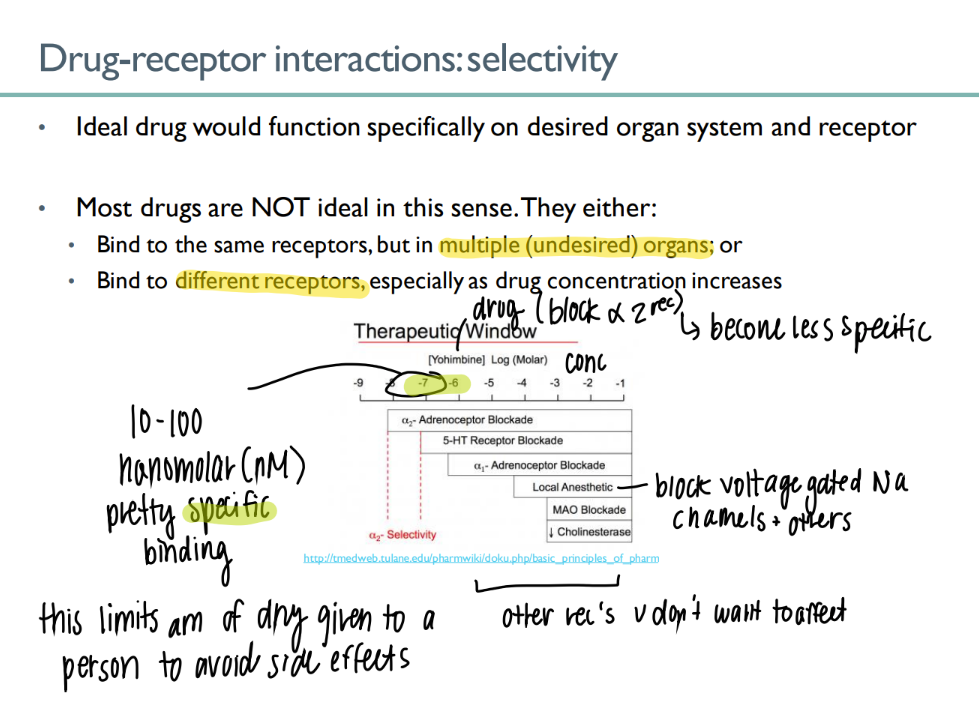
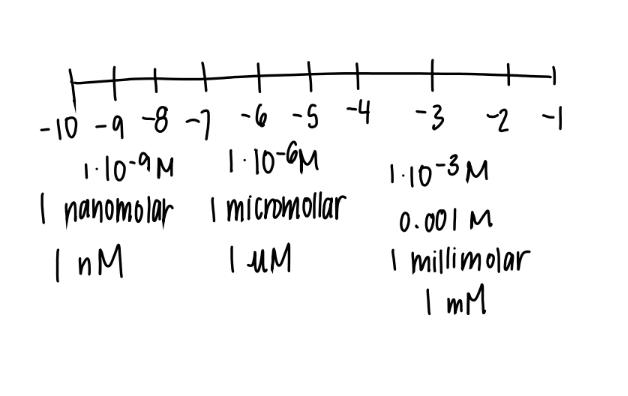
what’s an nM?
1 × 10^-9
nano-molar
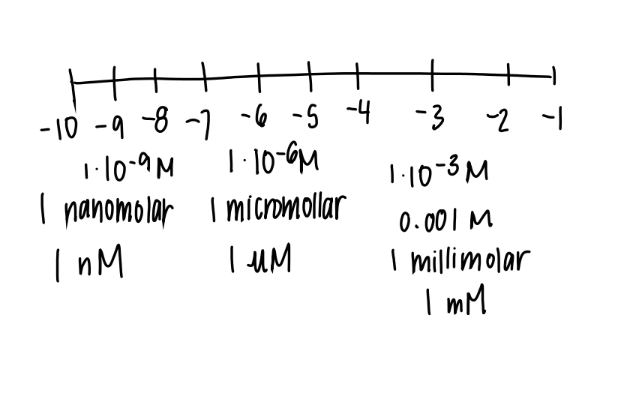
what’s a uM
1 × 10^ -6
micro-molar
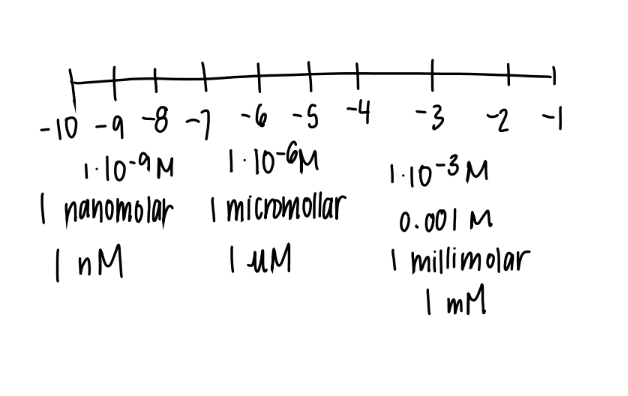
what’s a mM
1 × 10 ^ -3
milli-molar
wtf is the “kd”?
dissociation constant - drug conc where half the receptors are bound

low KD = ?
HIGH drug affinity
(don’t need much drug conc to bind to all those receptors)

wtf is drug “affinity”?
STRENGTH of bond btw drug and receptor.
what does a ligand-receptor binding curve look like?
y axis = LR/ R0
(bound ligand-receptors / total receptors)
x axis = drug conc
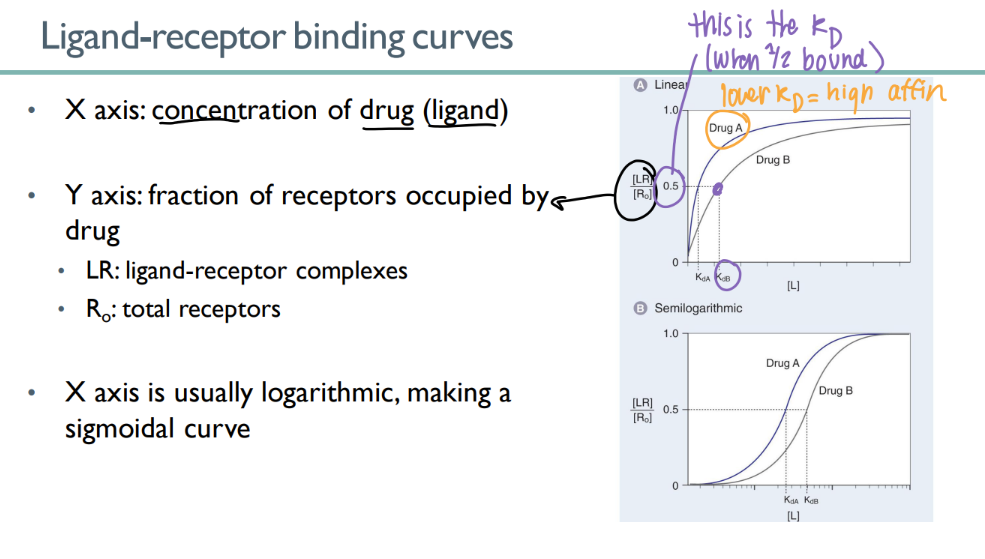
what does a ligand-receptor binding curve tell you?
the KD!
see where 50% of the receptors are bound.
lower KD = higher affinity
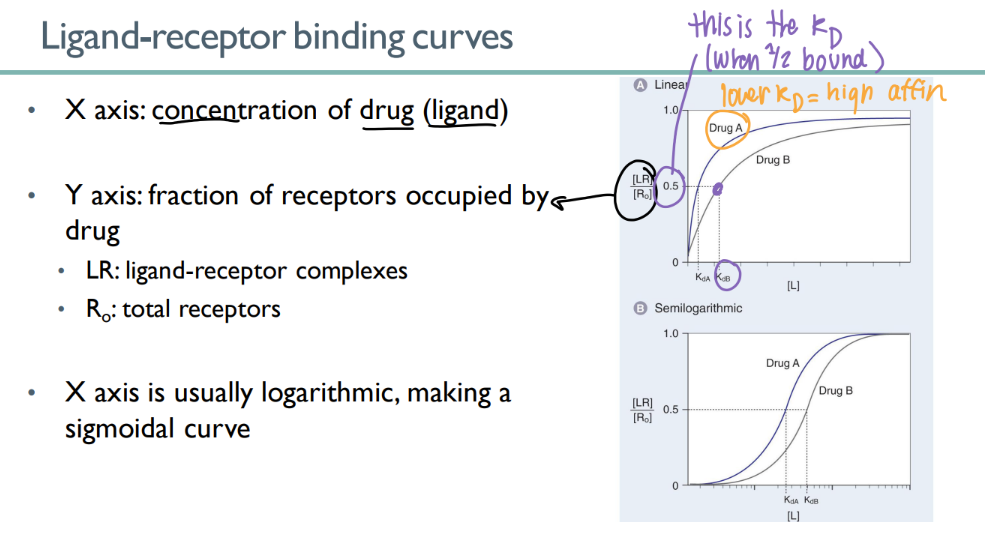
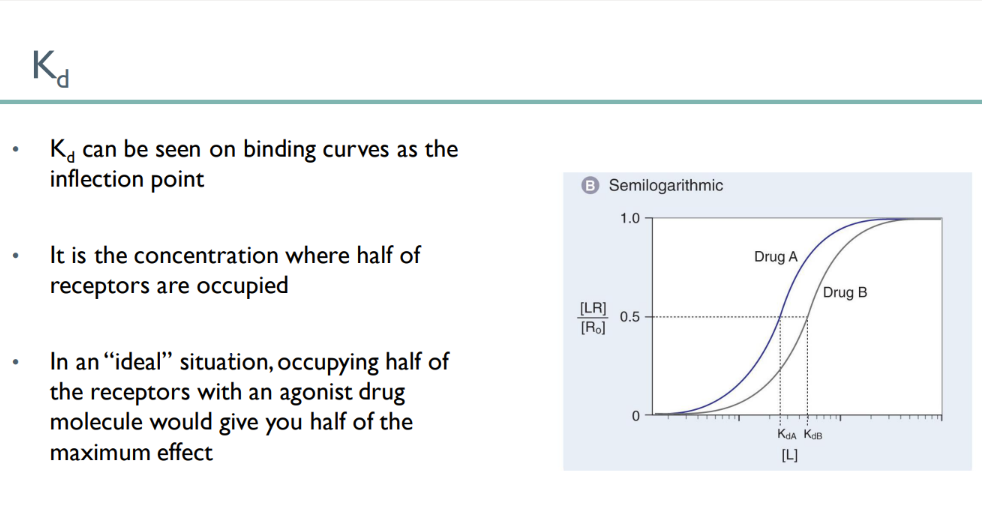
what does the KD look like on a receptor-binding curve?
in an ideal situation, what does meeting the KD mean?
the INFLECTION point
KD = 50% are bound, so 50% of maximum effect!
wtf is “intrinsic activity” ?
receptor STAYS active (not just TURNED active for a millisecond)

what’s a “dose response” relationship?
see the body’s response to a particular DOSE of a drug, rather than constantly taking blood samples to find the CONCENTRATION of the drug in the body.

wtf is the EC/ ED 50?
dose to get 50% of the max effect
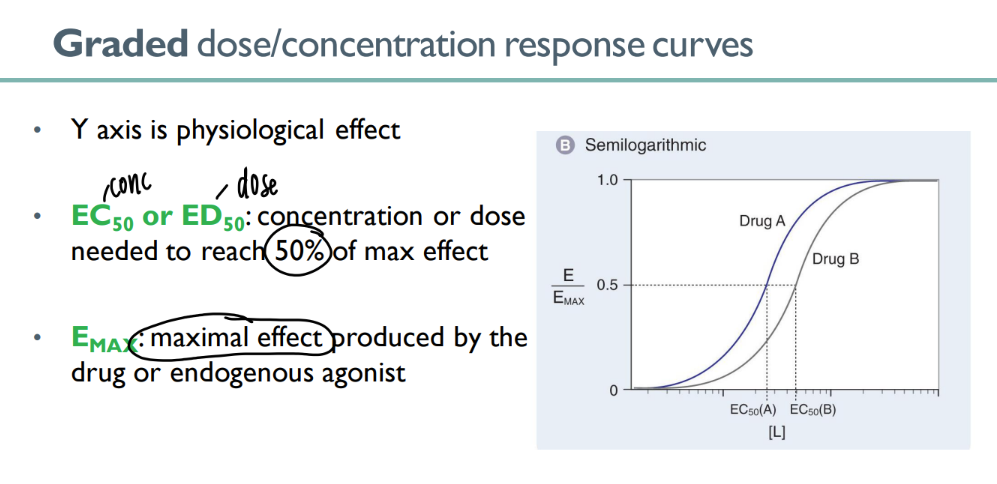
what’s the emax?
the MAXIMUM effect that can be caused by a certain drug (or endogenous agonist)
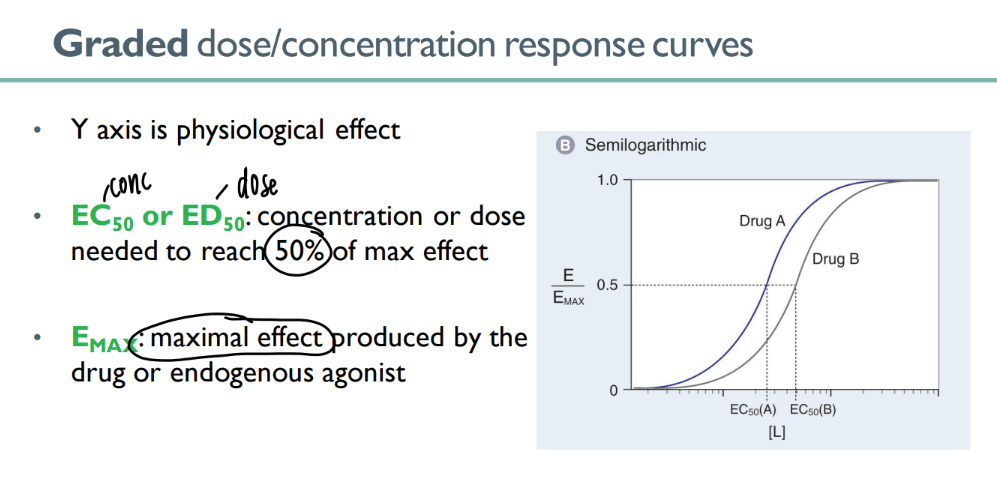
wtf does it mean for a drug to be more “potent”?
SMALLER dose to get to EC50 (certain level of effect)
how do spare receptors affect ec50 and kd?
ec50 (dose to get 50% maximum effect) < dose to get 50% of receptors activated
(obvi u need less drugs)

what is the concept of “spare receptors”?
why does it happen?
there are MORE receptors present than actually NEEDED to be occupied to elicit a certain response.
signal amplification via 2nd messengers!

