Trinity building a phenotype
1/89
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
90 Terms
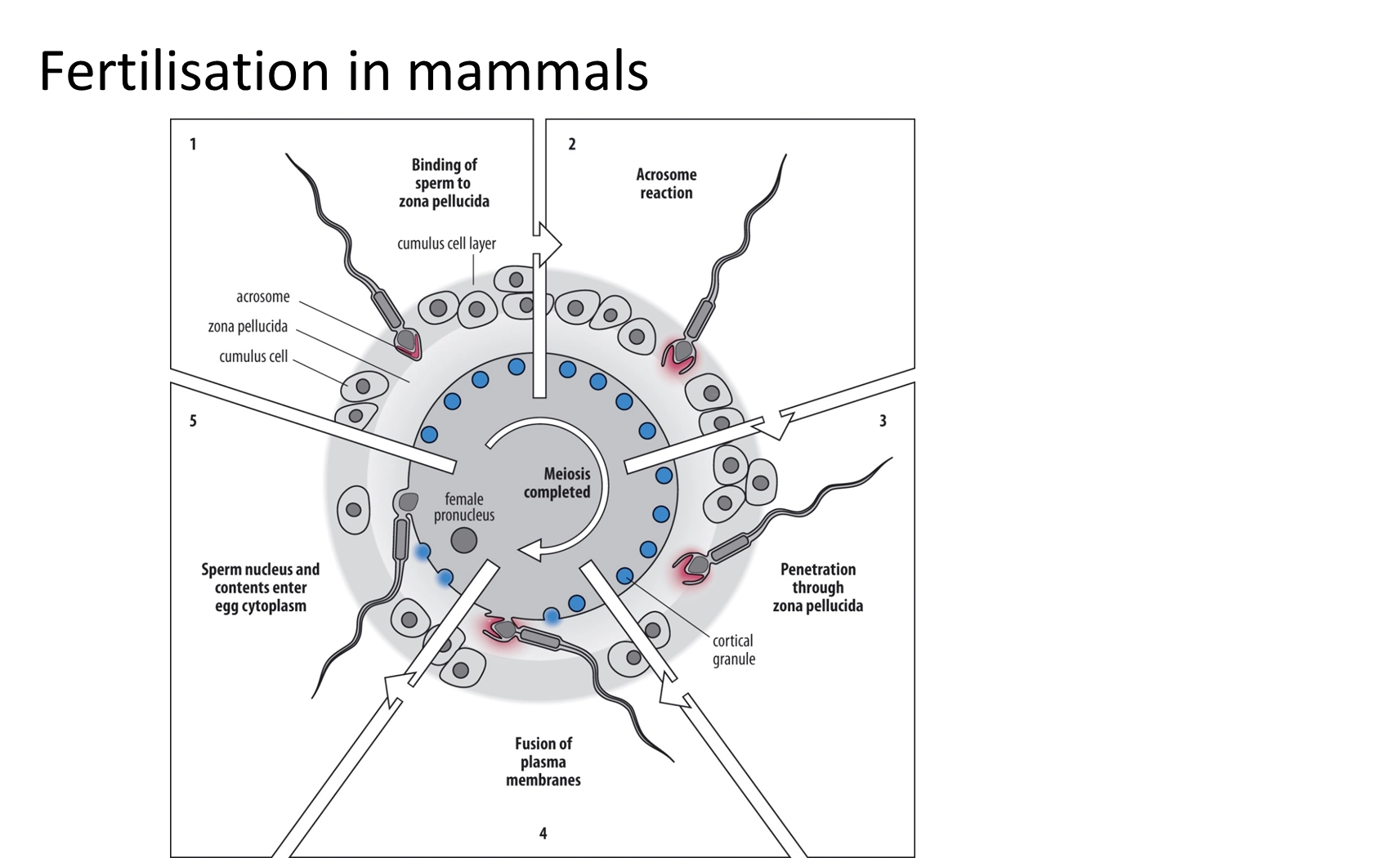
what are the mechanisms of fertilisation in mammals?
release of enzymes from the acrosome is triggered by the recognition event- when the sperm protein izumo on the acrosomal membrane binds to the egg receptor juno on the plasma membrane
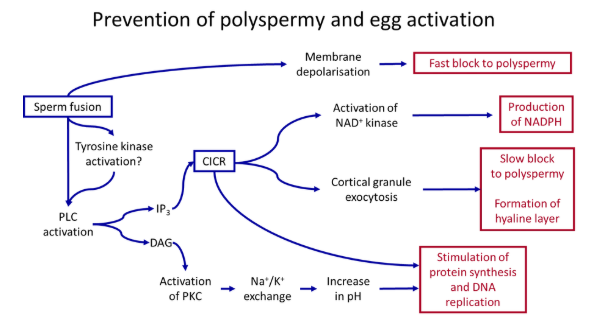
further fertilisation (polyspermy) is then prevented by the fast and slow blocks
how are multiple fertilisation events prevented?
the primary ‘fast block’- upon fertilisation, the membrane potential depolarises from -70 to +20, due to the influx of calcium (or sodium) ions
this propagates around the whole cell very fast, causing multiple different changes dependent on the species eg. deactivating juno sperm receptors
the secondary ‘slow block’- activation of phospholipase C in the phosphatidyl inositol signalling pathway causes calcium waves to propagate around the cell due to calcium-induced calcium release (positive feedback)
this causes the production of a physical barrier around the egg by cortical granule fusion- specialised vesicles fuse with the egg cell membrane, releasing enzymes that cause the hardening of the cells of the zona pellucida
what are the additional functions of calcium waves following fertilisation of the egg cell, other than preventing polyspermy?
activation of metabolism for divisions to occur
activation of protein kinase C to increase the cell pH for protein synthesis and DNA replication
activation of calmodulin-dependent kinase II to phosphorylate many different targets in an enzymatic network, including causing the degradation of cyclins to trigger meiosis II to complete
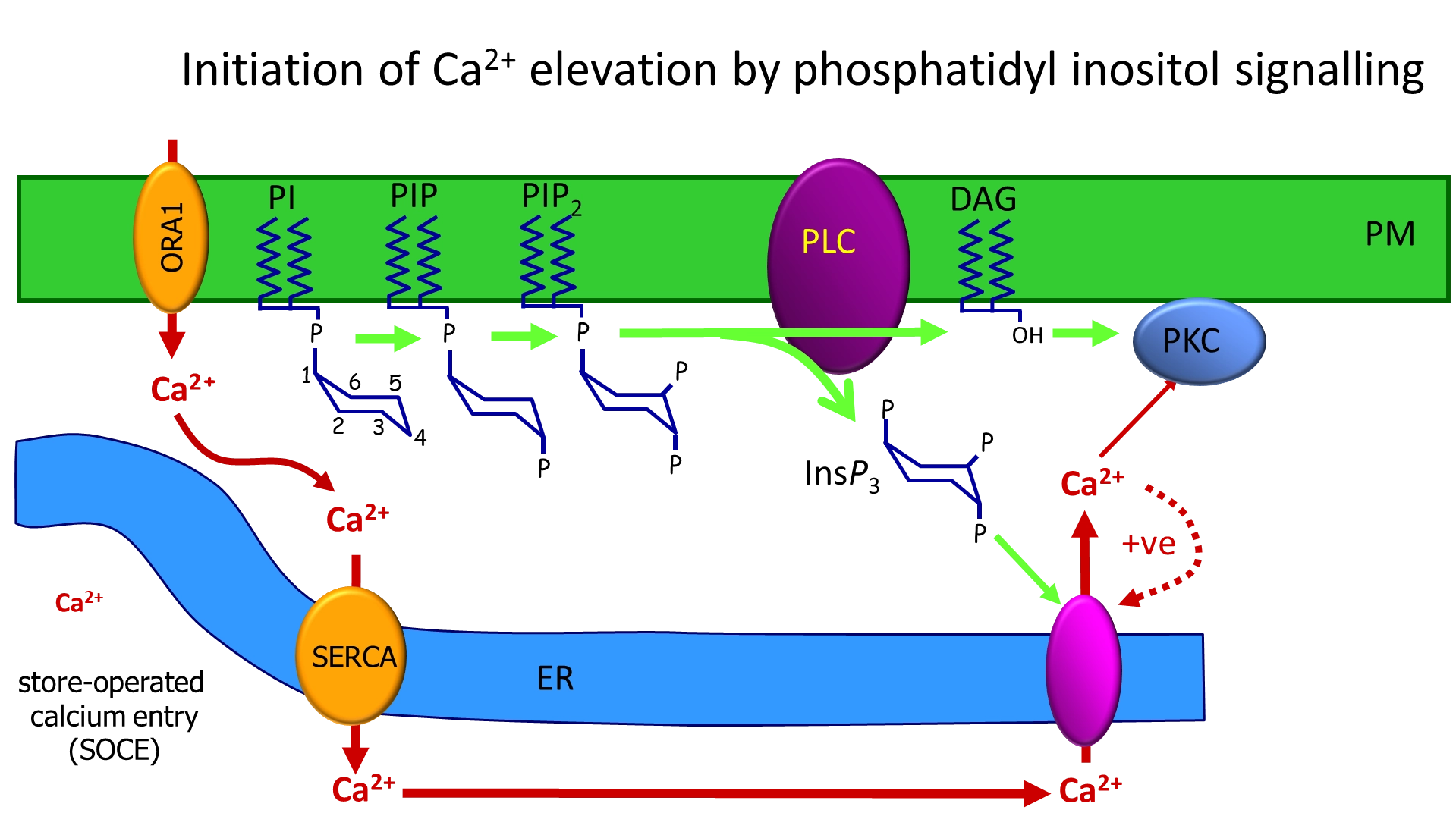
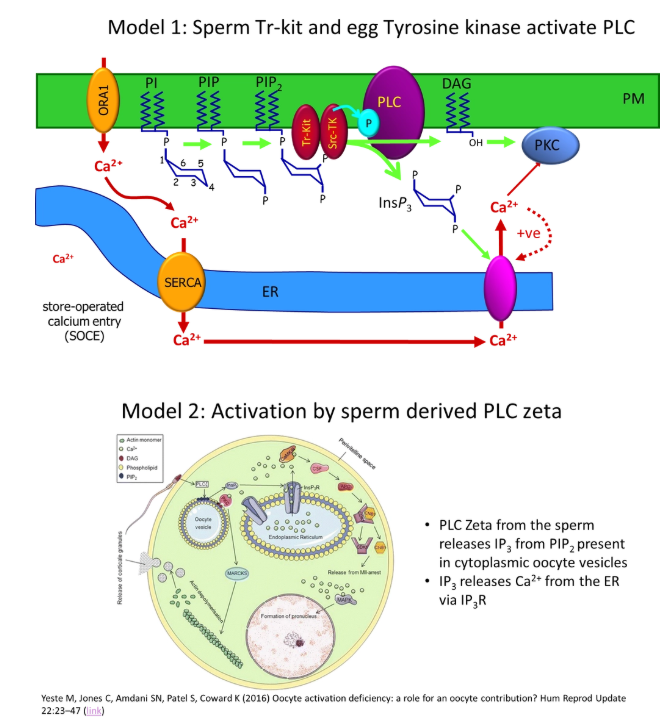
how does the phosphatidyl inositol signalling pathway cause the initiation of calcium waves following fertilisation of egg cells?
when the enzyme phospholipase C (PLC) is activated, it cleaves PIP2 (phosphatidyl inositol with two inositol phosphorylations) in the membrane
this releases inositol triphosphate (IP3) and leaves diacylglycerol in the membrane
IP3 binds to a calcium channel IP3 receptor in the endoplasmic reticulum
this causes calcium release, and further calcium-induced calcium release from nearby channels (positive feedback) to produce a calcium puff/spark
in an all or nothing response, multiple localised sparks are needed to reach a threshold for the regenerative calcium wave to propagate around the cell, which triggers many events in early development
this process depletes the ER of calcium, which needs to be replenished
a complex across the ER detects this depletion and links to a channel in the PM to allow calcium back in
what are the alternative models of how the egg cell phospholipase C is activated for calcium wave initiation?
different models are used in different species
egg-receptor mediated:
binding of a sperm factor (fertilin) to a receptor on the egg triggers PLC activation (however fertilin hasn’t been chemically identified)
sperm oocyte-activating factor (SOAF):
no PLC activation needed- IP3 in the sperm could initiate the calcium wave, but there would need to be loads of it, because there isn’t enzymatic amplification (therefore unlikely)
a kinase released from the sperm activates the egg PLC
the sperm brings in a specific PLC that is already active
how does the egg cell recognise the calcium wave following fertilisation?