what are the TD50 and LD50?
dose that would cause a toxic or lethal dose in 50% of people
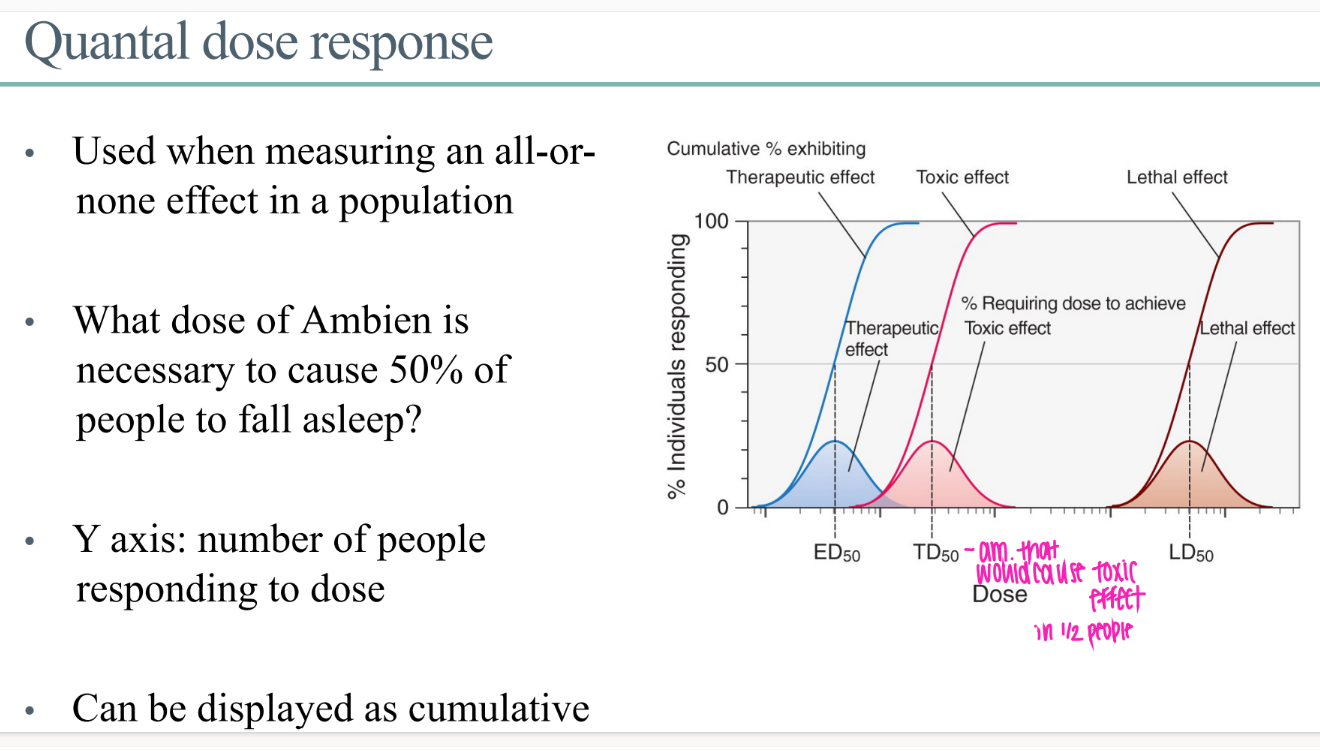
what does the quantal dose response GRAPH look like?
y axis = # ppl responding to dose
x axis = dose of drug
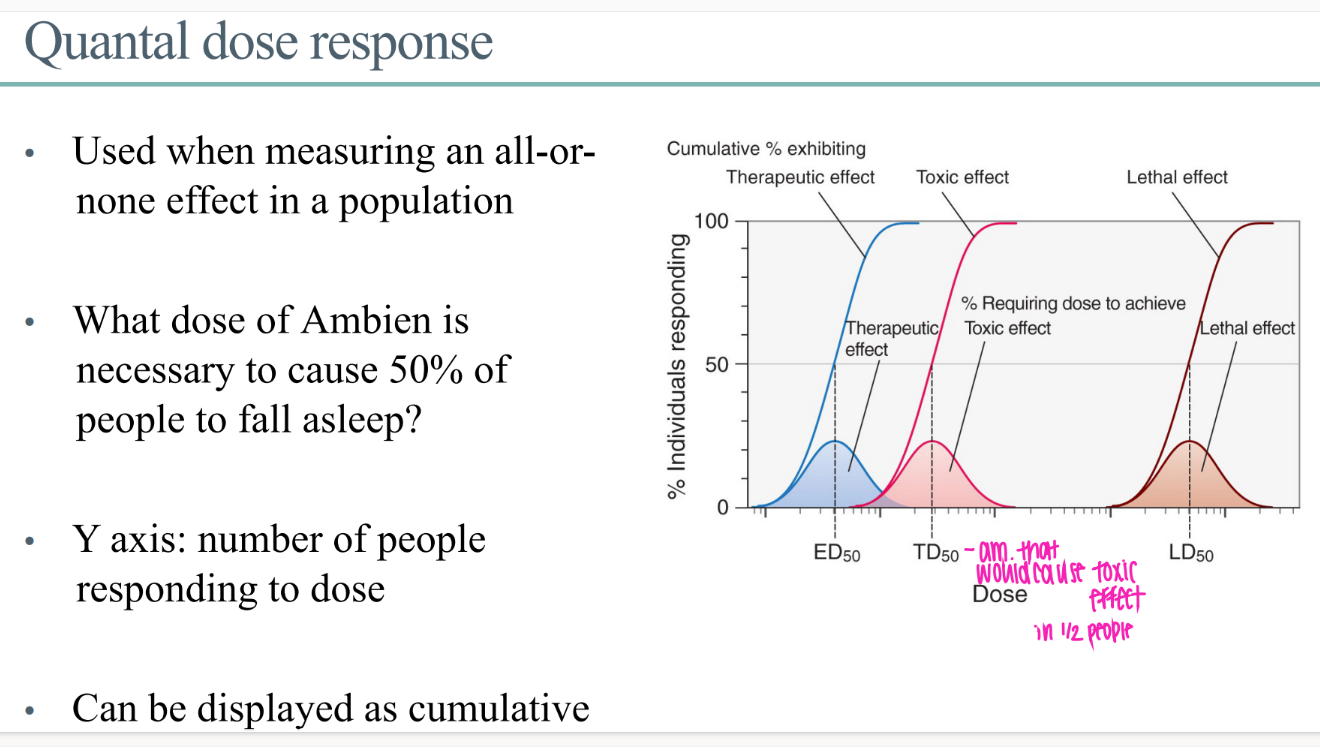
what IS a quantal dose effect?
for a drug effect that is ALL or NOTHING.
ie puts people to sleep or not.
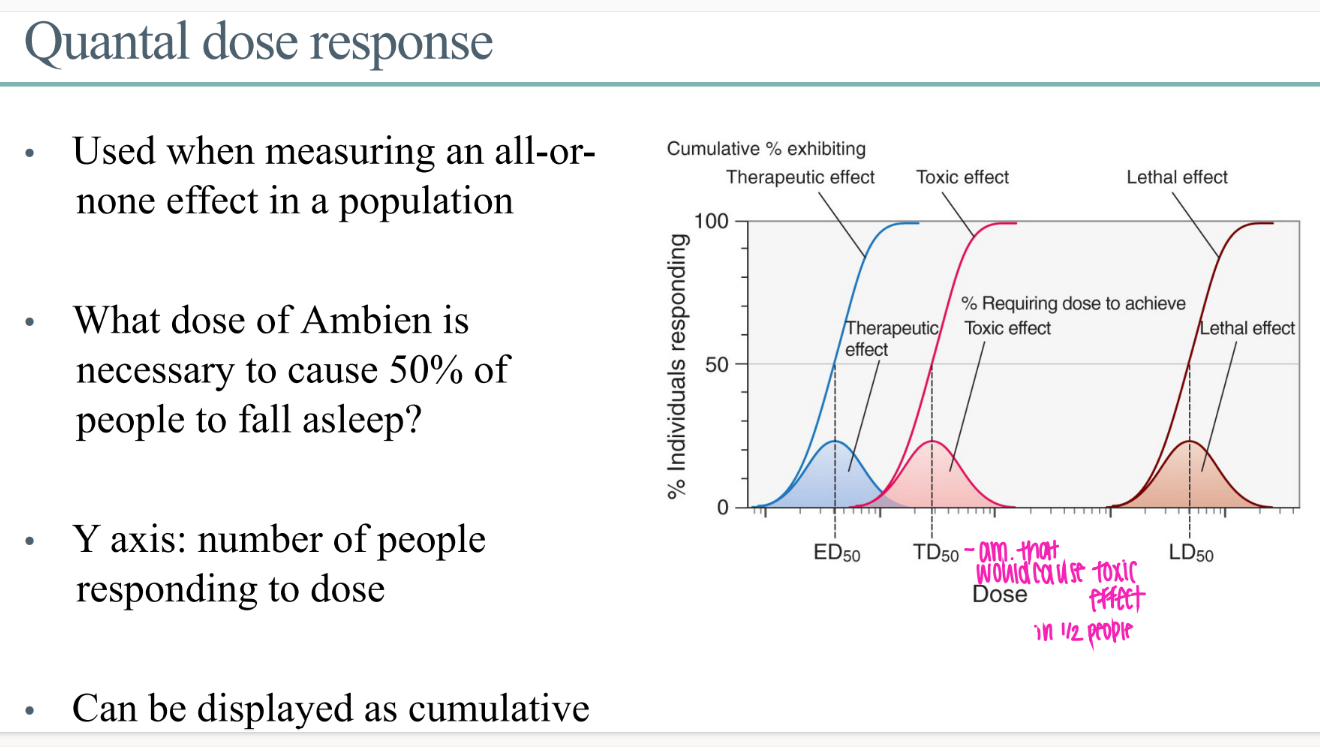
WTF is the ED50 in the context of quantal dose?
dose of drug for 50% of people to get whatever the beneficial drug effect is
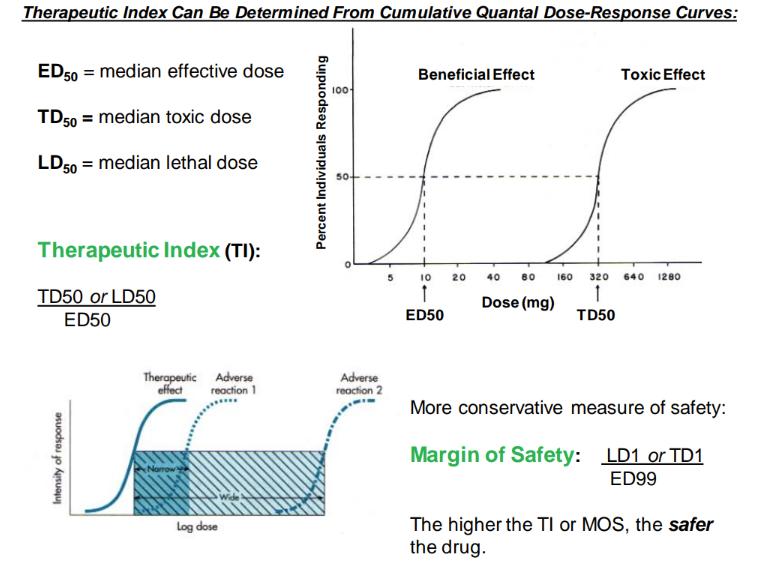
mathematically, what is the therapeutic index?
logically, what is it?
LD or TD / ED50.
aka, find out how many pills u need to take to die / experience toxicity.
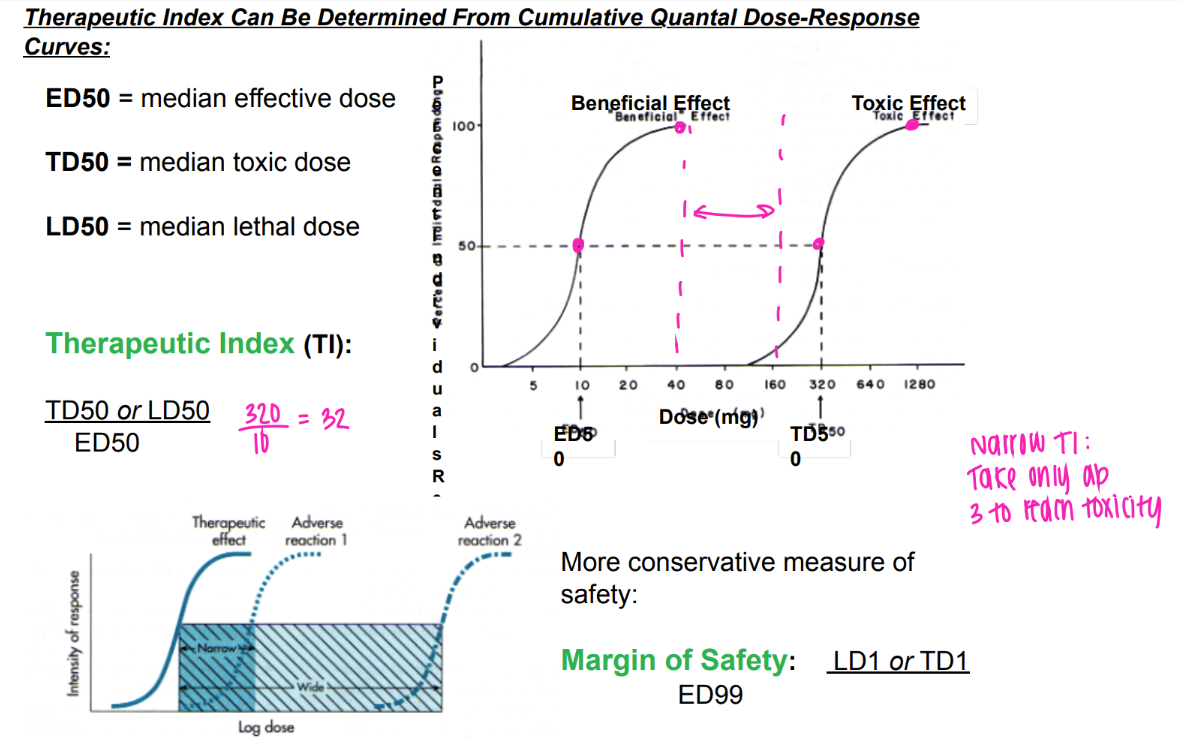
what’s the MOS?
mathematically?
logically?
margin of safety
LD1 or TD1 / ED99
(aka, perfect pill size where 99% of people have beneficial effect)
(being extra careful- 1% are experiencing toxic/ lethal effect)
aka the am of pills needed to intoxicate/ kill just 1% of people
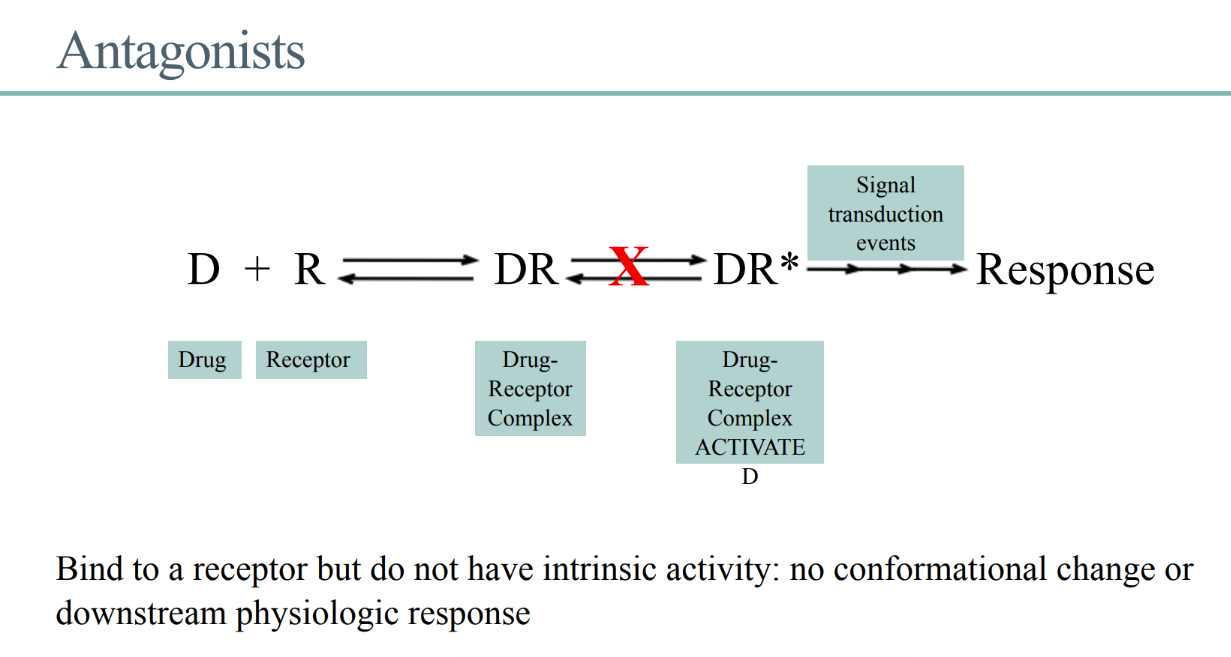
what’s the definition of an antagonist?
BINDS to receptor
NO intrinsic activity (turning of the key)
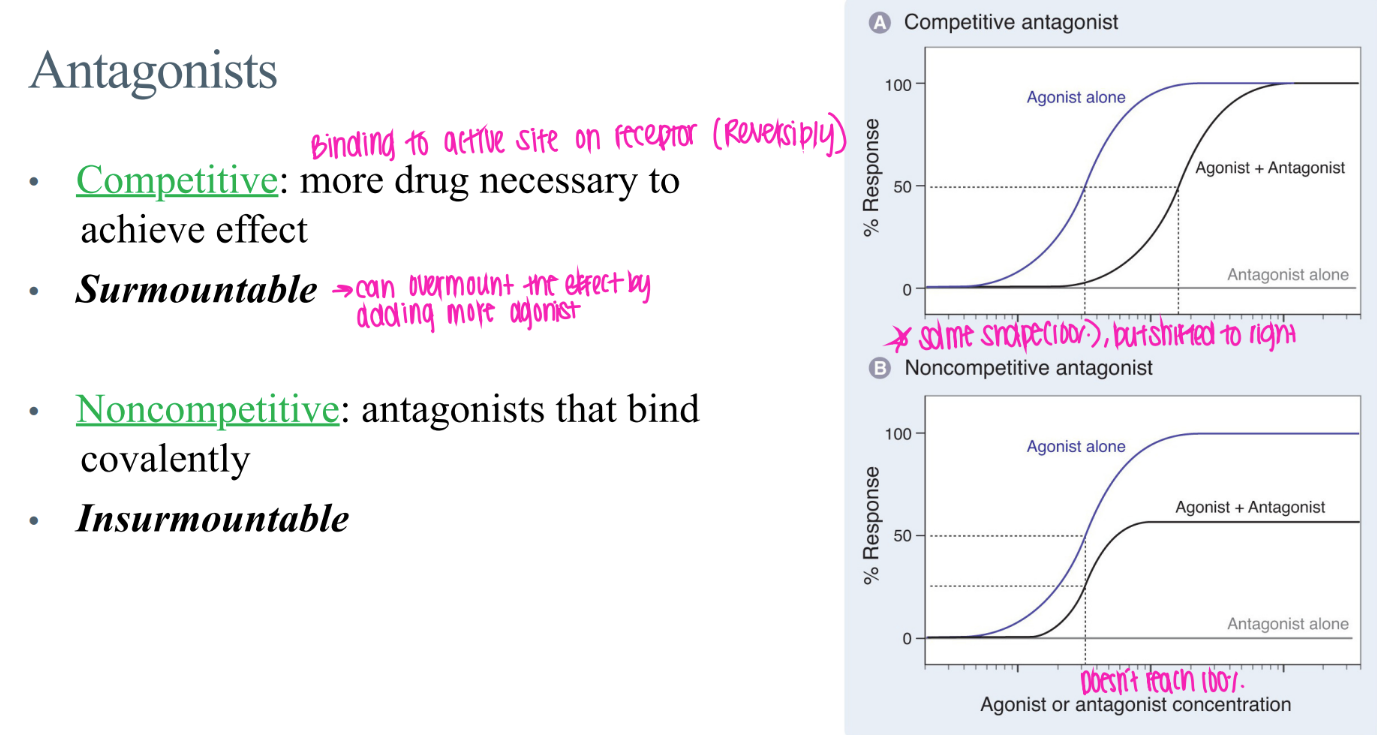
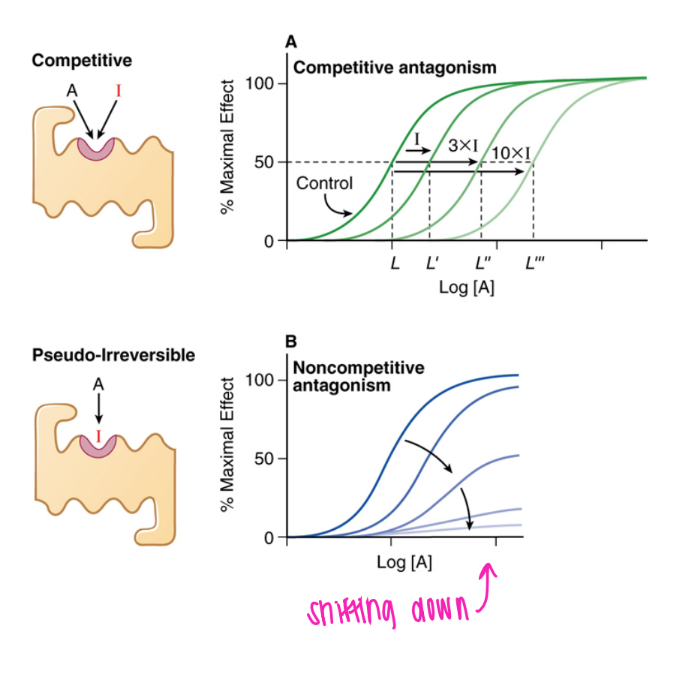
what’s the dif btw COMPETITIVE + non-competitive antagonists?
comp: reverisbly bound to active site → surmountable (just incr drug conc!)
non-comp: COVALENTly bound → insurmountable
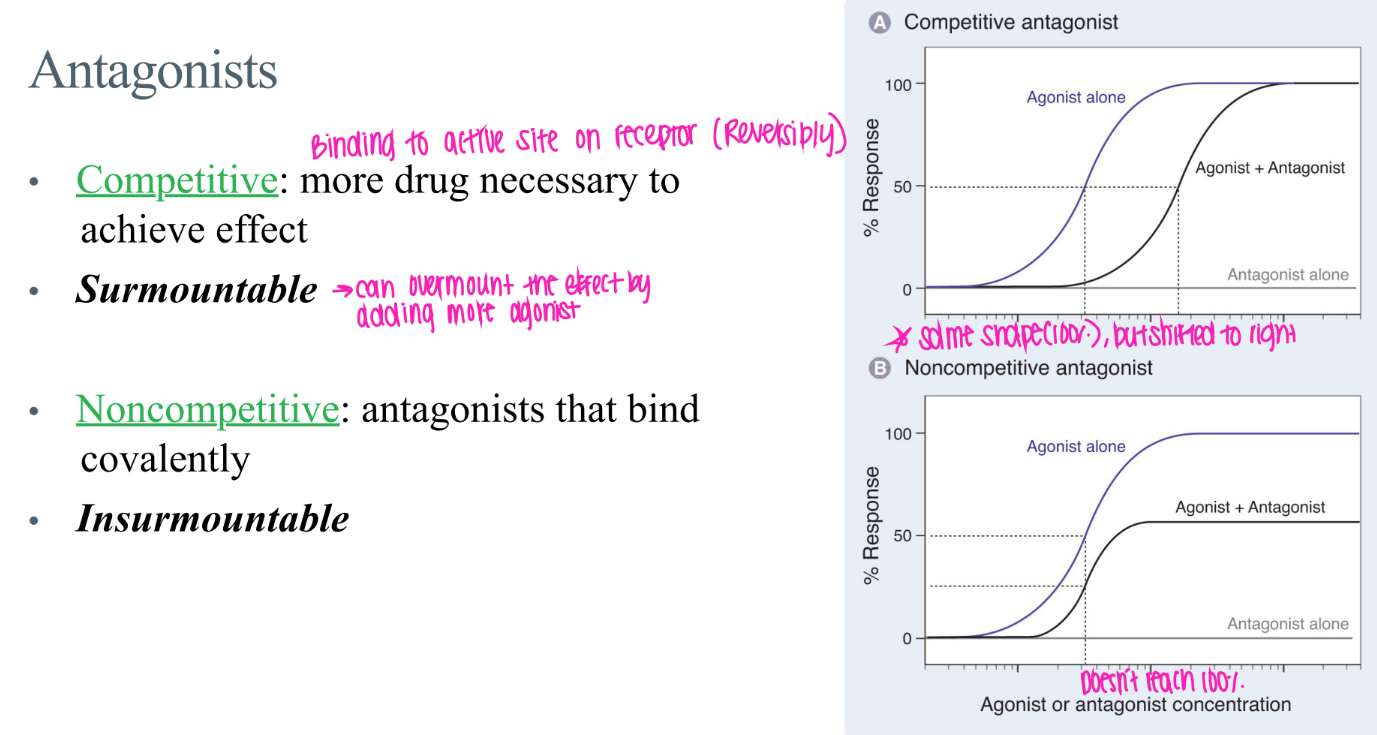
what’s the GRAPHICAL dif btw COMPETITIVE + non-competitive antagonists?
comp: curve shifted right (just need more drug)
non-comp: curve messed up, NEVER reach 100% activation
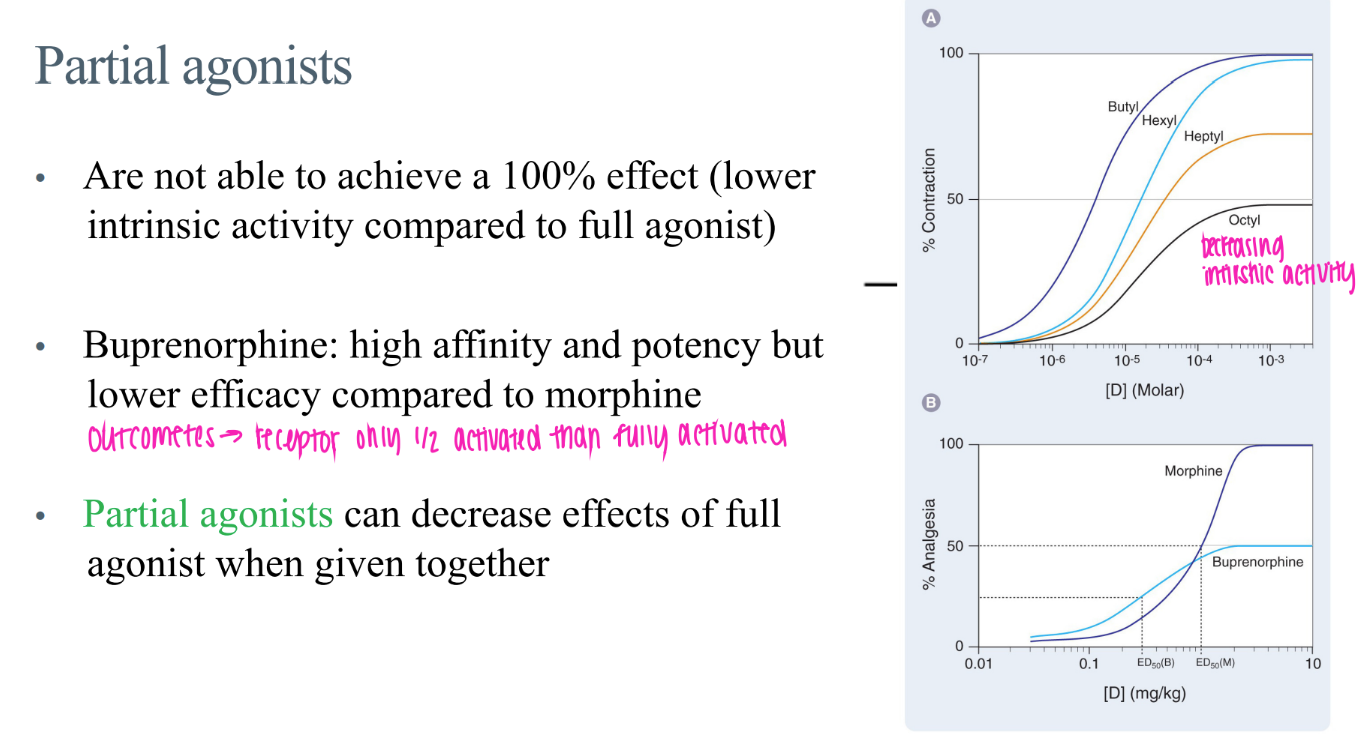
wtf are “partial agonists”?
bind BUT doesn’t activate receptor 100% / less intrinsic activity
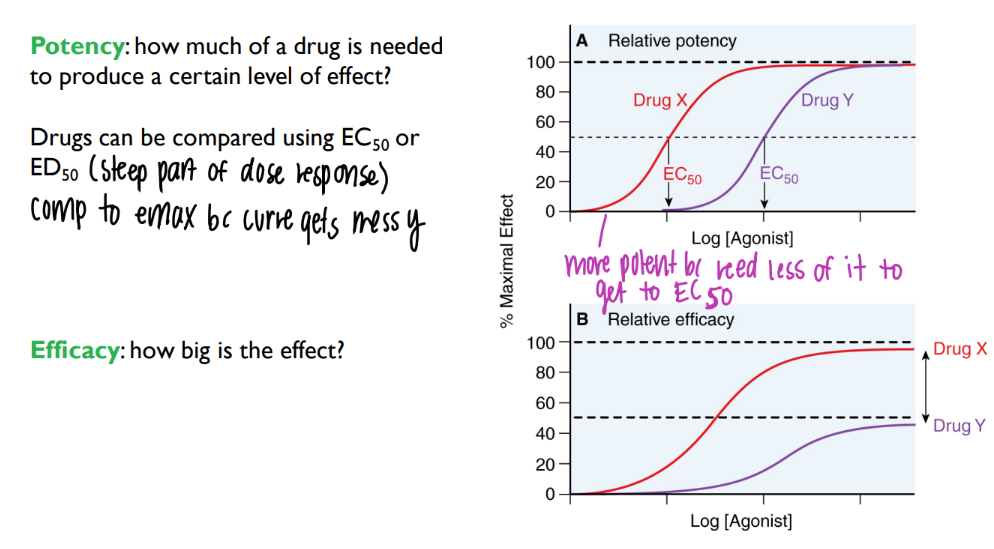
wtf is the difference btw drug affinity, potency, and effectiveness?
affin: strength of bond btw ligand + receptor
potency: am of intrinsic activity / activation of receptor
effectiveness: DOES the activated receptor ACTUALLY do smth to ur body