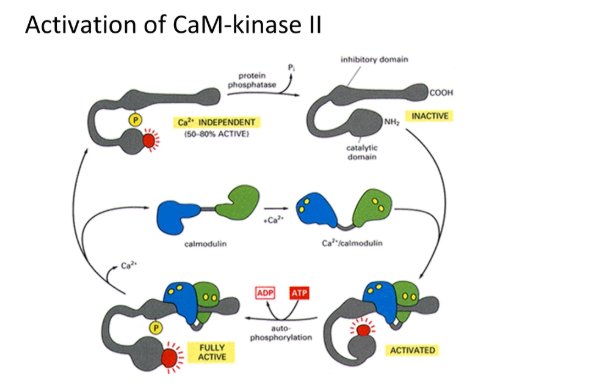
the protein calmodulin (CaM) binds to calcium at 4 binding sites by cooperative binding (sigmoidal kinetics = small response at low concentrations, large threshold response at high concentrations, unlike michaelis-menten)
binding of 4 calcium causes a large conformational change
the inactive calmodulin-dependent kinase II (CaM-kinase II) has an inhibitory domain that blocks its own active site
this is activated when this form of calmodulin binds and triggers auto-phosphorylation
calmodulin falls off but CaM-kinase II remains active and can phosphorylate many different targets in a large enzymatic network, including activation of the cell cycle
what are the different kinds of pathways in animal signalling?
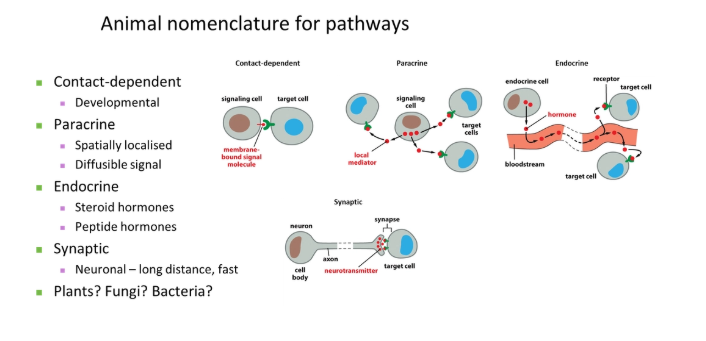
in order of increasing distance:
contact-dependent
paracrine- short-distance localised signal
endocrine- bloodstream-transported signal
synaptic
describe steroid hormone signalling and how it works
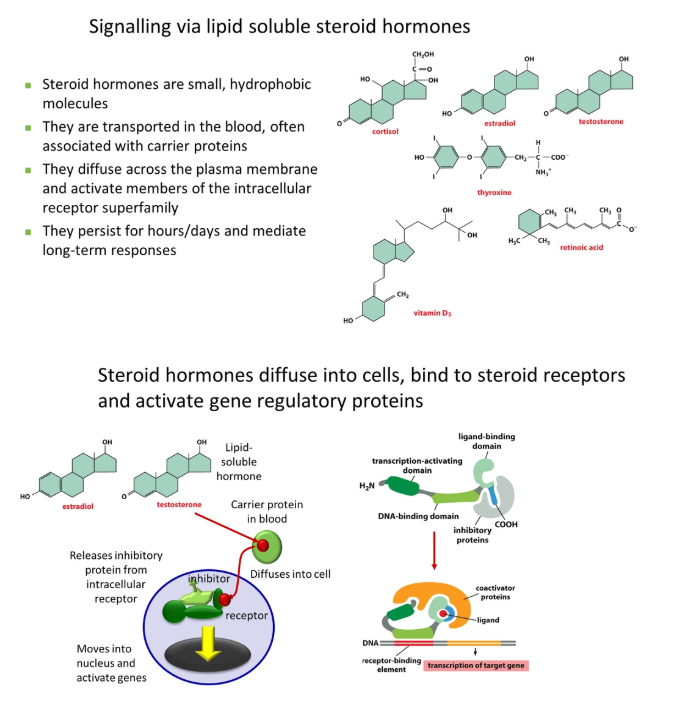
small + cheap to make (no N, P or S)
hydrophobic = able to cross membranes independently
persistent, generalised, long-term responses
the steroid hormone detaches from its carrier protein in the blood, diffuses into the target cell and binds to an intracellular receptor, often displacing an inhibitor protein
this activates the receptor (no signal amplification), exposing a DNA-binding site, so it can move into the nucleus and activate genes
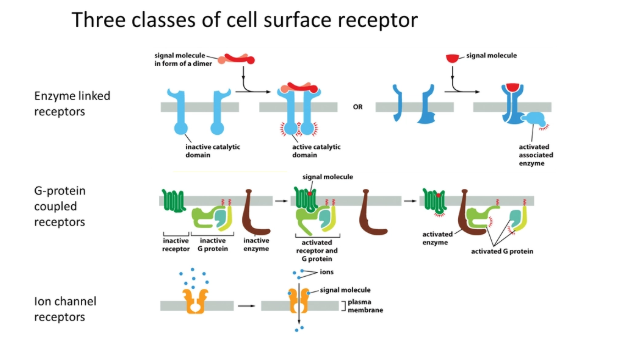
what are the three classes of cell surface receptor?
enzyme-linked receptors- binding of a ligand causes a conformational change in the receptor activating a catalytic domain, or recruiting/activating an enzyme eg. receptor tyrosine kinases (MAPKKK cascade)
G-protein coupled receptors- binding of a ligand causes a conformational change in the receptor, which activates a trimeric G protein intermediate, activating an enzyme in turn eg. cAMP signalling
ion channel receptors- binding of a ligand causes a conformational change that opens the channel for ion diffusion
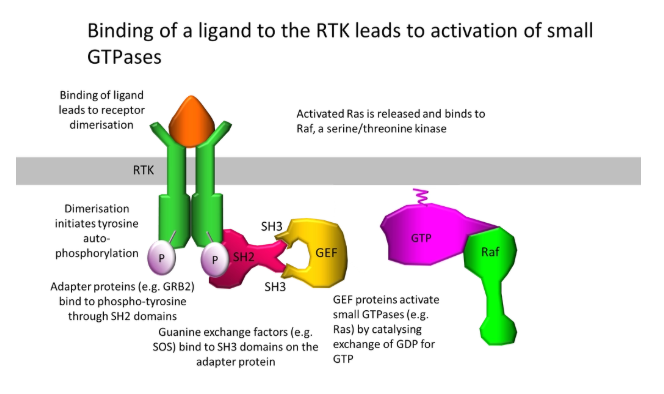
describe enzyme-linked receptor signalling and an example
enzyme-linked receptors- binding of a ligand causes a conformational change in the receptor activating a catalytic domain, or recruiting/activating an enzyme
most only have one transmembrane domain
example:
receptor tyrosine kinases, activated (eg. by growth factors), assemble as dimers and autophosphorylate their cytoplasmic tails at tyrosine residues
this scaffolding recruits a series of downstream components, including guanine exchange factors (GEFs), which temporarily activate small GTPases eg. Ras that swap GTP and GDP bound to proteins eg. Raf
Raf is a mitogen-activated protein kinase kinase kinase (MAPKKK), which phosphorylates MAPKK twice, in turn phosphorylating MAPK twice, which phosphorylates + activates transcription factors (an amplification module)
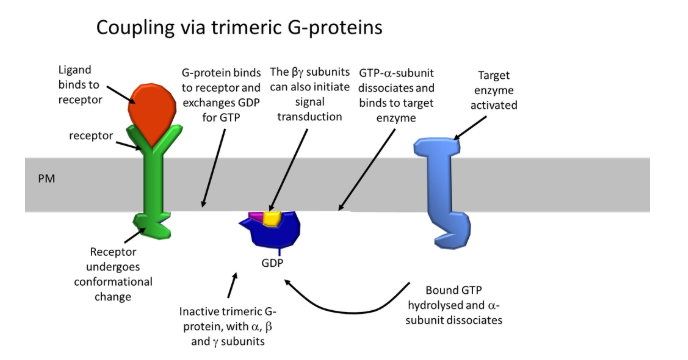
describe G-protein coupled receptor signalling and two examples
G-protein coupled receptors- binding of a ligand causes a conformational change in the receptor, which activates a trimeric G protein intermediate, activating an enzyme in turn
these receptors have 7 transmembrane alpha helix domains which bundle up
the N-terminus and extracellular loops bind the ligand, which causes a conformational change in the C-terminus and cytoplasmic loops, allowing them to interact with a GDP-bound G-protein
this changes its conformation so that it binds GTP (active)
the alpha subunit dissociates and binds to a target enzyme (eg. PLC), and hydrolyses the GTP to activate it
the other two subunits can also initiate signal transduction
example:
in cAMP signalling, adrenaline is secreted from the adrenal medulla into the bloodstream in response to stress
this binds to G-protein coupled receptors (sigmoidal binding) on the surface of muscle, liver and adipose cells
this causes the activation of adenylate cyclase, which produces cyclic AMP
this binds + activates protein kinase A (PKA), and releases it to phosphorylate phosphorylase kinase
this in turn phosphorylates glycogen kinase which initiates glycogenolysis
PKA also phosphorylates the receptor itself, inactivating + desensitising it
example:
in the inositol signalling pathway, the activated G protein is what activates the PLC (not in fertilisation)
this triggers calcium-induced calcium release to regulate many different factors
describe ion receptor signalling and two examples
ion channel receptors- binding of a ligand causes a conformational change that opens the channel for ion diffusion
examples:
the acetylcholine receptor is a pentameric ligand gated channel, with two acetycholine binding sites- ACh binding opens the channel to allow diffusion of ions through (primarily sodium because it is far from the equilibrium of the membrane potential)
the GABA receptor is a pentameric ligand gated channel with one GABA binding site- GABA binding opens the channel for chloride ion diffusion to resist depolarisation of the membrane
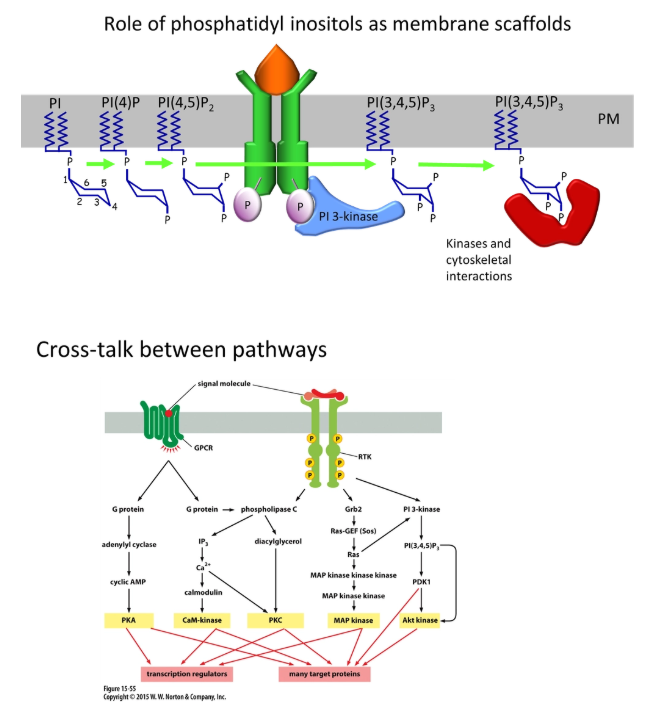
describe an example of cross talk between signalling pathways
different signalling pathways can interact:
eg. activated receptor tyrosine kinases (enzyme-linked receptors) assemble as dimers and autophosphorylate their cytoplasmic tails at tyrosine residues
this can bind and activate IP3 kinase from the inositol signalling pathway
this phosphorylates PIP2 to produce PIP3
PIP3 can act as a scaffolding protein for cytoskeletal interactions
why must the cell cycle occur with high fidelity and be tightly regulated?