what’s the dif btw potency + efficacy?
potency: how much DRUG needed to get certain LEVEL of effect (ie 50%)
efficacy: how BIG is the effect
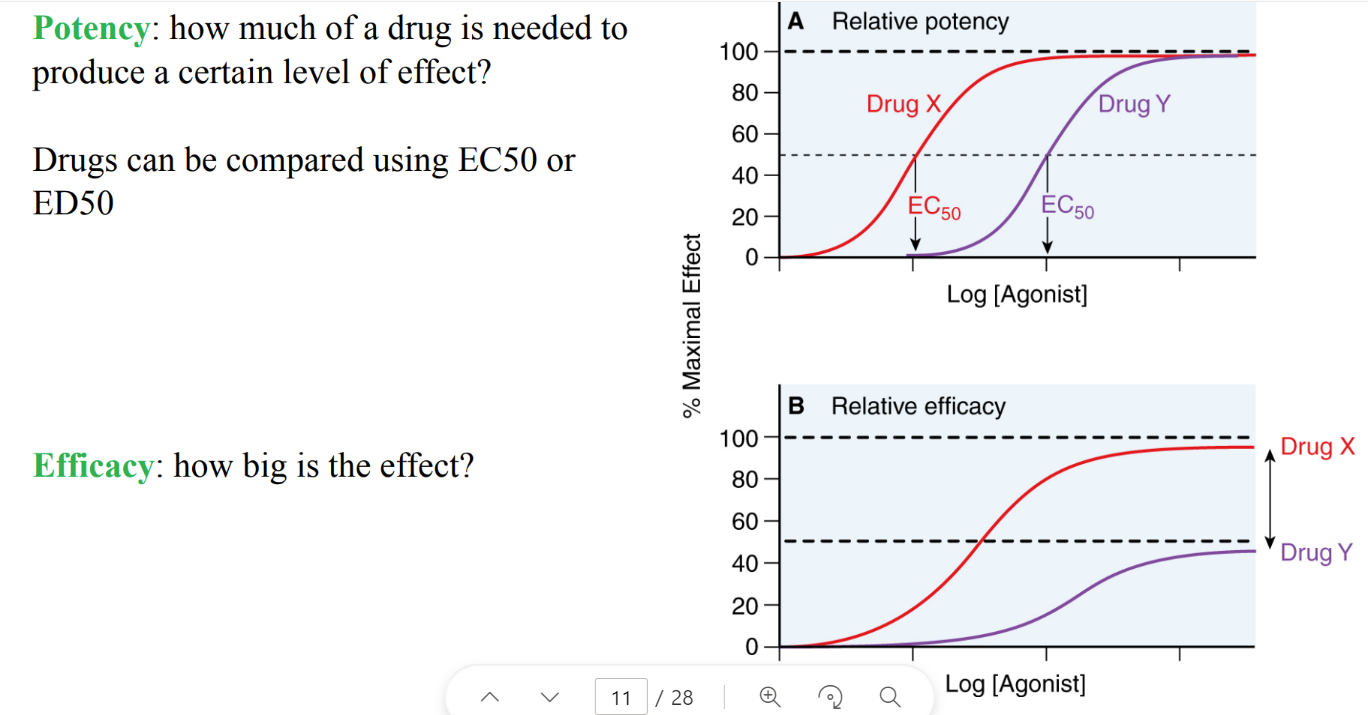
what are inverse agonists?
(versus ANtagonists?)
promote INACTIVE conformation → OPPOSITE effects of agonist.
antagonists: block active site so agonists can’t bind. → NO effects.
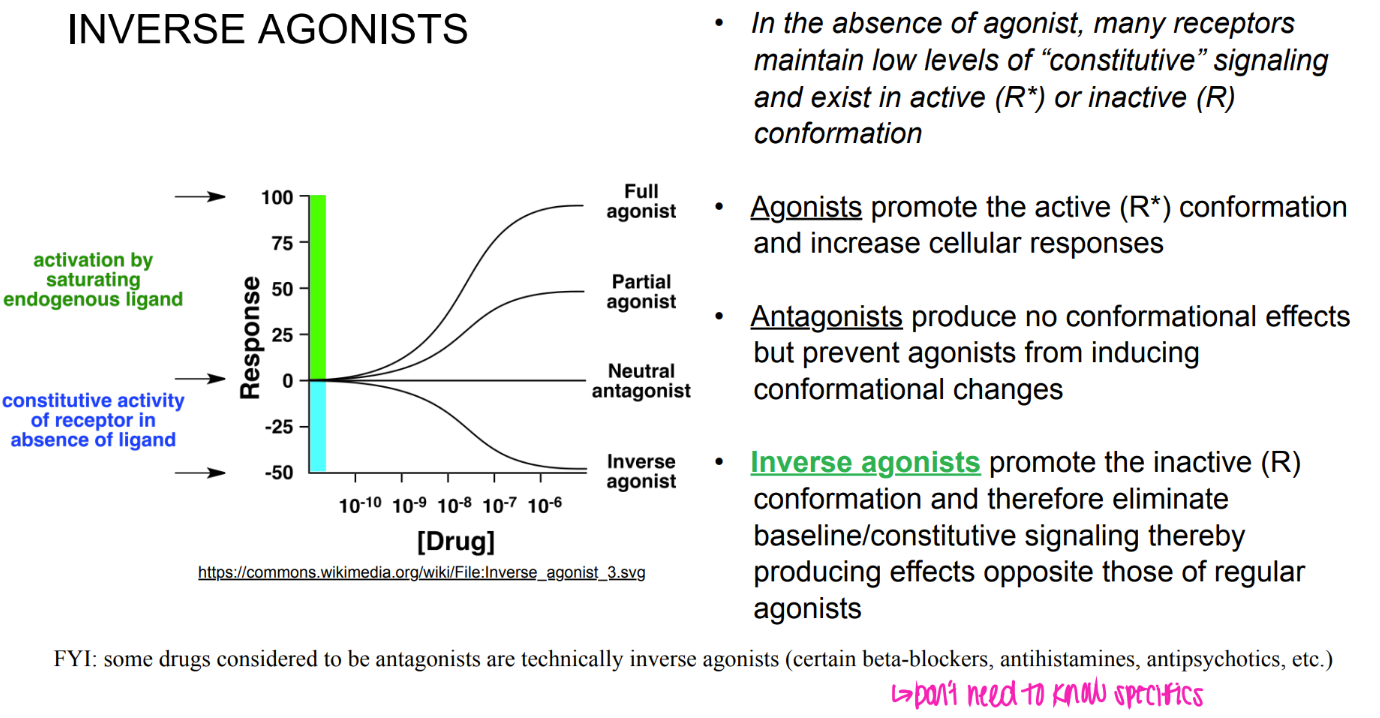
what’s the dif btw orthosteric + allosteric drugs?
orthosteric = binds to PRIMARY agonist site
allosteric= binds to secondary site, can incr OR decr agonist binding
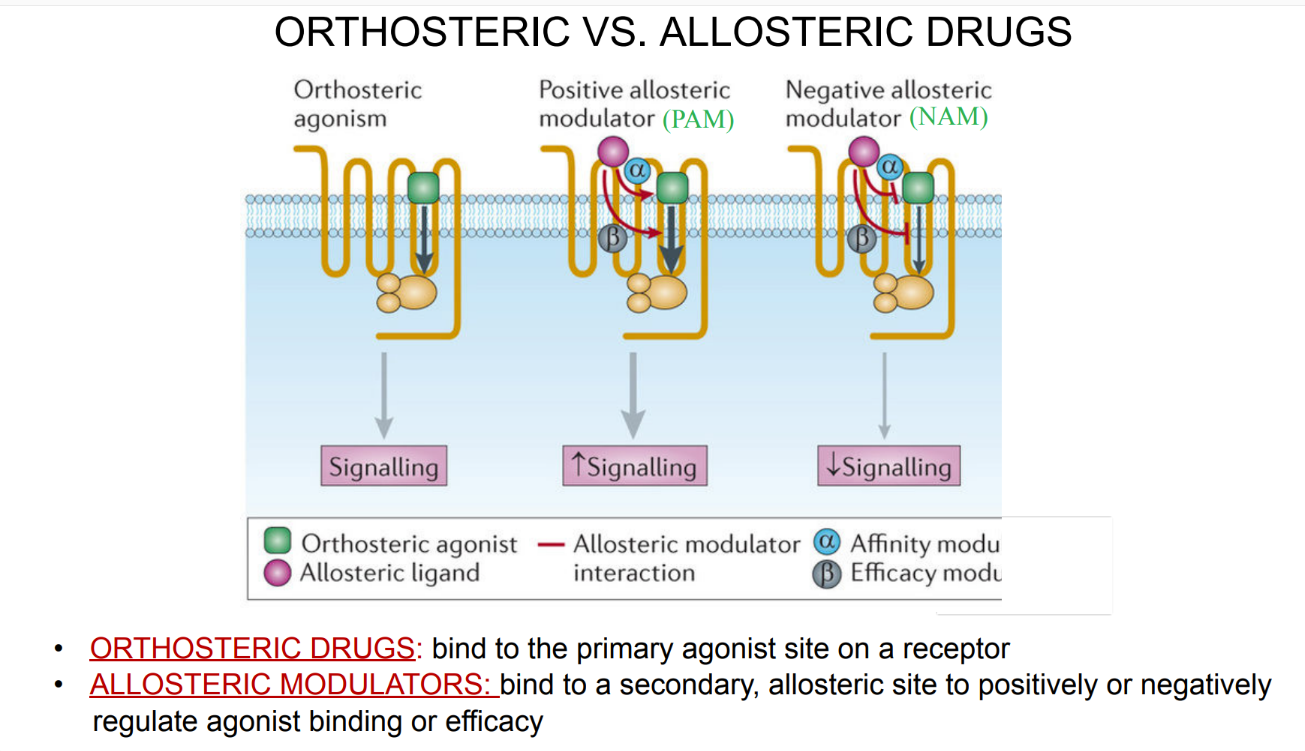
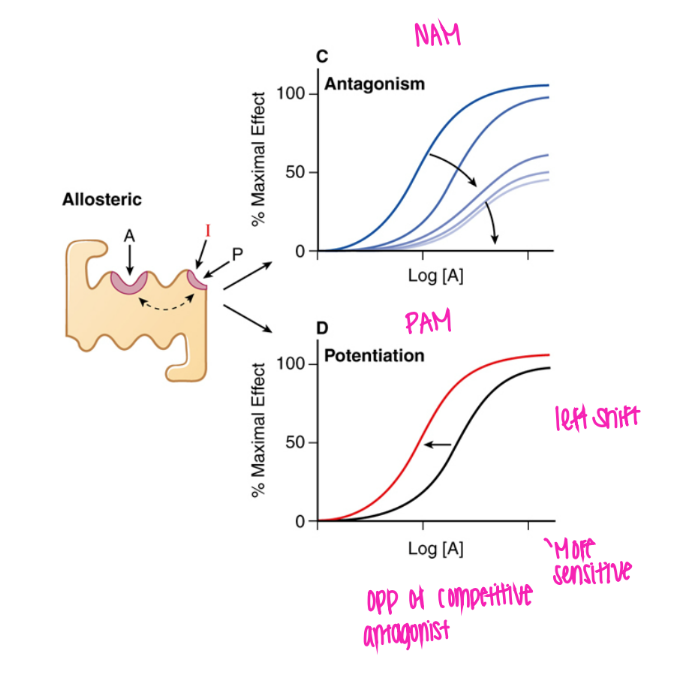
What does the NAM curve look like? why?
shifted right and DOWN.
bc decr maximum effect bc prevents agonist from binding
(negative allosteric modulator)
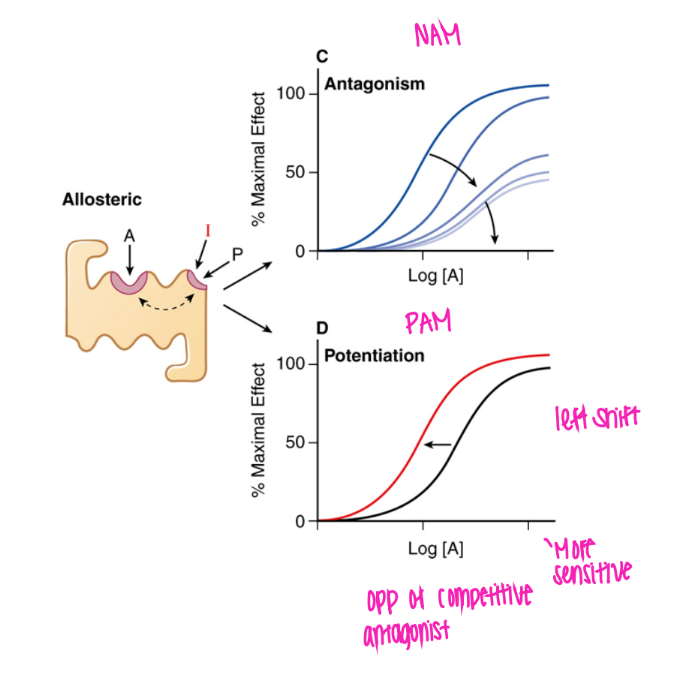
What does the PAM curve look like? why?
shifted left, can get to 100%
makes agonist bind more easily!
(positive allosteric modulator)
what does the curve look like for COMPETITIVE antagonism?
shifted right.
flood with more substrate + you’re gucci!
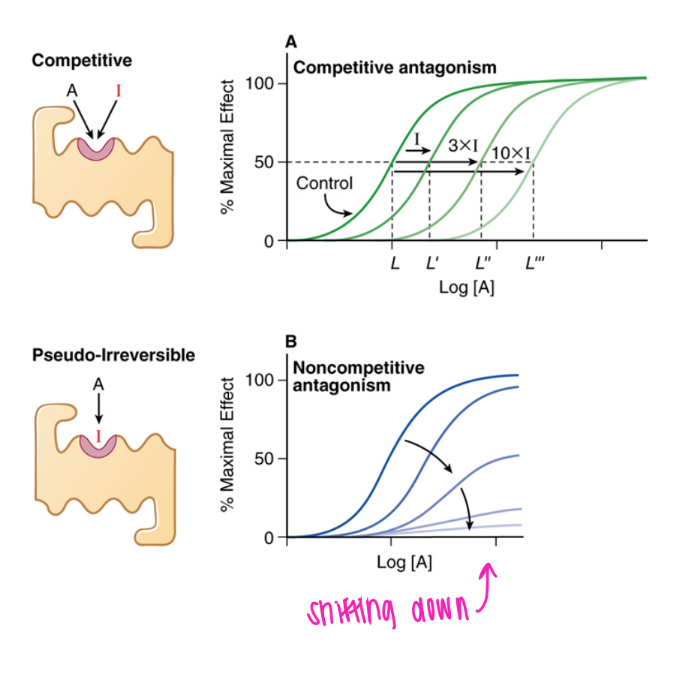
what does the curve look like for psuedo-irreversible antagonism?
shifted right and down; eventually gets to 0.
can out-compete until all the binding sites eventually all get blocked
wtf is “tachyphylaxis”?
decr drug response when SAME dose given multiple times