eg. parts of metabolism might be stopped during dividision to reduce mutations eg. to prevent the production of reactive oxygen species
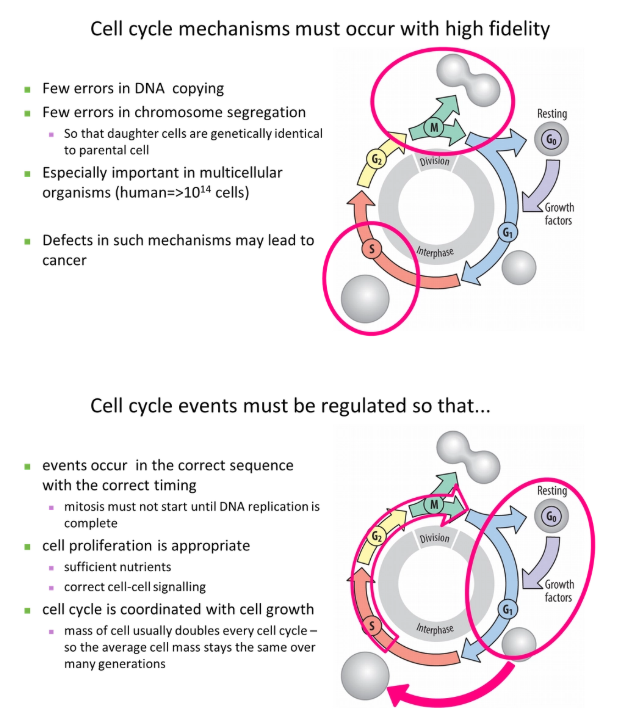
how is the first cell cycle activated?
MPF = maturation promoting factor (composed of the catalytic subunit cyclin dependent kinase and the regulatory subunit cyclin B)- conserved in all eukaryotes but have different names
this was chemically identified using temperature-sensitive mutations + recombination with different plasmids containing wild type genes
G1 and G2 can be omitted in the early embryo because it is faster, and the egg cell already contains loads of maternal resources for sub-division
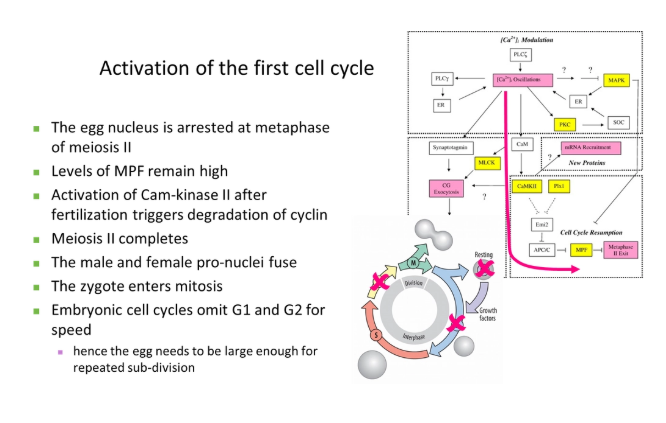
how is the cell cycle regulated?
the maturation promoting factor (MPF) is composed of the catalytic subunit cyclin dependent kinase (Cdk) and the regulatory subunit cyclin B
to progress into certain stages of the cell cycle, Cdk has to bind to the relevant cyclin to partially activate it
full activation of Cdk is also dependent on phosphorylation by Cdk-activating kinase (CAK), which is only carried out dependent on cellular conditions eg. sufficient size + lack of DNA damage
this allows the transition into the next stage by the phosphorylation of different target proteins
this cyclin is then degraded, deactivating Cdk- this is a ratchet that prevents the cycle moving backwards
Cdk activity is under additional regulation, allowing control at different checkpoints:
inhibitory phosphates can also be added to Cdk to block its active site and prevent progression into the next stage
additional Cdk inhibitor (CKI) proteins can block its activity eg. these get transcribed following detection of DNA damage
positive feedback loops in this process cause massive amplification to activate multiple Cdks, including those that degrade the cyclins
different cyclins (and in mammals, different Cdks) function at different stages of the cell cycle, dependent on the species
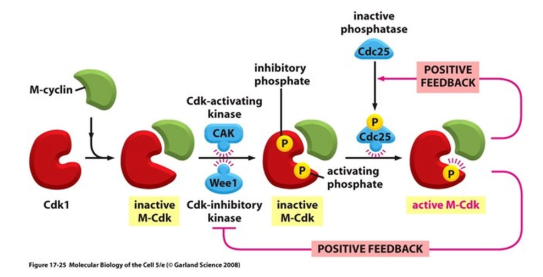
what is the centrosome cycle?
in animals:
during the S phase, the pair of centrioles (perpendicular to each other) self-replicate to form two pairs of daughter centrioles
they begin to produce spindle fibres
during prophase they begin to move to the poles by pushing away from each other using antiparallel spindles (plus ends together)
following mitosis, one centrosome (pair of centrioles) is distributed in either daughter cell
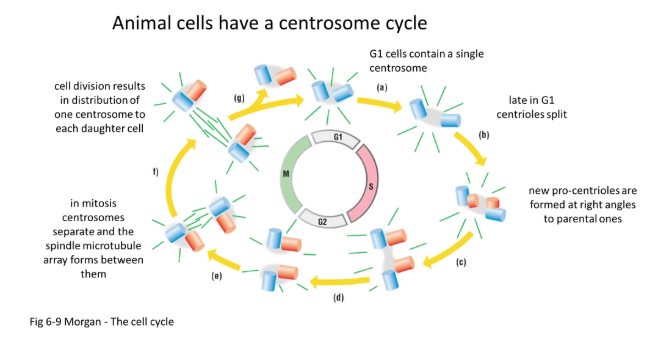
how are spindle fibres set up?
double-headed minus-end motors link adjacent microtubules together, drawing them in to focus at the minus pole
plus-end motors are anchored at the centrosome to tether the focus pole
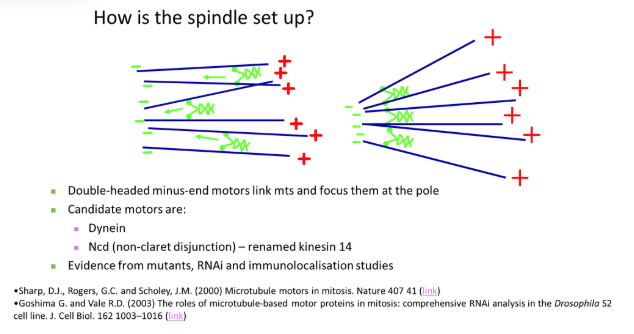
how do centrosomes separate to the poles of the cell in prophase?
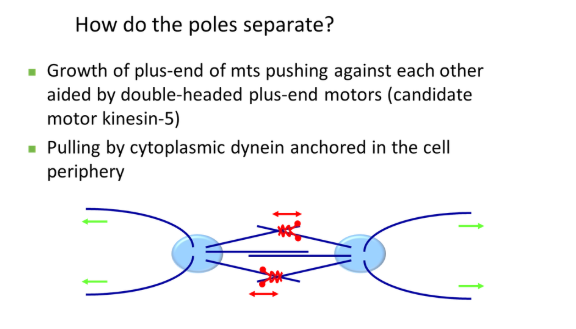
the pair of centrosomes produce spindle fibres that overlap antiparallel (plus ends together) and push each other away
this is carried out by double-headed plus-end motors that walk towards the overlapping region
also, cytoplasmic dynein (minus-end motor), anchored at the plasma membrane, pulls the centrosome away from the centre via astral microtubules
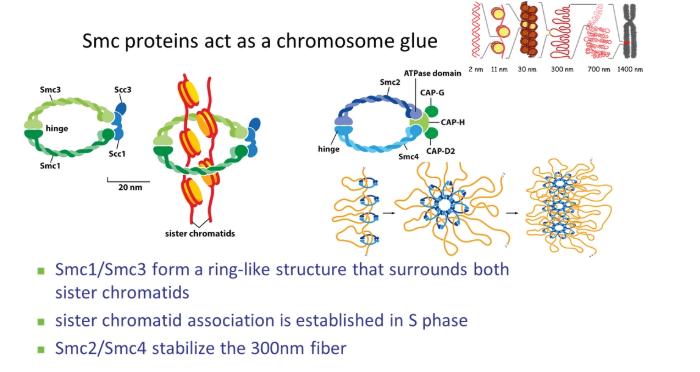
how does chromosome condensation happen in prophase?
Cdk phosphorylates histones
SMC (stable maintenance of chromosomes) proteins condense the chromosomes using their hinged ring structures
condensin stabilises the 300nm fibre by organising nucleosome loops
cohesin clamps the two sister chromatids together
how is nuclear envelope breakdown initiated in pro-metaphase?
the mesh of nuclear lamins (intermediate filaments) on the inner side of the nuclear membrane is phosphorylated by Cdk
this causes the filaments to dissociate from each other and break down the nuclear membrane by vesiculation
how do microtubule dynamics change in metaphase?
in metaphase, spindle fibres have to try find the chromosomes
instead of growing out long distances and catastrophising when they reach the plasma membrane, which is very inefficient, their dynamics change
they grow out short distances, then break down, in an exploratory effort to find the chromosomes
this is because plus-end motors increase catastrophe frequency
this means microtubule turnover and dynamic instability increases greatly
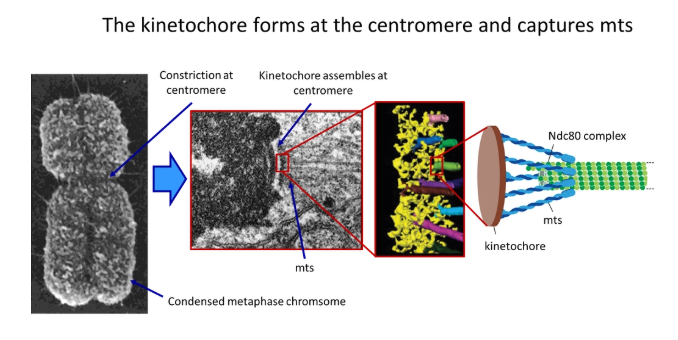
how do microtubules attach to the centromere? what process does this allow?
two kinetochores (protein complexes) assemble at the centromere of each chromosome, which grab onto the microtubules upon contact
this is carried out by a cluster of anchored minus-end protein motors called the Ndc80 complex
this holds around the sides of each microtubule so the end is still free to depolymerise and polymerise
this allows the poleward flux (while tubulin subunits are lost from the minus end, they are added at the plus end- constant length but still dynamic)
how are the chromosomes maintained along the metaphase plate?
tension from spindle fibres would pull all chromosomes into the centre of the cell- something has to keep them arranged along the metaphase plate
while the centromeres are pulled towards the centrosomes by minus-end motors at the kinetochore, the chromosome arms are pushed towards the metaphase plate by plus-end motors on the attached spindles
this is called the polar ejection force
how is correct centromere attachment by spindle fibres detected?