what happens in de-sensitization of a drug receptor?
receptor STILL phosphorylated on membrane
receptor binds b-arestin

what happens in drug receptor DEACTIVATION?
receptor REMOVED from membrane

chronic exposure to a drug leads to?
what is this called?
chronic exposure to drug leads to…
DEGRADATION of receptors
decr gene expression (don’t make as many)
down regulation / up regulation

what is pharmacodynamic tolerance?
receptors are down regulated → effect of drug DECREASES over time

A weak acid drug with pKa of 6.2 is taken orally. In the stomach, it will be what percent ionized?
100% NONionized.
bc pKa of drug is HELLA higher than pKa of stomach (~1).
so the drug doesn’t want to dissociate

what is upregulation? what causes it?
MORE receptors
CHRONIC antagonist exposure

wtf is “internalization” ? when does it happen?
internalizing receptors (bring into interior of the cell)
DOWNREGULATION
how is transcription related to receptors?
transcribe genes to make new receptor proteins
(takes foreverrrr)

what are the FOUR ways that the body treats drugs?
absorb
distribute
metabolize
EXCRETE/ eliminate
ADME

what are the 2 ways drugs are absorbed?
most diffuse acr lipid bilayer
carriers/ endocytosis (rare)

what is the partition coeff of a drug? what does it mean for it to be higher?
am dissolved into oil vs WATER
higher = more lipid soluable = good = can pass thro membrane
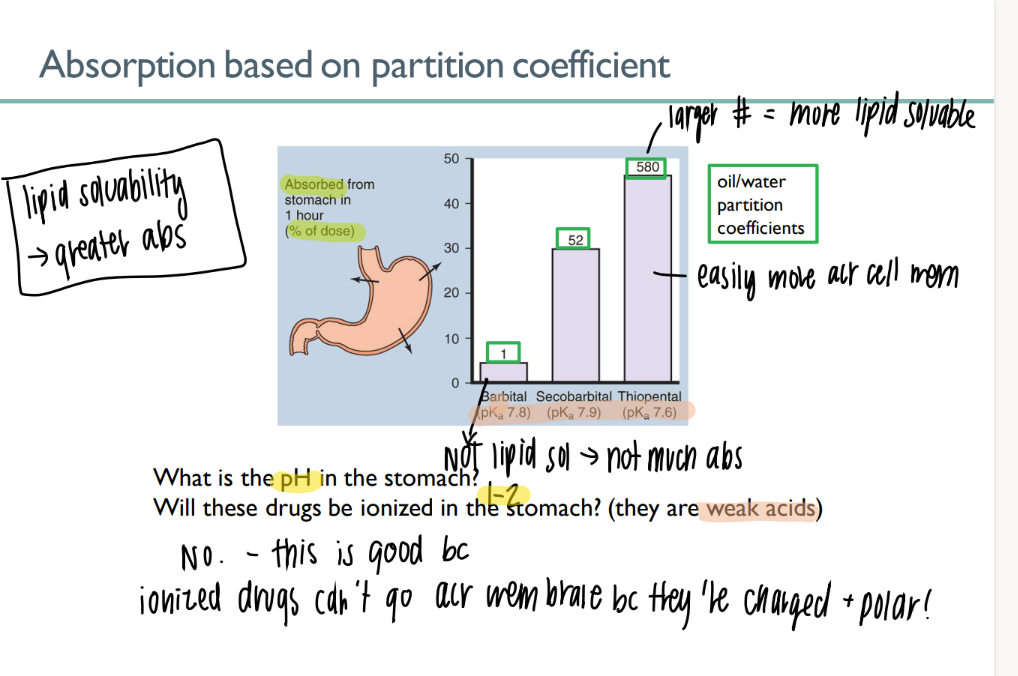
most drugs are what? why?
WA/WB
will NOT ionize in acidic environ → CAN cross the lipid bilayer
(charged + polar ions can’t cross bilayer)
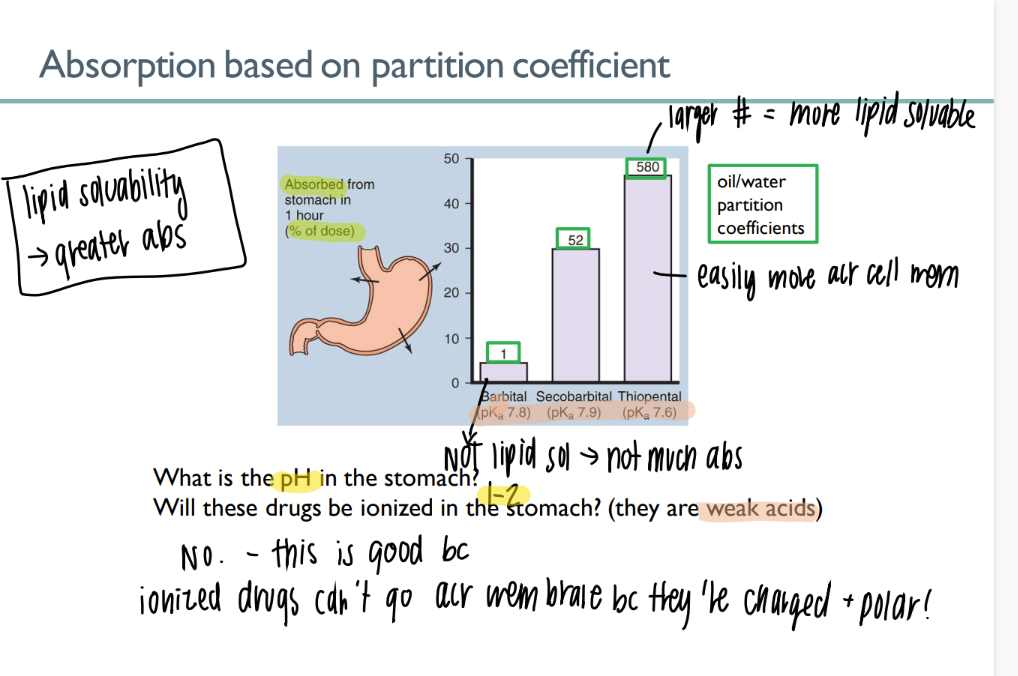
what’s the ph of blood?
7.4

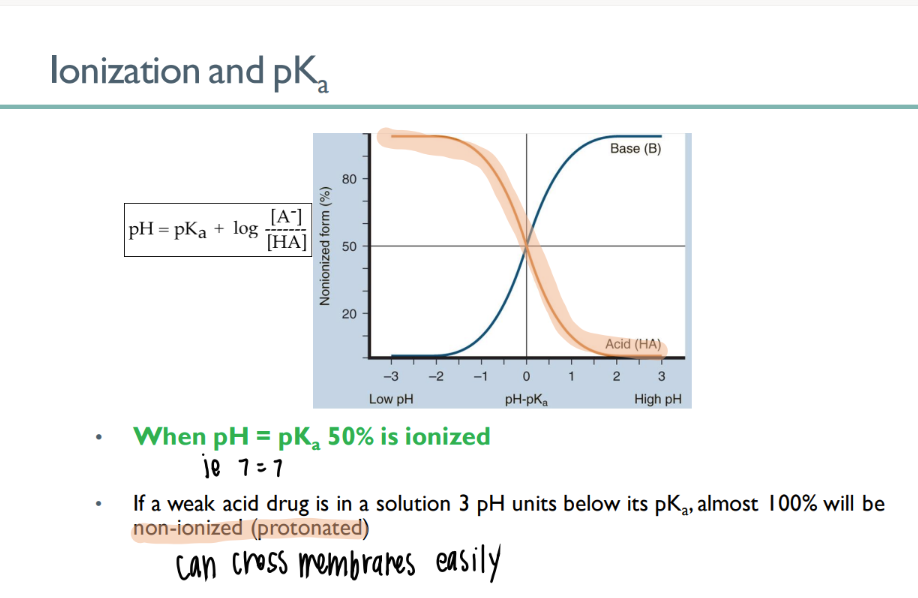
when pH = pKa….
50% of drug is IONIZED!
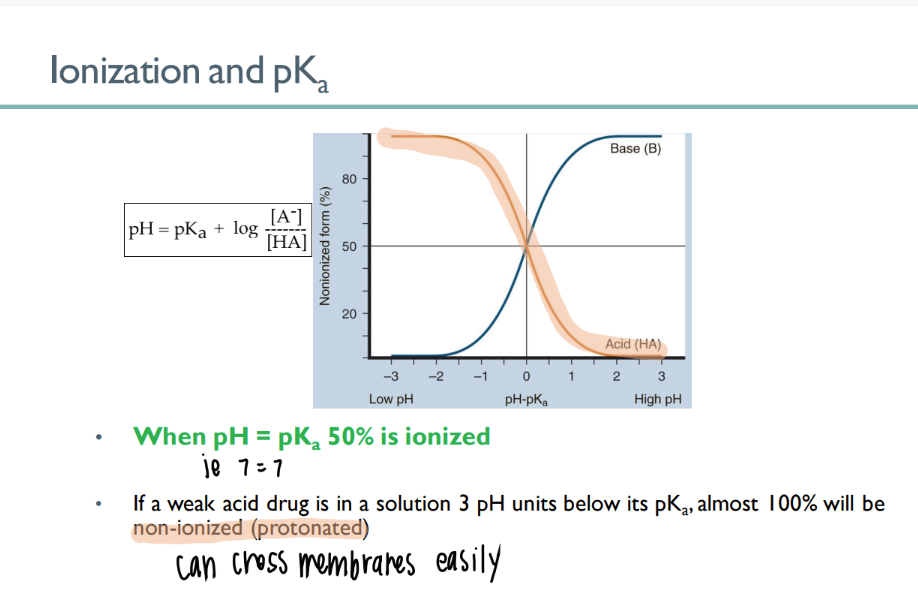
If pH < pKa…
aka….
100% stays per usual
(NOni-ionized = good)
WANT the drug to be in an ACIDIC environment
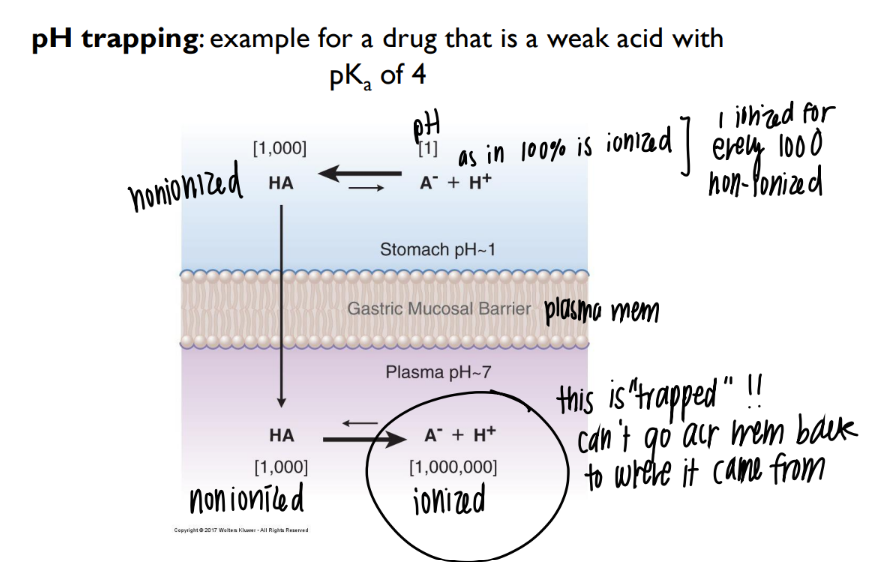
what happens in pH trapping??
acidic environ OUTSIDE cell → drugs stays intact, crosses membrane
neutral environ INSIDE → drug IONIZES and can’t leave the cell 🙂
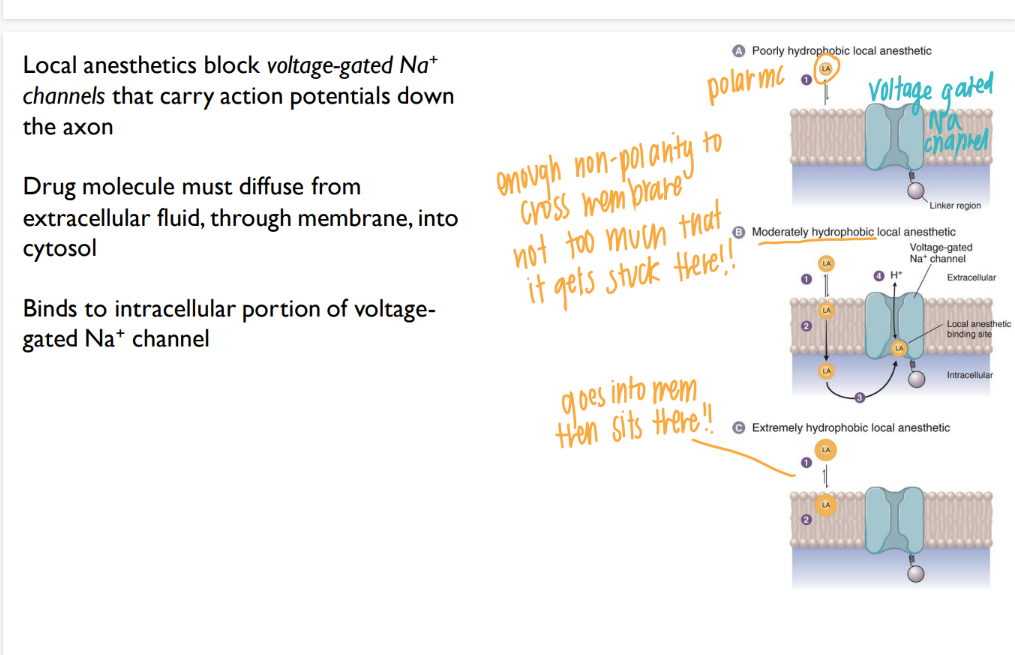
why do you need a moderately hydrophobic local anaesthetic?
hydrophillic (np) = bounces RIGHT off the membrane
pure hydrophobic (polar) = so lipid soluable, it goes into membrane and SITS THERE.
moderate = goes all the way THROUGH membrane, then binds to Na+ channel
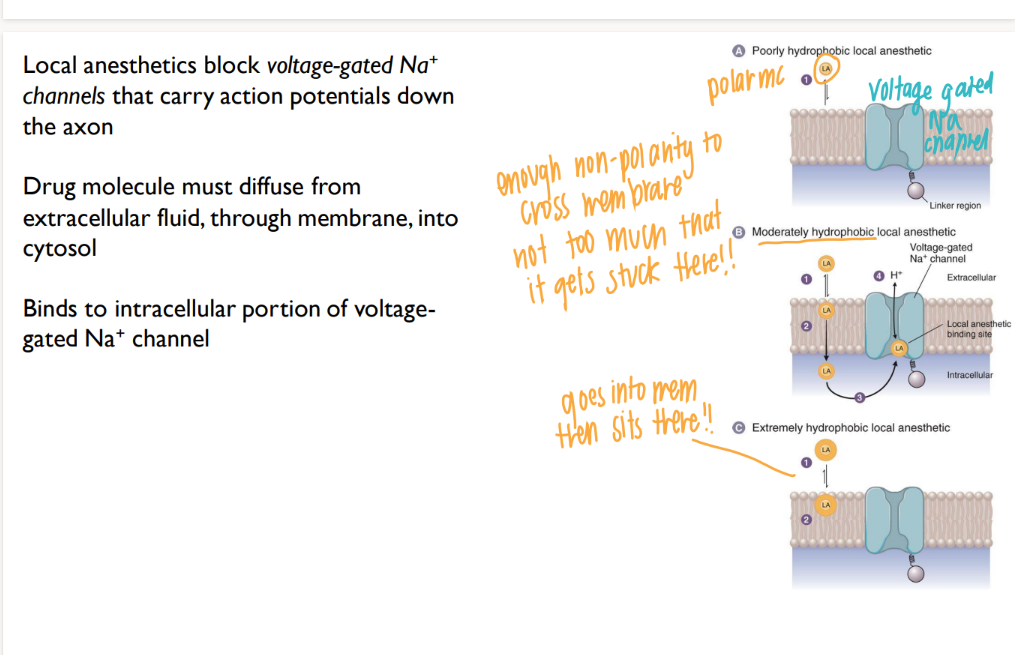
how do local anaesthetics work?
go into cell, BLOCK Na+ channels → no APs! down the axon
summarize: for optimal absorption, what do you need?
drug trapped = acidic on outside (intact) neutral on inside (ionize)
somewhat hydrophobic (np)
what’s the dif in junctions btw MOST capillaries + blood-brain barrier ones?
so, how does the drug need to be to get inside the BRAIN?
most = leaky, brain = TIGHT junctions
lipid soluable!
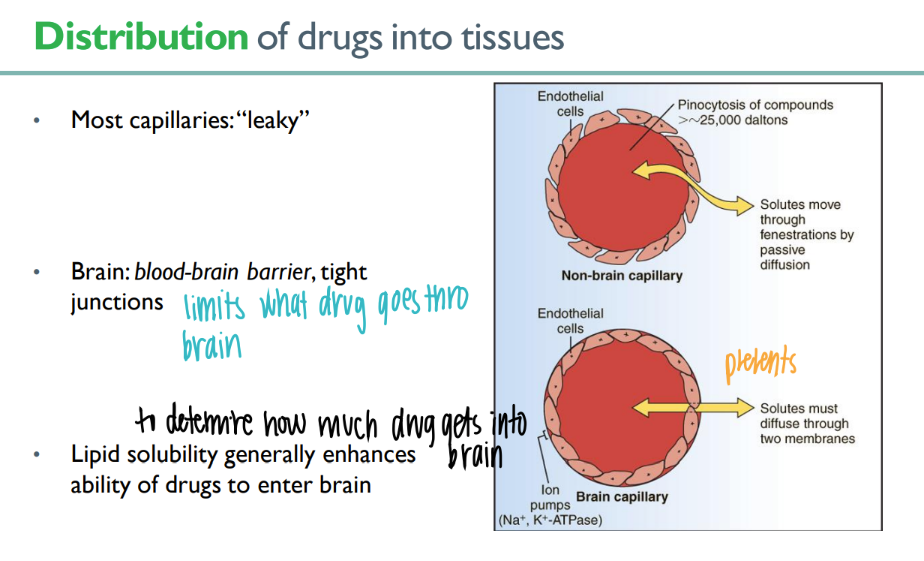
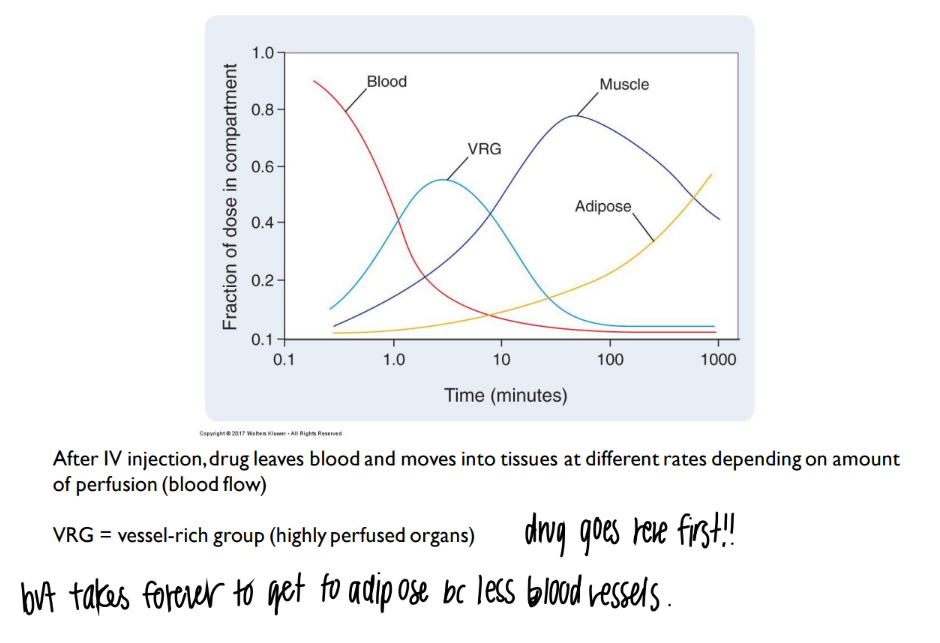
what’s the link btw blood + drug distribution?
more blood vessels = more drug delivered there
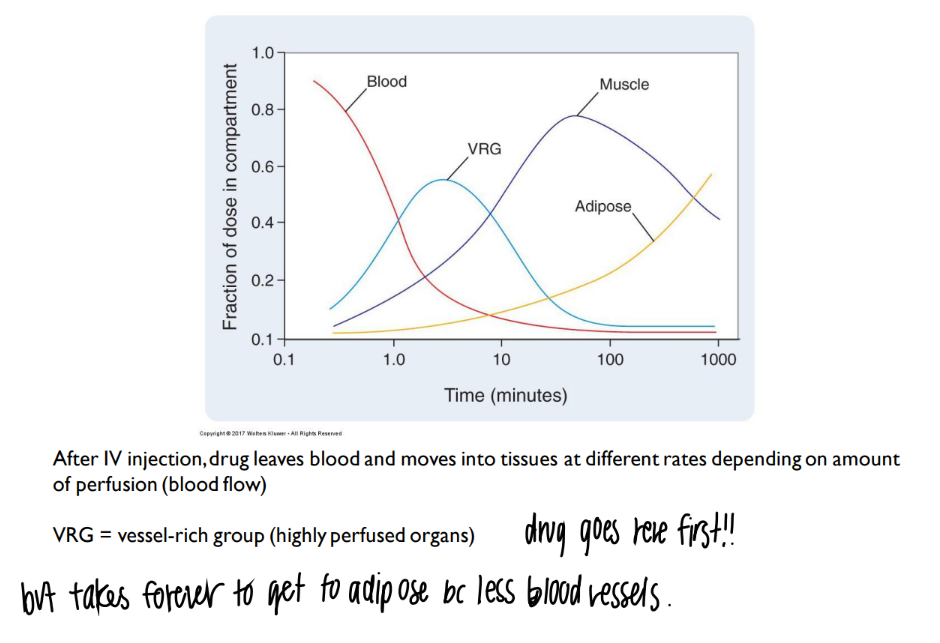
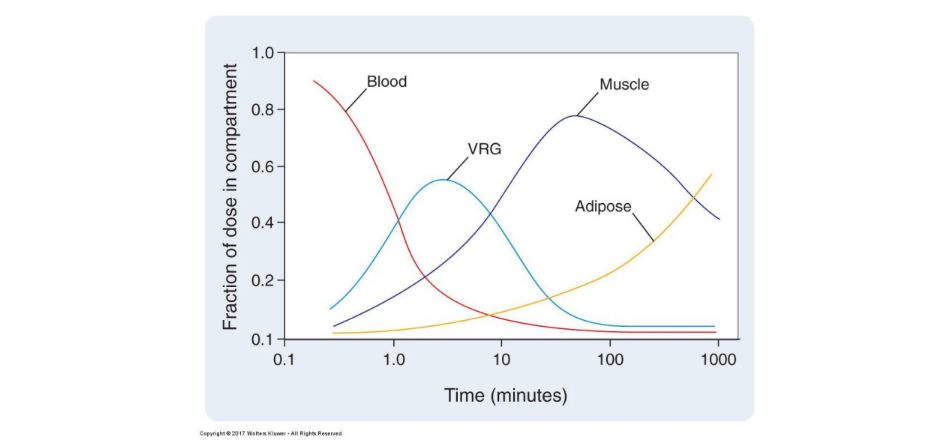
explain this chart.
what is VRG?
more blood vessels = more drug distributed.
(ie fat/ adipose doesn’t have much blood vessels = not much drug delivered there)
vrg = vessel rich group = organs with TONS of blood vessels
what is responsible for the “finite” duretion of drug action in the body?
metabolism
excretion

what happens in metabolism ? what’s another term for it?
BIOtransformation
drug is INACTIVATED by enzymes

what organ is primarily responsible for drug METABOLISM?
liver!

what 3 organs are responsible for EXCRETION of drug?
kidneys
liver (using bile salts)
lungs ??

what are the 2 forms in which drugs are EXCRETED?
UNCHANGED active drug
inactivated drug METABOLITES!