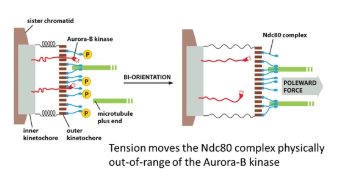
tension from spindle fibres on both sides of the chromosome is the only stable arrangement- this has to be sensed for every kinetochore
in the inner kinetochore, the Aurora-B-kinase enzyme is anchored, which phosphorylates the Ndc80 complex of the outer kinetochore (which tethers the microtubule)
however, when the kinetochore is under tension, the inner and outer layers are pulled apart and the Aurora-B-kinase is pulled out of range to phosphorylate Ncd80
this works because phosphorylation of Ndc80 reduces its affinity for microtubules, meaning it promotes the release of incorrectly attached microtubules
unattached kinetophores are bound by the mitosis arrest deficient protein (Mad), which prevents chromosome separation (by blocking securin ubiquitination by the APC) if any one kinetochore is not attached/aligned
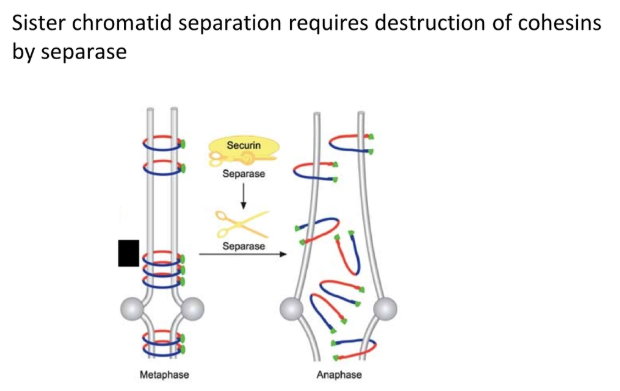
how is chromosome separation triggered and prevented?
loss of the cohesin SMC proteins that bind sister chromatids together allows for chromosome separation
when the anaphase-promoting complex (APC) is activated by Cdk, it ubiquitinates the securin protein
this tags securin for degradation by protease, releasing the separase enzyme, which destroys cohesin
the APC also degrades cyclin B (by ubiquitination → protease break down) to deactivate Cdk so that mitosis can’t be reinitiated
unattached kinetophores are bound by the mitosis arrest deficient protein (Mad), which blocks the APC if any one kinetochore is not attached/aligned
this prevents chromosome separation if there is a single error (positive signal)
what drives chromosome separation at anaphase?
there are multiple redundant mechanisms to ensure correct, complete chromosome separation:
plus-end motors anchored at the centrosomes pull the microtubules in
minus-end motors at the kinetochore pull the microtubules towards the centrosome
the microtubules depolymerise at both ends
the late stage of anaphase is driven by plus-end motors pushing the overlapping spindle fibres away from each other at the centre (these are only activated at the M/A transition)
why are microtubules used for spindle fibres? why are intermediate filaments used for nuclear lamins?
spindle fibres- even though microtubules are more energetically expensive to produce than actin, they are used for spindle fibres because actin isn’t stable under compression (it buckles)- whereas microtubules can be pushed away from each other to separate the poles
nuclear lamins- intermediate filaments are persistent for a longer period of time than microtubules and actin filaments, because they are less dynamic
why are membranes capacitors?
a capacitor is any component which allows the accumulation of a charge difference
lipid bilayers prevent ion movement, so charge will not reach equilibrium across a membrane without ion channels etc
capacitance (C) is equal to the charge separated (Q) x the voltage (V)
biological membranes have a capacitance of 1 μFcm-2 (= 10 fFμm-2)
this can be measured using a black lipid bilayer
this trait means that not very many ions need to move to change the membrane potential
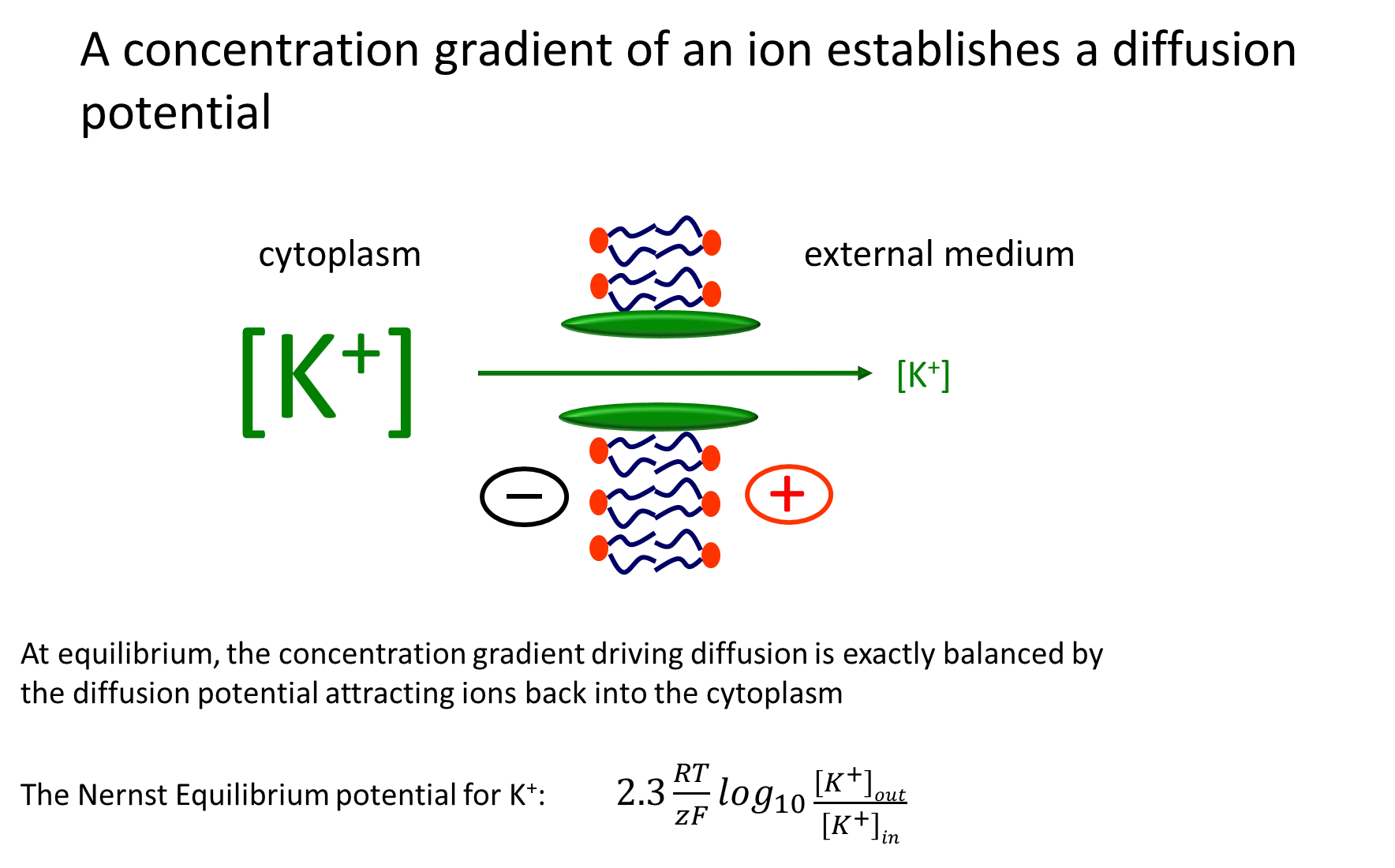
what is the potassium diffusion potential?
as sodium potassium pumps exchange 3 sodium ions for 2 potassium ions into the cell, the concentration of potassium ions increases and the membrane potential becomes more negative in the cytoplasm
a potassium ion channel allows potassium to diffuse out of the cell, making the cytoplasm more negative
this is opposed by the increasing membrane potential attracting positive ions back into the cytoplasm
at equilibrium (different concentrations but electrochemical potential is 0), the two forces are balanced
this is described by the Nernst equilibrium formula
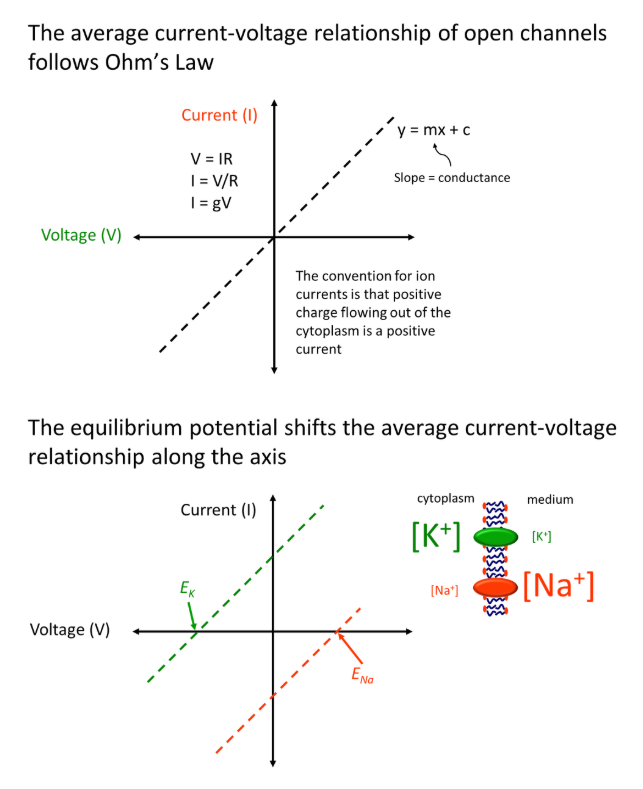
how do equilibrium potentials change the average current-voltage relationship?
for a membrane where three sodium ions are exchanged for two potassium ions in by a sodium-potassium pump, where all channels are always open:
the linear current-voltage graph shifts to the left with a potassium ion gradient across a membrane, because the equilibrium potential is reached when the flow of ions out of the cytoplasm (due to concentration gradient) is equal to the flow of ions in (due to the charge difference, negative inside the cell)- so the equilibrium is reached at a negative potential difference
the graph shifts to the right for a sodium ion gradient across a membrane, because the equilibrium will only be reached when enough sodium ions diffuse through to make the cytoplasm positively charged (and so balance the flow of ions in/out)
the shift of the relationship for any ion is dependent on its concentration gradient
however, these relationships become non-linear in normal systems, where we observe voltage gating
the current will remain at 0 until a particular voltage is reached, at which point it increases/decreases until reaching the linear line
this ambiguous increase/decrease region is due to the channels closing and opening intermittently (the time-averaged current is dependent on the frequency and duration of channel opening events)
how is ion channel selectivity achieved?
channel width:
potassium is larger than sodium, so it is easy to produce a channel that is selective for sodium because it is too small to allow potassium flow
however, this doesn’t allow the production of a channel selective for potassium (that doesn’t let sodium through)
charge density:
the positive charge of these ions attracts water in a hydration sheath around the ion, which must be removed (at least partially) to allow transport through the channel
removal of water requires the input of hydration energy, which is retrieved upon water re-binding after
because potassium is a larger ion, it has a lower charge density, and so its hydration energy required is lower
this means that a potassium selective channel can have charge groups sufficient to remove water from potassium but not from sodium
(a water filled cavity in the ion channel allows fast movement from the selectivity filter out of the channel because water molecules are readily available)
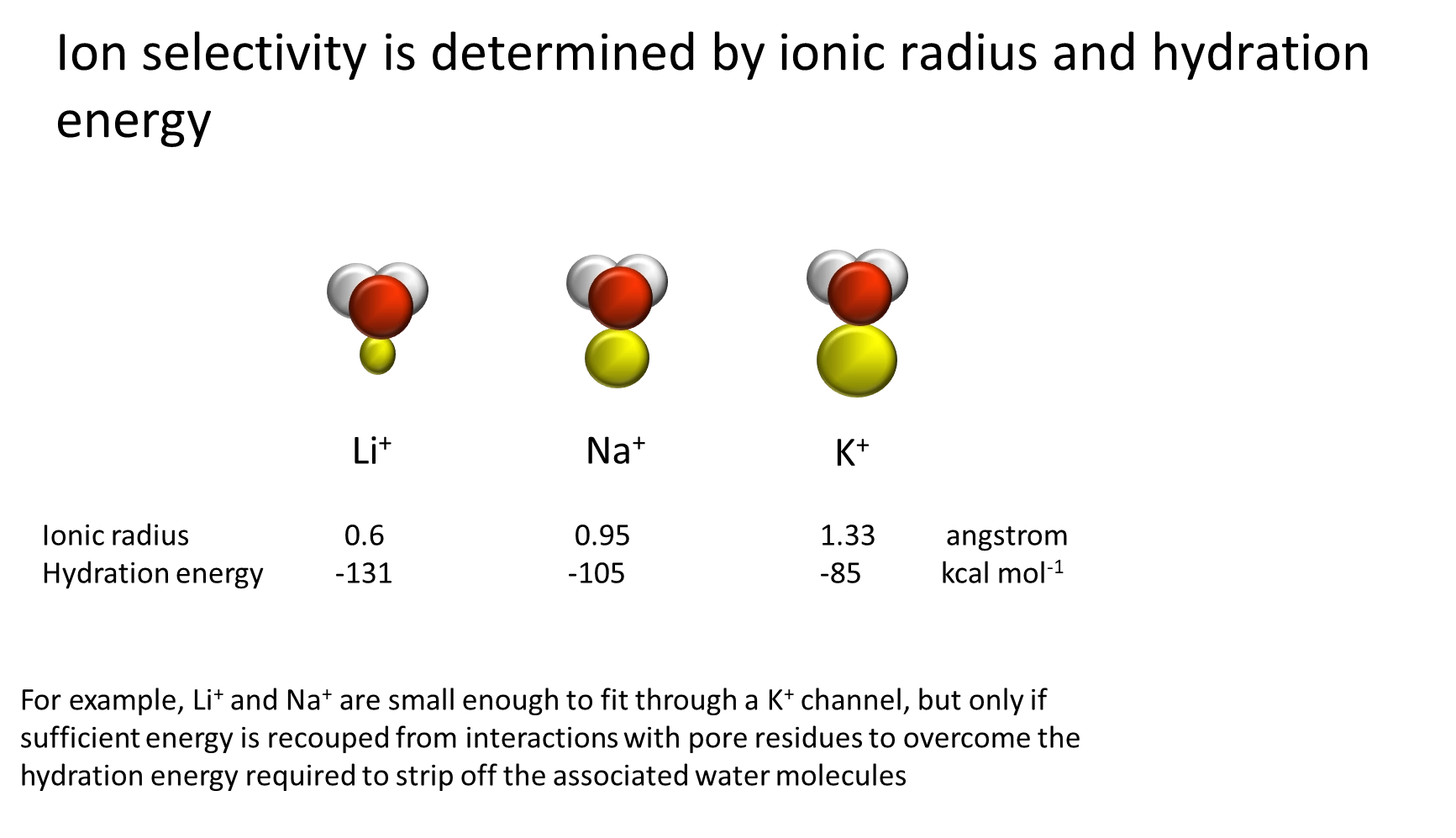
how is voltage gating of ion channels achieved?
most voltage-gated channels have 6 membrane-spanning domains per subunit
the fourth domain (S4) has positively charged arginine/lysine aa.s every third residue of the helix, that make it responsive to membrane potential changes
this is because applying a membrane potential will encourage the charges to try to move
this enforces a conformational change in the protein, causing the channel to switch between open and closed
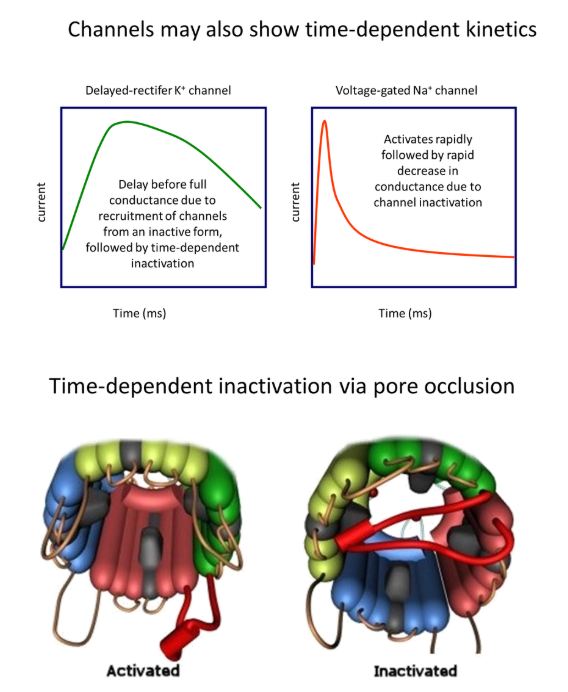
how is time-dependent kinetics of ion channels achieved?
pore occlusion:
cytoplasmic loops of the protein are able to block the ion channel
these are out of the way when the channel is active, but it changes conformation with time to block the channel
the kinetics of the ion channel are dependent on the speed dynamics of the cytoplasmic loop
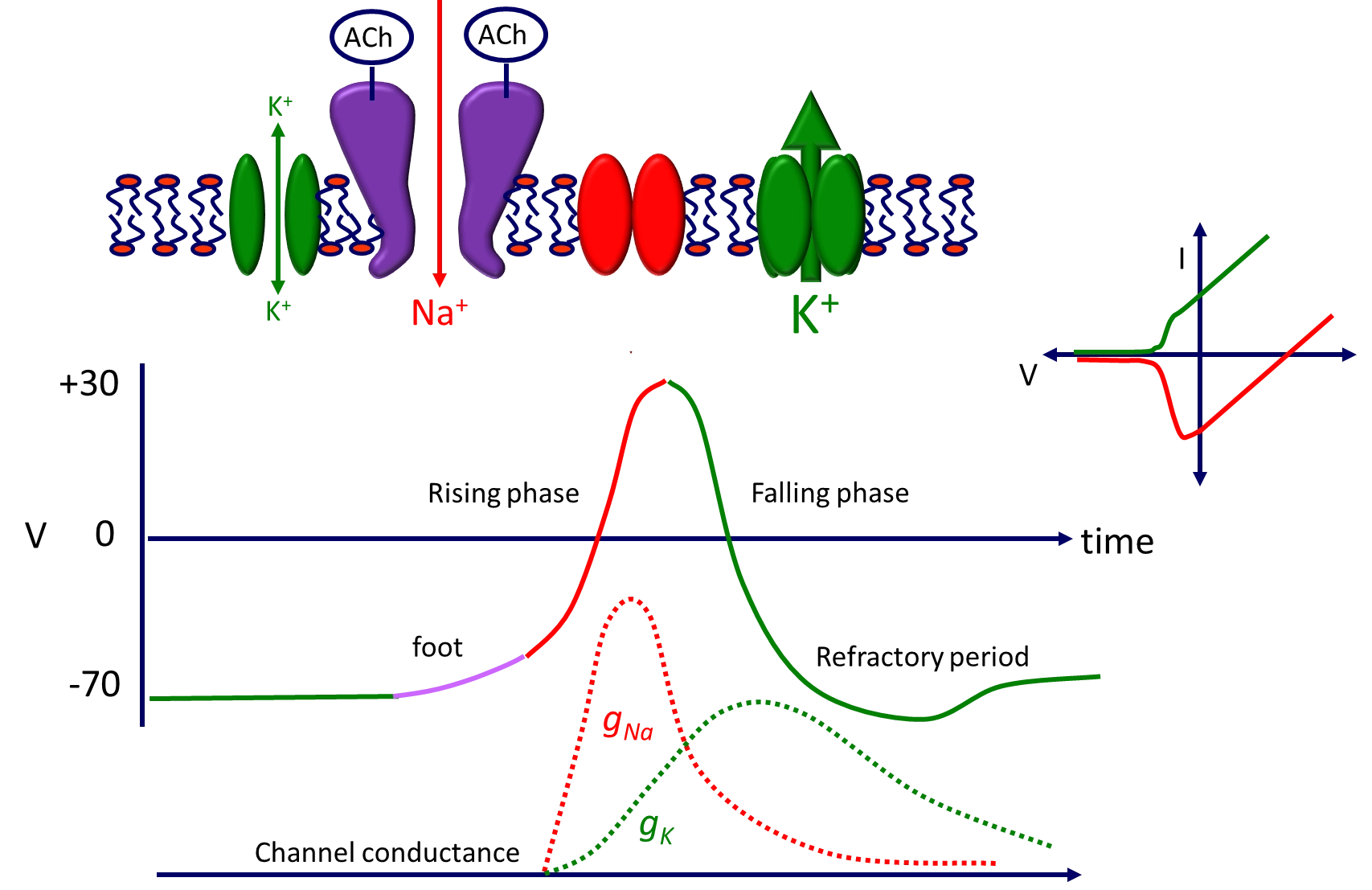
describe the generation of an action potential at the post-synaptic membrane
potassium diffuses through the leak channel towards equilibrium- the channel is too small to reach equilibrium at -90 but stays at -70
when acetylcholine is released into the synaptic cleft it binds to the pentameric ion channel at two binding sites
potassium is close to equilibrium but sodium is not, so sodium will diffuse through the channel to bring in positive charge (start of depolarisation)
following this, the sodium channel rapidly opens due to voltage-gating, letting even more sodium in, until reaching the sodium equilibrium potential, which then closes the sodium channel
at this point the potassium channels slowly open, and potassium floods out to repolarise the membrane until the potassium equilibrium is reached (-90, causing the refractory period)
the membrane is no longer active until the potassium channel closes (slowly)
what are excitatory and inhibitory post-synaptic potentials?
many different dendrites are present on the cell body of one neurone
this allows for the spatial and temporal summation of action potentials at synapses
for example, one action potential from a synapse is not sufficient to generate a new action potential in the dendrite- meaning temporal/spatial summation is needed (all or nothing response) eg. cholinergic synapses for sodium/calcium
this summation can also occur in inhibitory synapses eg. GABA receptors that allow chloride ion influx to prevent membrane depolarisation

how do the directions of growth define plant shapes?
apical growth is elongation up and downwards
branching enables the lateral exploration of space
radial growth allows secondary thickening
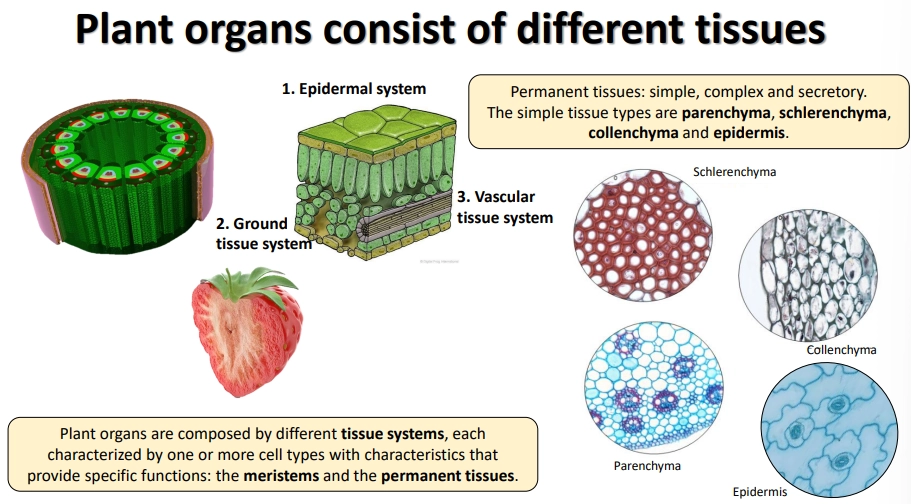
what are the different permanent plant tissue systems and what are they made up of?