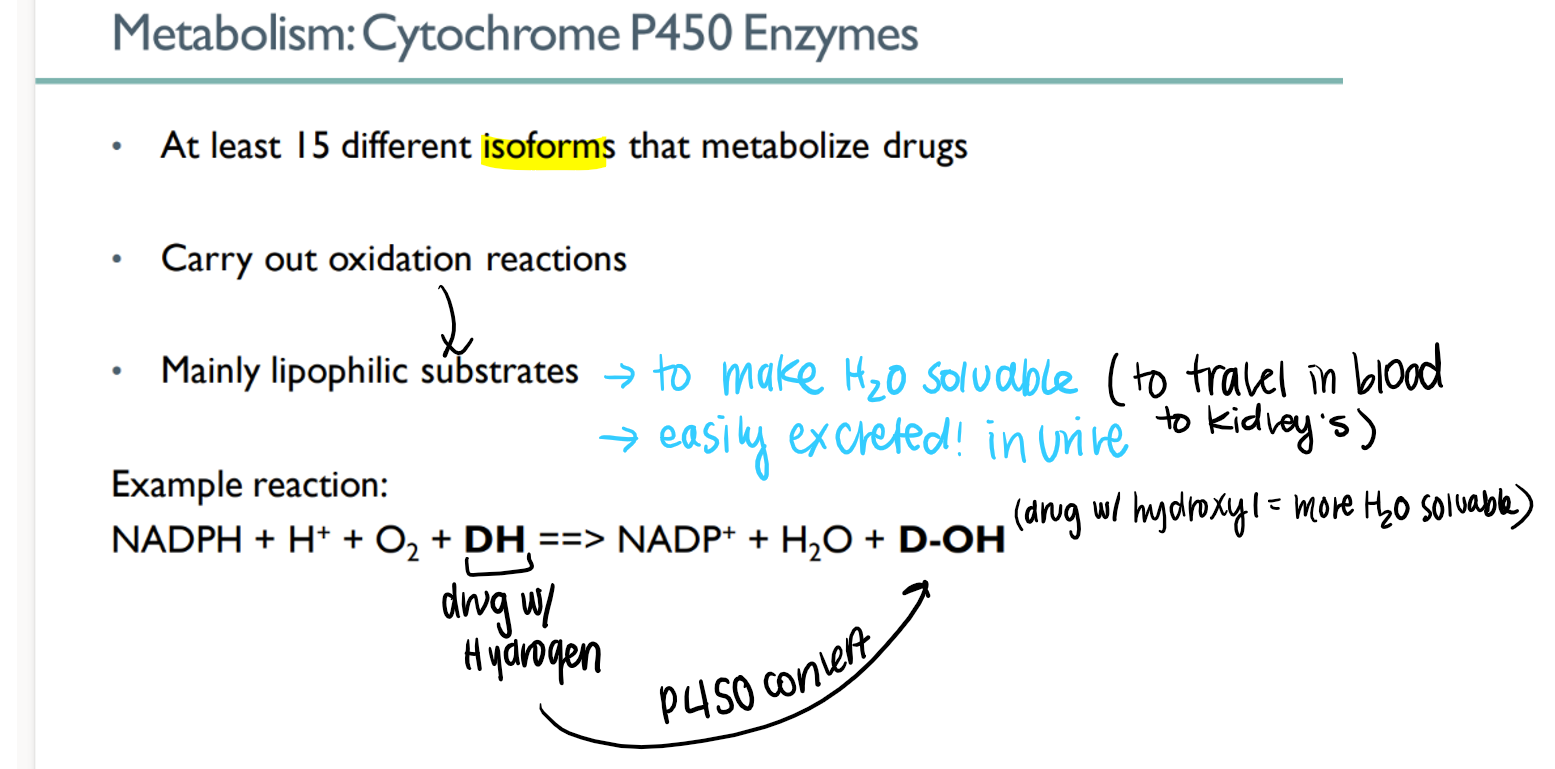
how many dif types of cytochrome p450 enzymes are there? what do they do?
15 isoforms!
METABOLIZE drugs to make water soluable and excrete-able
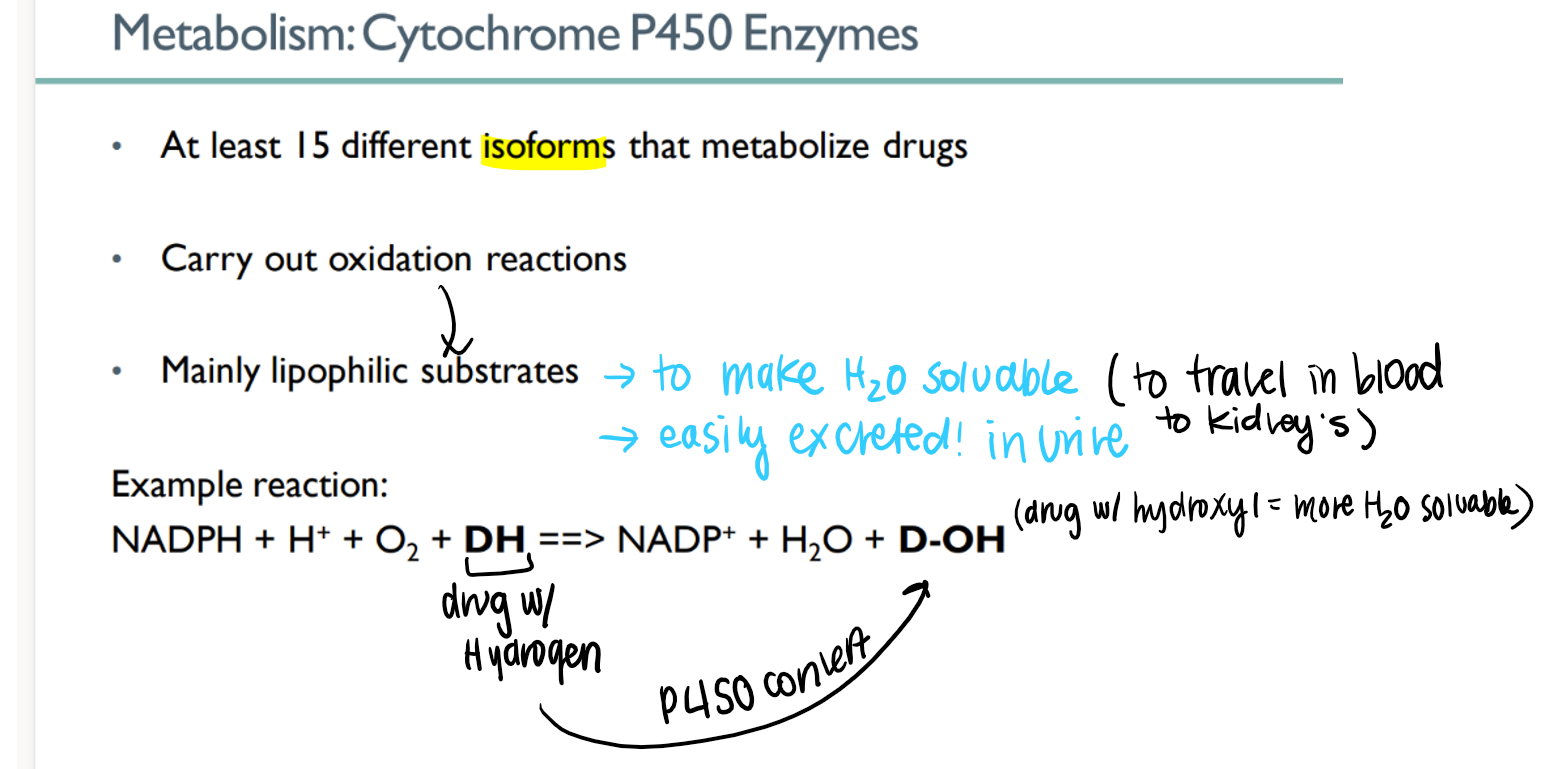
how do p450 enzymes work?
oxidize lipophilic drug (H) to make hydroPHILIC (OH)
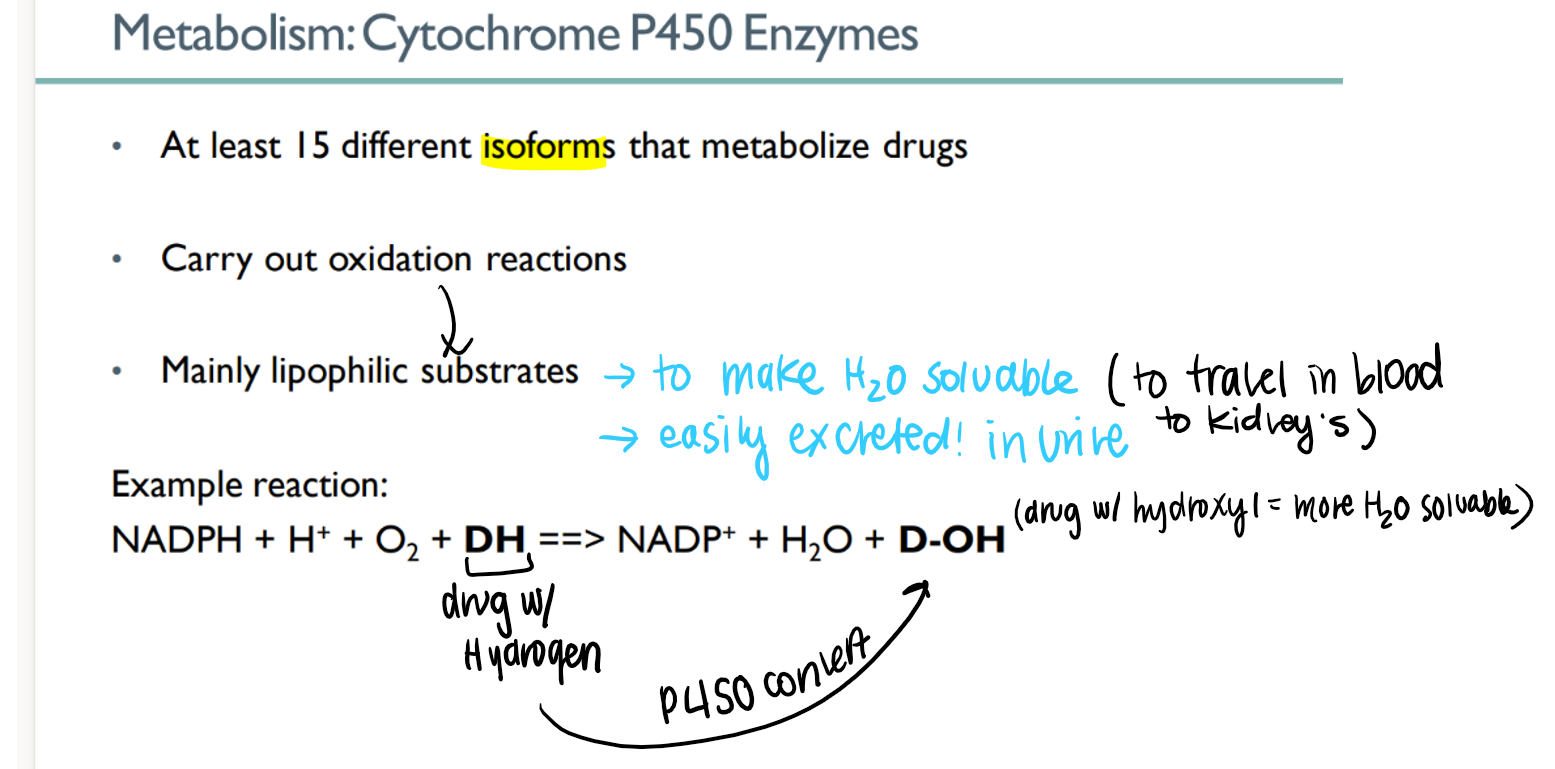
how is having a hydroPHILIC (D-OH) drug better for METABOLISM?
water soluable →
travel via blood to kidneys →
excreted in watery urine!
how many cytochrome P450 families are there?
3

what indicates the family of a CYP enzyme? what about the specific enzyme?
family = LETTER
enzyme = NUMBER

each P450 enzyme can …
metabolize MANY types of drugs

a single drug can be metabolized…
which leads to…
by MANY different cytochrome p450 enzymes
different metabolites! (can be a problem)