the permanent tissue systems (non-meristem):
epidermal system- protective, reducing water loss
vascular tissue system- transport
ground tissue system- everything else
these are made up of simple, complex and secretory tissues
different regions of ground tissue are made up of the simple tissues:
schlerenchyma- thick lignified secondary cell walls + cell death for structural support
colenchyma- live cells with primary thickening of the cell wall at corners- strength without sacrificing flexibility/growth (mostly in young organs)
parenchyma- thin cell walls + loosely packed- photosynthesis + storage (eg. cortex + pith of stems, cortex of roots, mesophyll of leaves, endosperm of seeds)
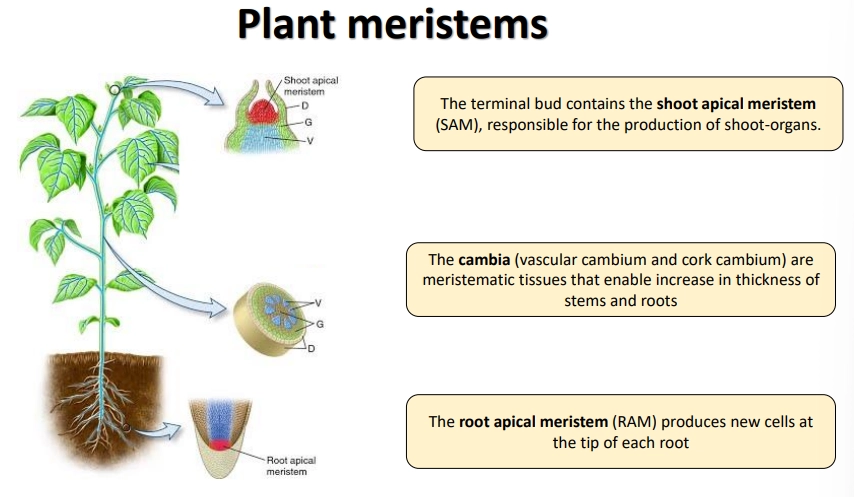
where are plant meristems found?
the shoot apical meristem (SAM)
the vascular cambium of the stem
the root apical meristem (RAM) + lateral root meristem
meristems are also produced at regular intervals in the stem that remain inactive- these allow regeneration following damage from herbivores, wildfires, etc, compensating for their inability to move, e.g., adventitious roots
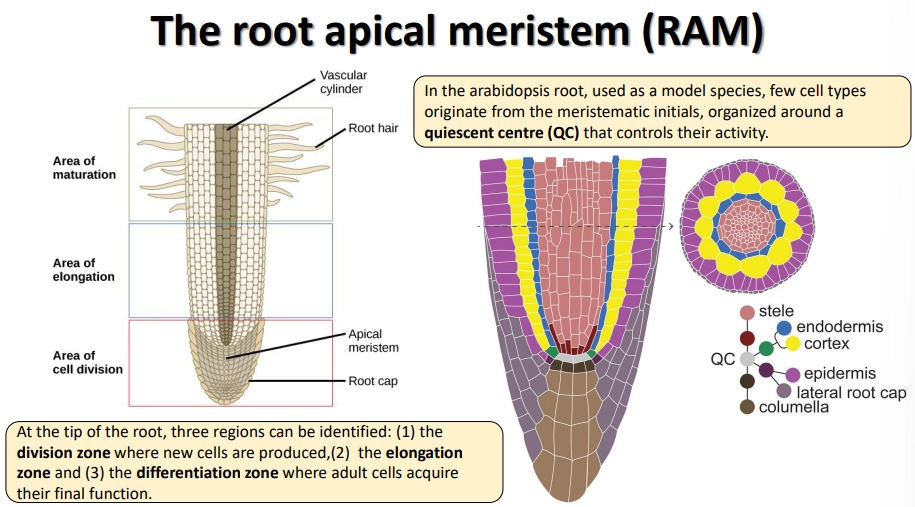
what is the structure of the root apical meristem?
at the tips of roots, there are three regions: the division zone, the elongation zone and the maturation zone (for the development of secondary features)
at the dividion zone of Arabidopsis, the rate of meristem cell division is controlled by a cluster of 4 cells called the quiescent centre
the root cap is made up of the lateral root cap itself and the columella, a cluster of cells at the tip that contain amyloplasts used to sense the direction of gravity
the new cells then differentiate into the stele, endodermis, cortex and epidermis (including root hairs)
root hairs are produced at epidermal cells that are in contact with two inner cortex cells, due to receiving sufficient diffusible signals from the edges of these cells
lateral root primordia (LRP) are inactive meristems at regular intervals that don’t activate until the cells from the outermost layer of the stele (pericycle) begin to divide and break through, due to auxin peaks
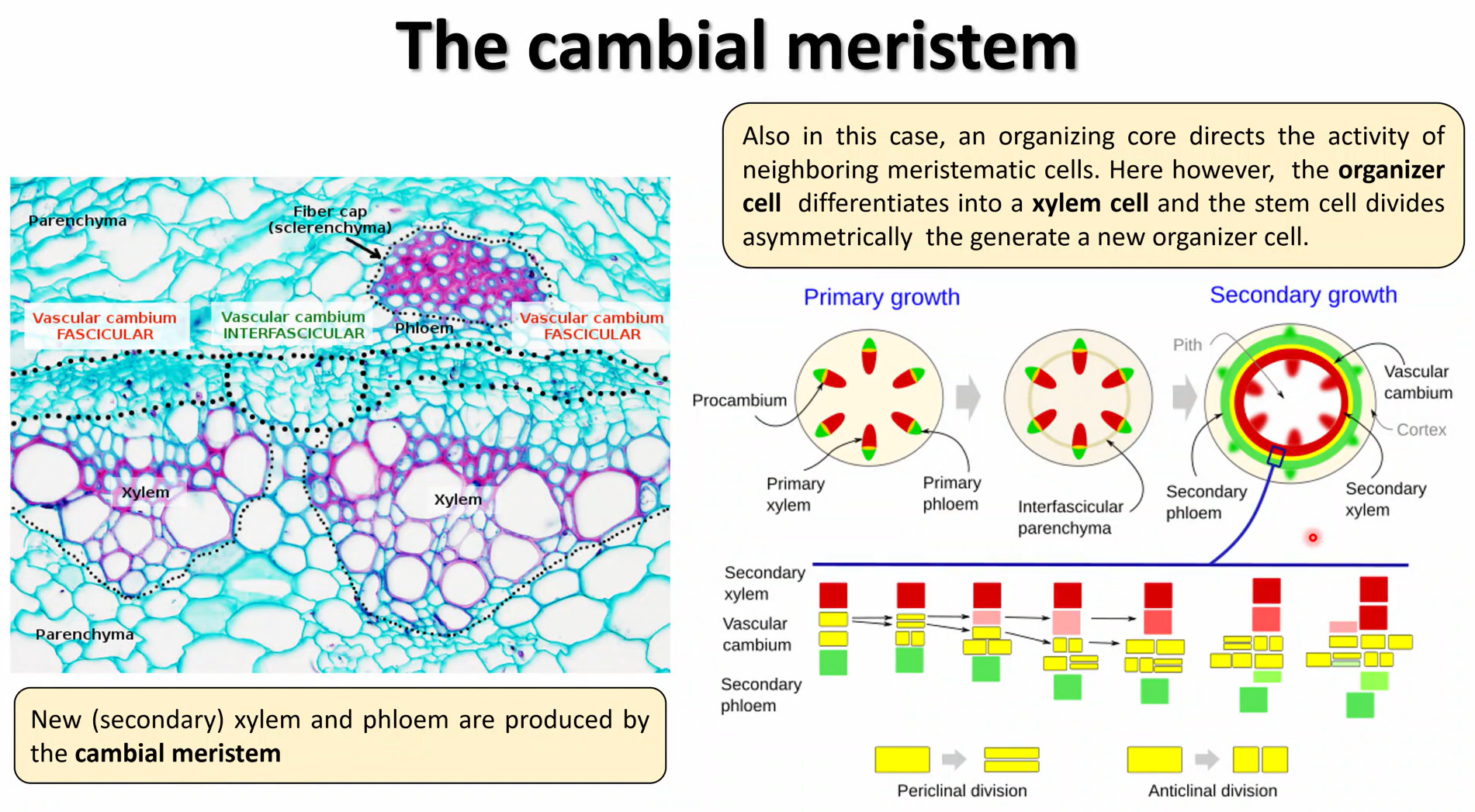
how does the cambium cause secondary thickening?
division is activated when the interfascicular parenchyma ring is formed
the cambium cells close to the xylem side divide periclinally (horizontally) and differentiate early
the cambium cells close to the phloem divide anticlinally (vertically) and do not differentiate as early, to increase the number of cells per layer
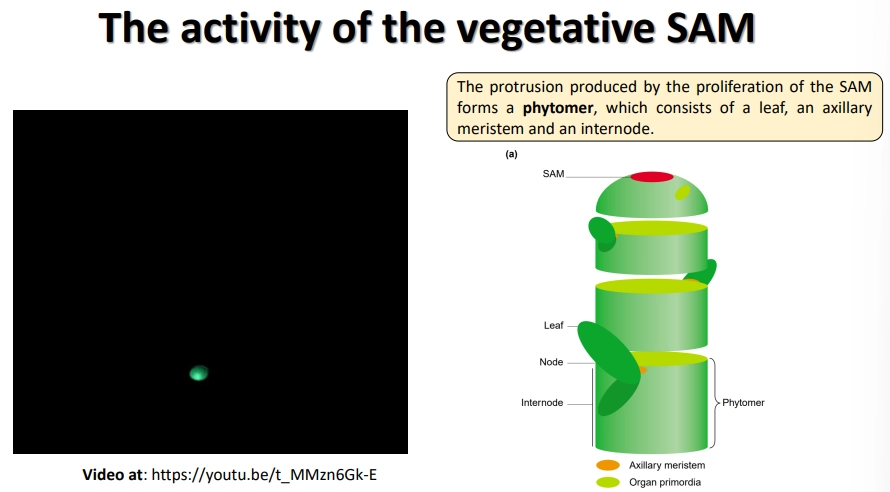
what is the structure of the shoot apical meristem?
the shoot apical meristem is highly protected in layers of leaf primordia
the inner meristem dome can be separated into three layers of meristem cells
the organising centre is a pool of up to 100 cells, which determines the speed of division + identitiy of neighbouring cells
the SAM differentiates the stem in regular units called phytomers, which each contain a primordial leaf at a node, an inactive axillary meristem and an internode region
how are leaves produced at the shoot apical meristem?
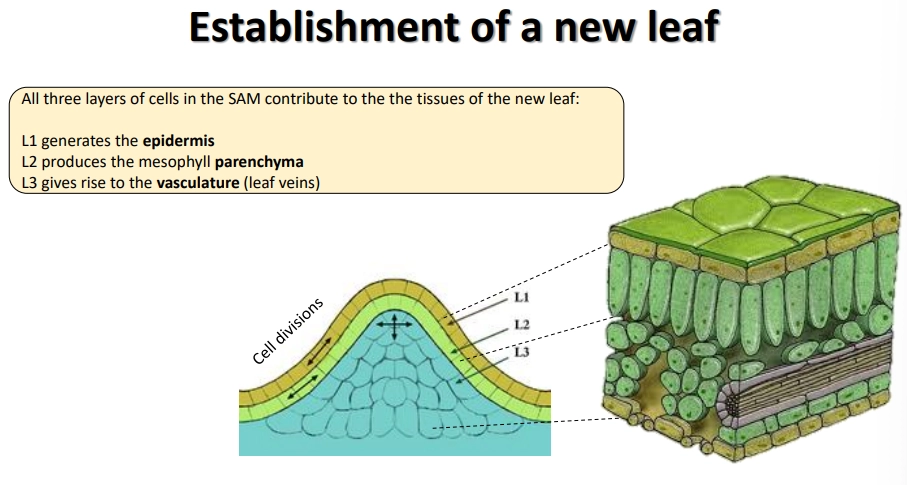
the inner meristem dome of the SAM can be separated into three layers of meristem cells
to produce leaves, the 1st layer generates the epidermis, the 2nd produces the mesophyll parenchyma, and the 3rd produces the vascular tissue
the adaxial (upper) side is optimised for light capture and the reduction of water loss, while the abaxial (lower) side is optimised for gas exchange- the polarity of leaf primordia is established early, defined by which side is closer to the SAM
once the leaf cells have stopped dividing, they normally continue to replicate their DNA, because polyploidy supports more active metabolism
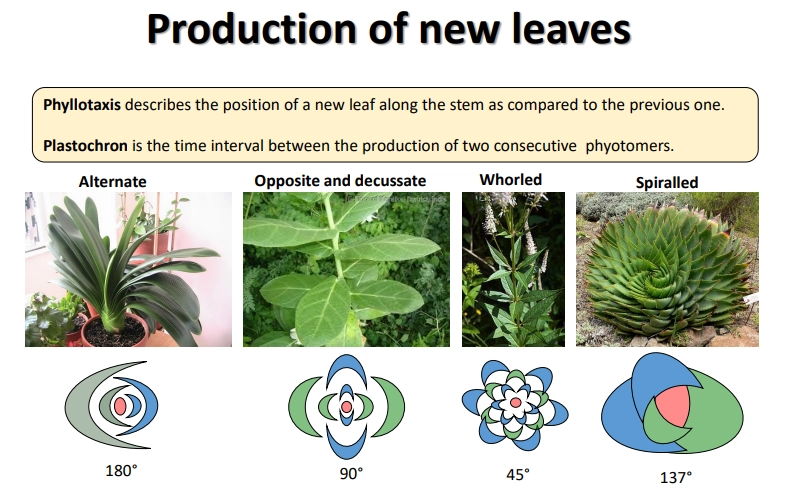
what is phyllotaxis and what are the different forms?
spiralled is the most common- less likely to shade previous leaf
how are reproductive structures produced at the shoot apical meristem?
the SAM transitions from vegetative to reproductive (it becomes the inflorescence meristem)
the infloresence meristem loses phyllotaxis and produces many determinate flower meristems, that produce concentric whorls of new organs
these whorls are the sepals (together form the calyx), the petals (together form the corolla), the stamens, the carpel (multiple carpels form a pistil)- (the sepals + petals are the perianth)
shoot architecture is dependent on: if the vegetative meristems along the stem also turn into reproductive meristems, if the reproductive meristems terminate with flowers or continue producing more meristems, and how the terminal flower meristems branch
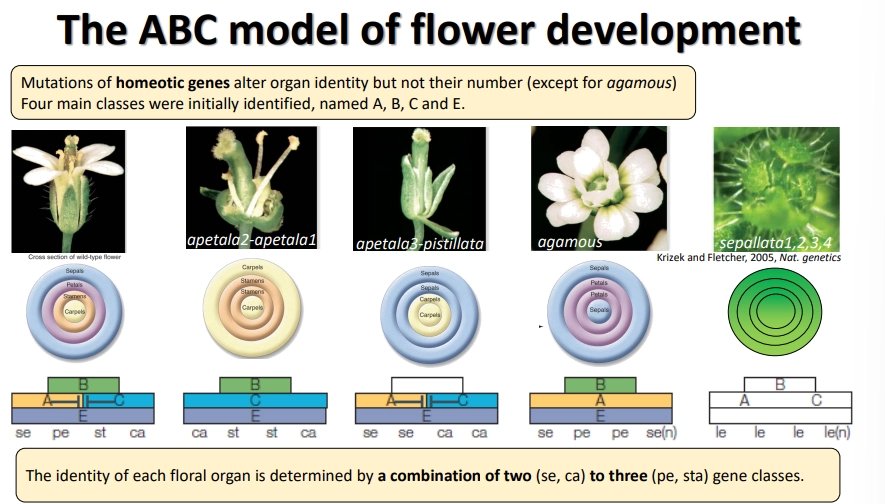
how is flower development controlled by genes?
five main classes of homeotic flower genes were found in Arabidopsis: A, B, C, D and E
the development of each floral organ in the whorl is dependent on a combination of two or three gene classes that get expressed
A and C mutually repress each other’s expression
all organs need E class genes to be present
eg. sepals need A and E genes- if A is lost, they will have C and E genes and become carpels
the D class was not identified for a long time due to high gene redundancy- redundancy is also present within the other classes in different species
class C genes are also responsible for terminating the meristem, so C gene homeotic mutants produce an indeterminate number of organs
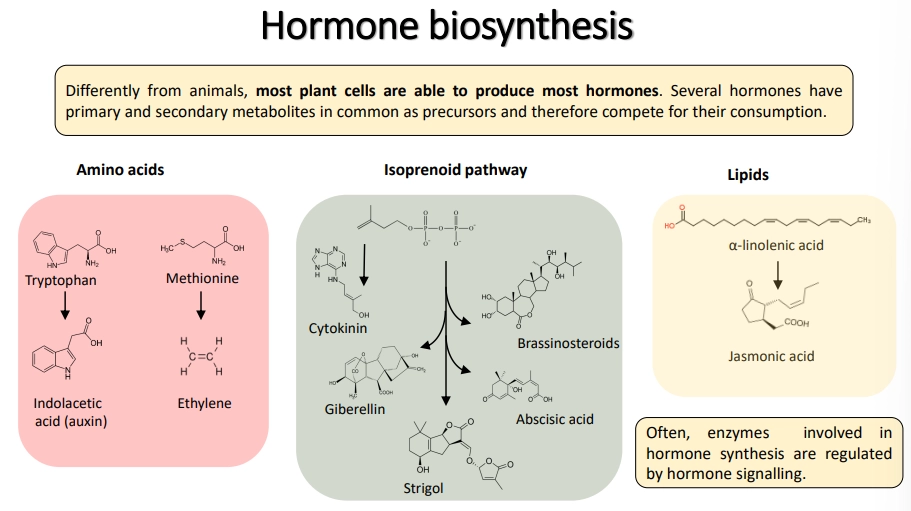
how are hormones synthesised in plants and synthetically?
hormones tap into the same pool of precursors as many other biosynthetic/metabolic pathways, so they compete for their consumption and have to be regulated to stop them from negatively affecting growth
synthetic hormones do not necessarily have the same structure, but they bind to the same receptors- often these cause stronger responses because plants are not equipped to degrade/deactivate them
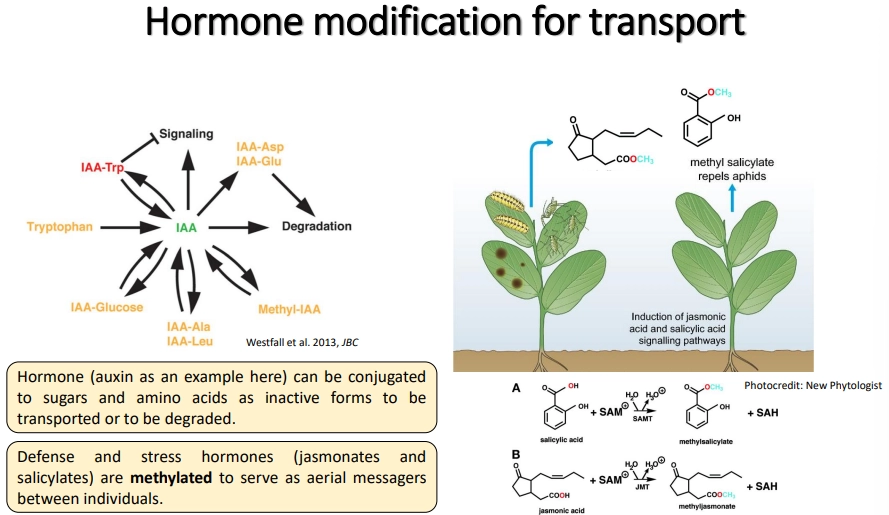
how are plant hormones modified for transport?
it is important that hormones are temporarily inactivated during transport and only activated upon arrival
this can be done by conjugation to sugars/amino acids
these signals can also sometimes trigger enzymatic degradation
methylation of defence and stress hormones (jasmonates/salicylates) reduces interaction with the aqueous environment to facilitate emission as volatile signalling compounds
hormones are transported by the xylem (root to shoot) and phloem (source to sink)
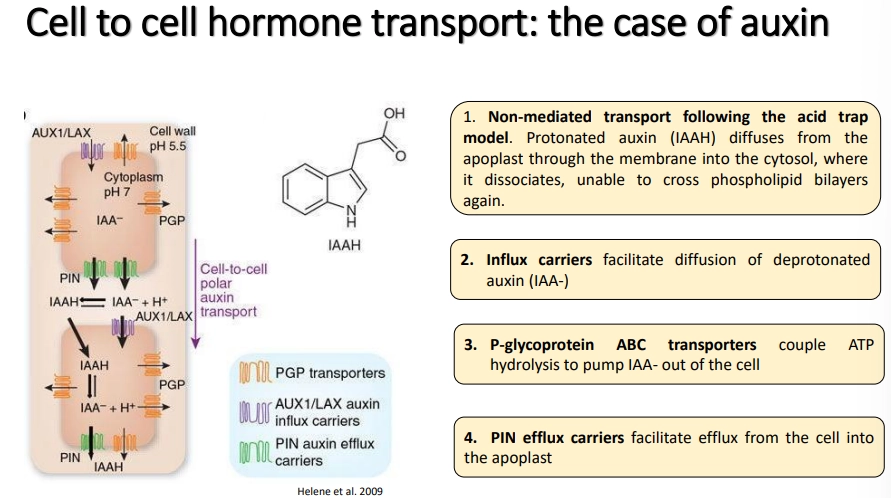
how are auxins transported?
into cells:
non-mediated- protonated auxin in the apoplast (low pH, amplified by high auxin concs) can cross the membrane into the cytosol, where it is deprotonated and gets trapped
influx carriers facilitate the diffusion of deprotonated auxin (from neutral apoplast) into the cell
out of cells:
ATP hydrolysis-coupled transporters act as pumps to export auxins outside (regular distribution)
PIN efflux carriers facilitate efflux of auxins in an asymmetric distribution around the cell dependent on cell type, to create polar gradients- this determines the identity of stem cells at root auxin maxima and these gradients allow auxins to act as concentration-dependent morphogens
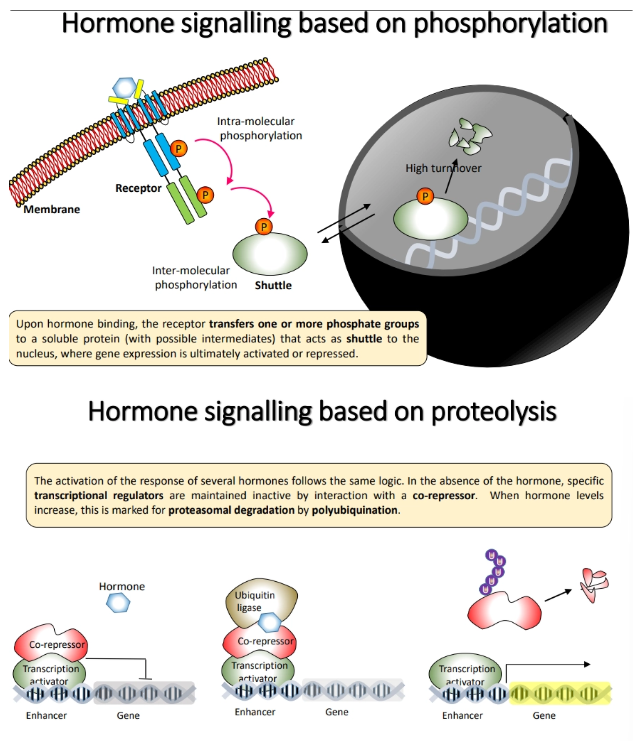
how are hormones perceived in plants and how do they change gene expression?