what’s the MOST common METABOLIC enzyme involved in drug interactions?
CYP3A4

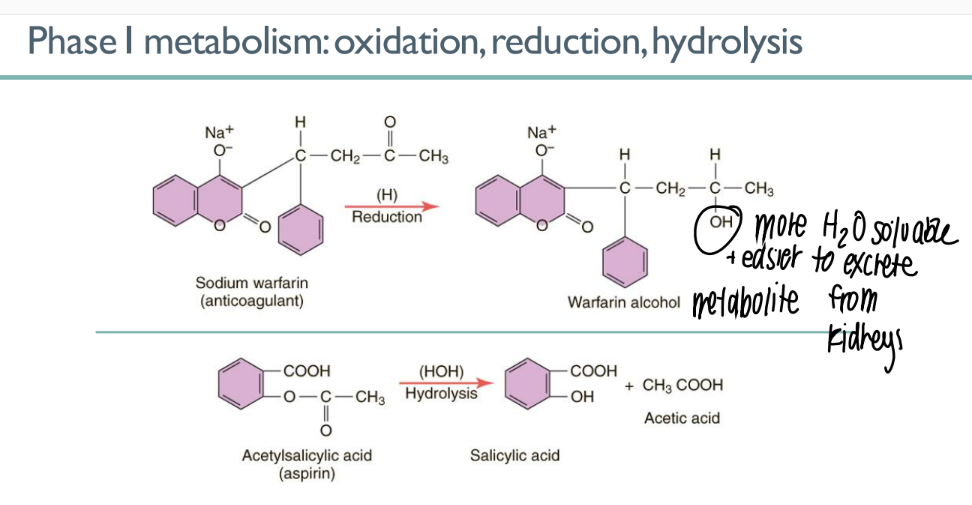
what’s the PURPOSE of phase 1 metabolism?
what are the 3 processes that can happen? what do they do?
make drug metabolite HYDROPHILLIC
oxidation, reduction, hydrolysis (all add an OH group)
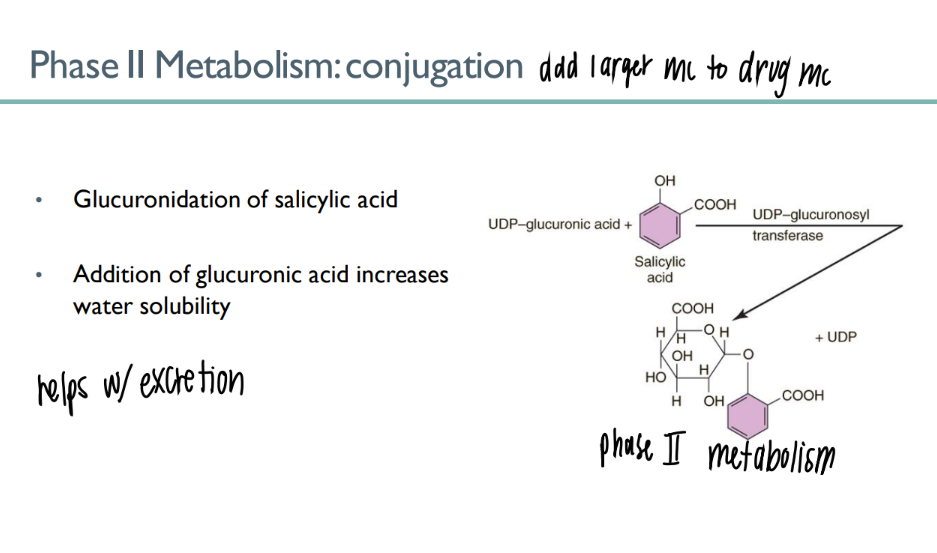
what’s the PURPOSE of phase 2 metabolism?
conjugation: attach drug to larger mc so more easily secreted in URINE
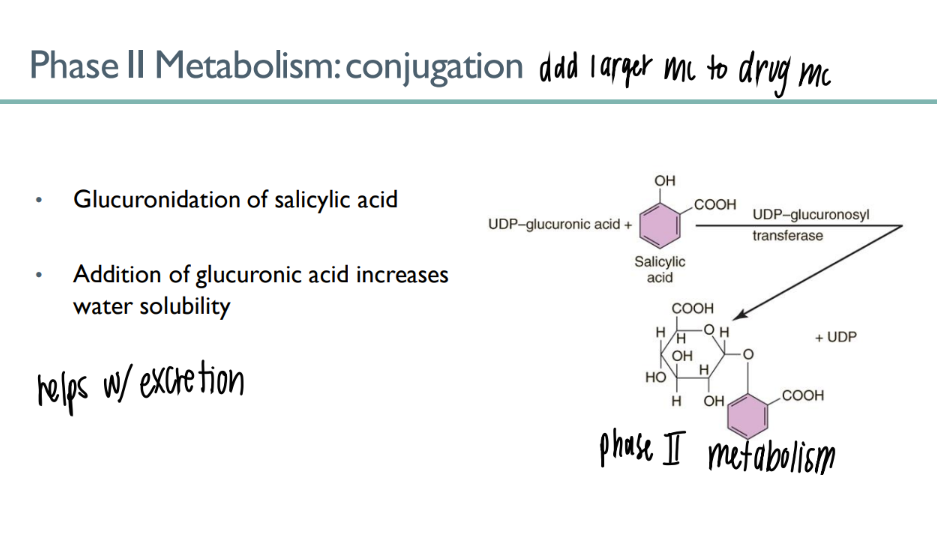
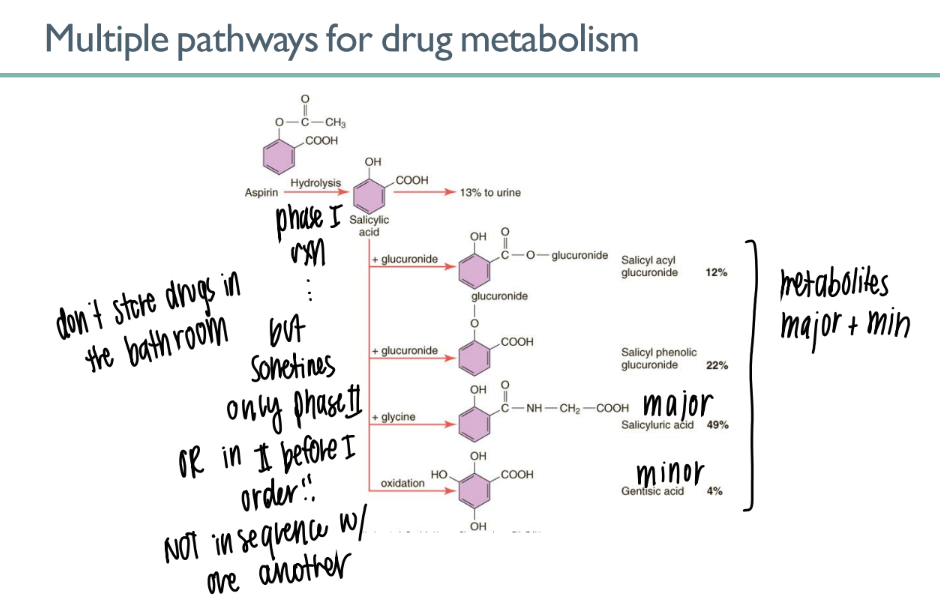
glucuronidation of salicyclic acid is an example of what TYPE of process?
what’s the purpose of it?
phase II metabolism (conjugation)
adding glucuronic acid mc to drug → INCR water soluability → better EXCRETION in URINE
what’s responsible for the MULTIPLE pathways of drug metabolism?
only phase I
only phase II
phase II before I
phase I before II
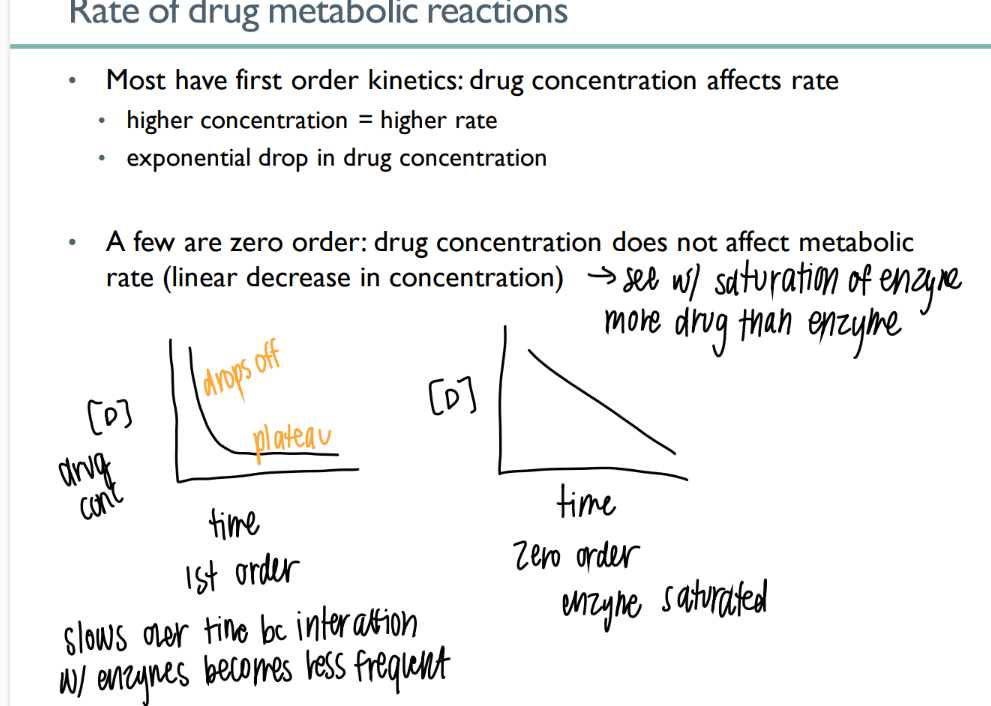
are most drugs 1st order or 0 order metabolism?
what does this mean?
how does it look on a graph?
FIRST order
drug conc AFFECTS rate (get rid of drug until no more drug)
EXPONENTIAL
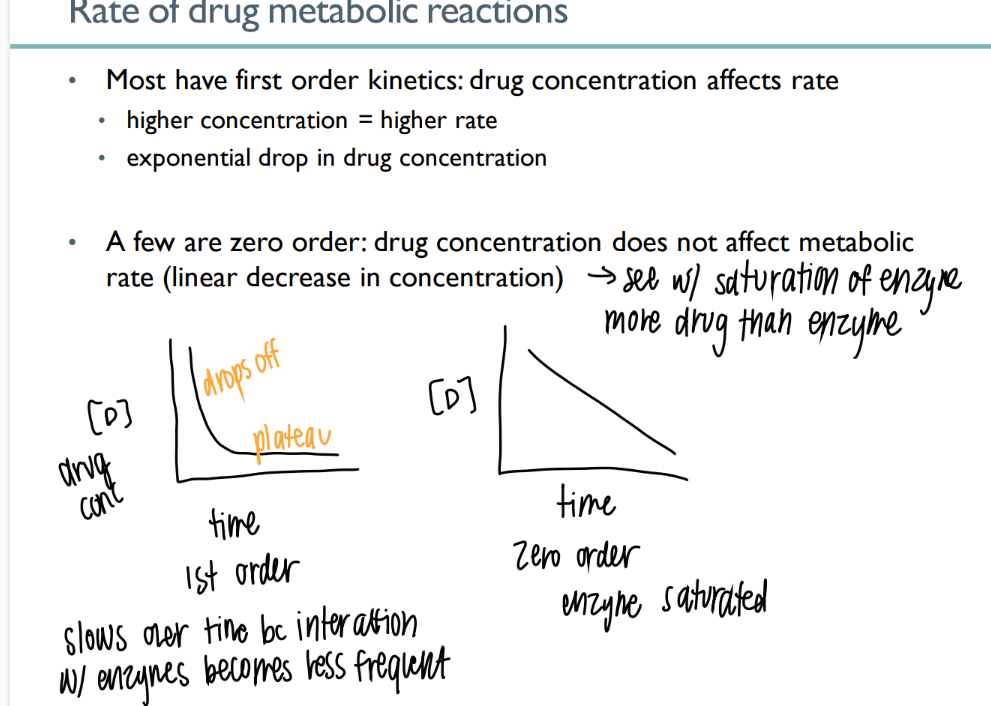
what does 0 order drug metabolism mean?
what does it look like on a graph?
drug conc does NOT affect the rate. limited number of enzymes available
LINEAR decr
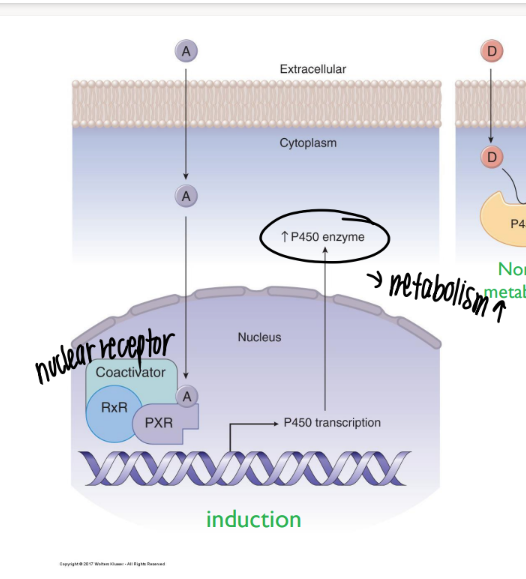
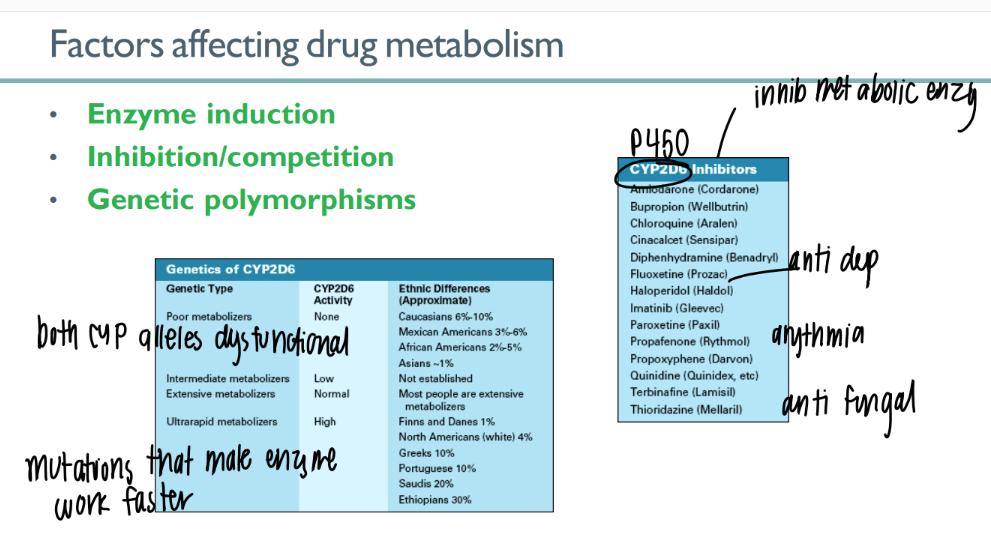
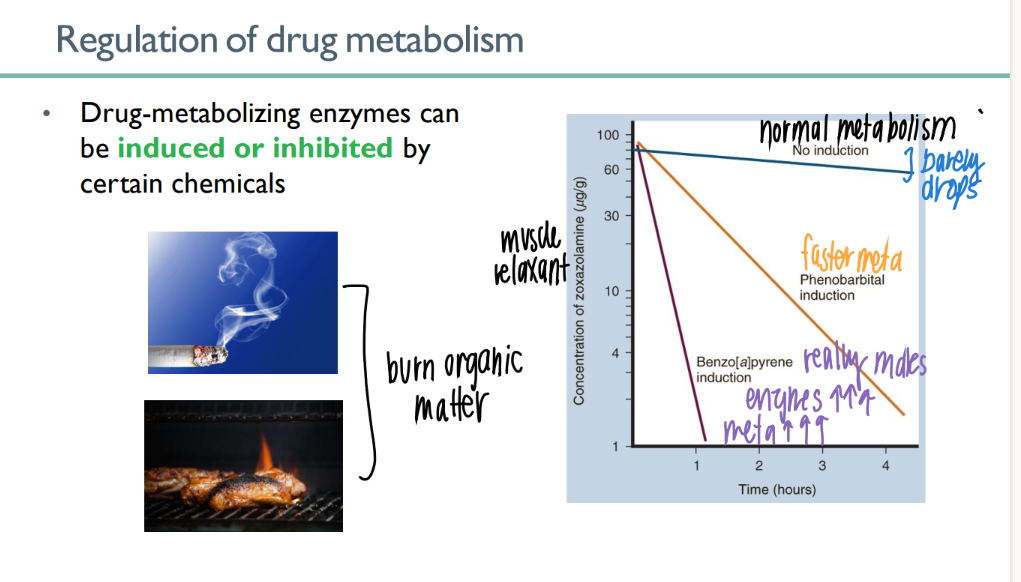
how does enzyme induction affect drug metabolism?
certain chemicals INCR # p450’s that are made → incr meta
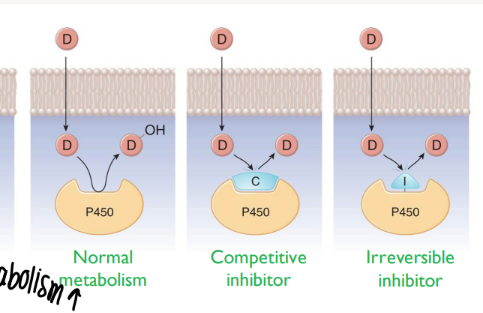
what are the 2 types of inhibition that decr drug meta?
competitive inhib (outcompete)
irreversible inhib
how can genetic polymorphisms affect drug meta?
CYP (450) alleles dysfunctional
CYP enzymes work faster