membrane-associated receptors bind to the hormone and activate a signalling cascade mediated by phosphorylation
a shuttle protein is eventually phosphorylated, which activates/represses gene expression in the nucleus eg. cytokinins
soluble receptors bind to the hormone and change their conformation and hence protein interactions, to cause a signalling cascade mediated by proteolysis
the hormone binds to and activates ubiquitin ligases (a proteasome), which polyubiquitinates a bound corepressor, tagging it for degradation so that the transcriptional regulator is unblocked and able to act eg. auxins
how do plant hormones allow interaction with other organisms?
to fight biotrophic pathogens (don’t kill the plant)- plants trigger localised apoptosis of cells to trap the pathogen in dead tissue- by salycilates
to fight necrotrophic pathogens (kill the plant)- plants trigger production of toxins, secondary metabolites, against the pathogen- by jasmonates
to mediate symbiotic interactions with mycorrhizal fungi, plants use strigolactones for signalling- however, parasitic plants have evolved to recognise these compounds as a signal of a host plant, to trigger germination
how are plant hormones used in agriculture?
auxins and cytokinins are used for in vitro regeneration from explants in tissue culture
calluses are formed by auxin + cytokinin application, shoots are induced by cytokinins, and roots are induced by auxins
auxins are the main component of rooting powders for propagation
ethylene is used to control fruit ripening
changes include softening by cell wall changes, pigmentation changes (breakdown of chlorophyll + synthesis of carotenoids/anthocyanins), aroma release, conversion of starch to sugars
low oxygen inhibits ethylene production in transport, release of ethylene promotes uniform ripening
how is embryo polarity established in plants?
the vacuole and nucleus are asymmetrically distributed in the early embryo, with an apical nucleus, leading to asymmetric division
this immediately defines the apical-basal body axis
the long basal cell (the suspensor) is the producer of auxins and supplier of nutrients from the maternal tissue
the apical cell is the receiver, in which auxins trigger anticlinal (vertical) and periclinal (horizontal) divisions to produce the main embryo body
following multiple divisions, the upper layer of cells in the dermatogen (16-cell) becomes the source of auxin
phosphorylation cascades downstream of auxin are responsible for determining cell fates
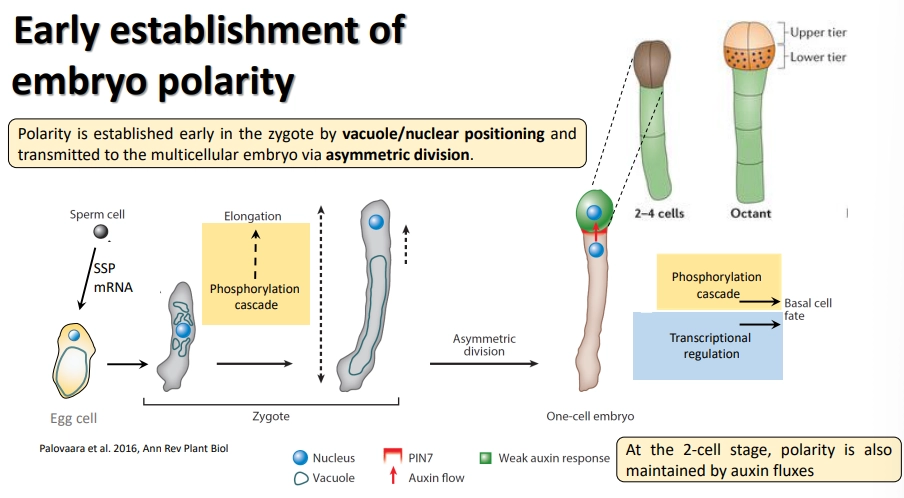
how are the shoot and root apical meristems defined in plant development?
the SAM and RAM are marked in the embryo by the expression of homeotic genes in the organising centre
shoot and root development mainly require joint auxin and cytokinin activity
activity of the RAM is regulated by small RNA, transcription factor, signalling peptide and auxin gradients, which can regulate gene expression
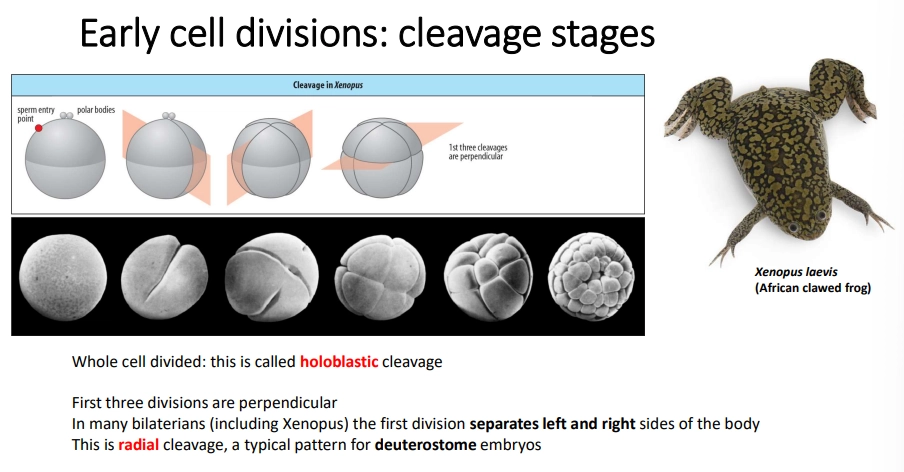
how is the oocyte initially divided in most bilaterian animals?
most animals undergo holoblastic cleavage
bilaterians typically go through three initial divisions
polar bodies (small cells with a discarded nucleus) determine the top
radial cleavage is seen in deuterostomes, where the three divisions are perpendicular
spiral cleavage is often seen in protostomes (ecydosozoa + lophotrochozoa)- the third division is twisted, and the divisions may be equal or unequal, to produce different cells
some species, eg. teleost fish, undergo teleoblastic (instead of holoblastic) cleavage, where cleavage is restricted to just part of the egg due to a high amount of yolk
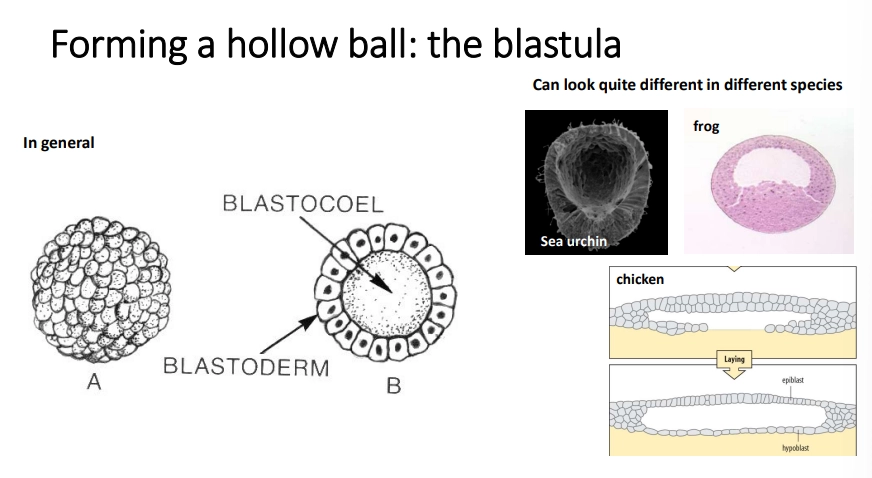
what is the blastula?
bilaterian animals enter a blastula stage during early development, where the zygote has a fluid-filled blastocoel space in the middle surrounded by a layer of cells called the blastoderm
this undergoes gastrulation to move cells into the blastocoel
this produces three cell layers: the ectoderm, mesoderm and endoderm, filled by the archenteron, with a blastopore (mouth/anus dependent on protostomes/deuterostomes)
this movement of cells is an example of morphogenesis (another being neural crest cell migration)
what are the three processes involved in development in animal cells?
differentiation- expression of different transcription factors that activate certain groups of genes (eg. myogenic genes controlled by MyoD) and repress others
pattern formation- lineage dependent mechanisms (programmed asymmetric cell division) + organising fields of cells (using intercellular signalling/morphogen gradients- Wolpert’s french flag model eg. maternal bicoid with mRNA localised at anterior + protein diffusion gradient)- along the three embryo axes, in many cases using particular intermediate Hox genes that encode homeodomain TFs
movement/morphogenesis- gastrulation + neural crest cell migration
what are the two main routes to identifying the genes that control animal development?
biochemistry:
studying compounds that have been extracted from tissues
this relies on fractionation and repeated assay to identify the protein
this works best with intercellular signalling systems, because they can be applied topically
not very useful with intracellular proteins/TFs
forward genetics:
observing the phenotype change from a mutant genotype
most of the genes identified were either for transcription factors or intercellular communication
these are very highly conserved across animal species (ancestral trait)
how do embryonic axes get determined in animal development?
A/P signalling is controlled by Wnt localisation (generally, but also bicoid in drosophila)
bilaterians- Wnt localised at the posterior end
non-bilaterians- Wnt localised at the oral end (they have an oral/aboral axis)
D/V signalling is controlled by Bmp localisation
deuterostomes- at the ventral side
protostomes- at the dorsal side
this is developmentally equivalent, mostly due to our labelling of dorsal and ventral according to gravity
how did the developmental gene toolkit evolve?
tandem gene duplication- during meiotic recombination, mistakes in crossing over cause one chromosome to have two copies of a section of DNA, while the other has none
lack of the DNA is likely to be fatal in the inheriting offspring
however the extra copies aren’t necessarily deleterious in the resulting offspring (leads to copy number variants)
whole genome duplication occurred twice in early vertebrate evolution
these events lead to redundancy
these permit the evolution of genes with new developmental functions by mutations (because redundancy means the mutation doesn’t affect the other copy of the original gene)
eg. changing the amino acid sequence of the homeodomain of Hox genes changes the DNA sequence it binds to
how the gene is expressed in different cells can also be changed
eg. changing the position of a particular Hox gene changes how far along the A/P axis the corresponding trait is expressed
repurposing of genes for new functions, eg. using Hox genes to control limb patterning
mutations in gene enhancers can affect gene expression eg. to allow binding of a new TF, prevent binding of an existing one, or create a new enhancer region for a TF
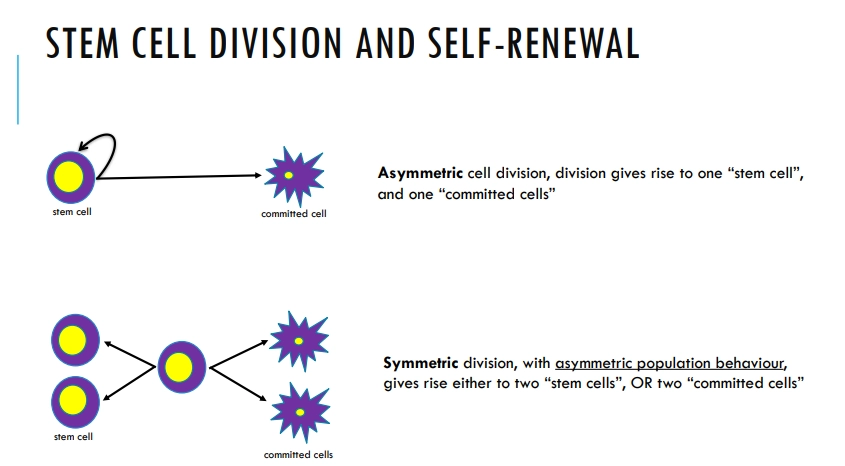
what are the two methods of self-renewal in stem cells?
later in development the stem cells create a lineage of transit amplifying cells that are responsible for most divisions
the stem cells themselves divide infrequently and slowly, to protect their genomes from mutation, as they are retained for life
transit amplifying cells are non-differentiated but also don’t self-renew
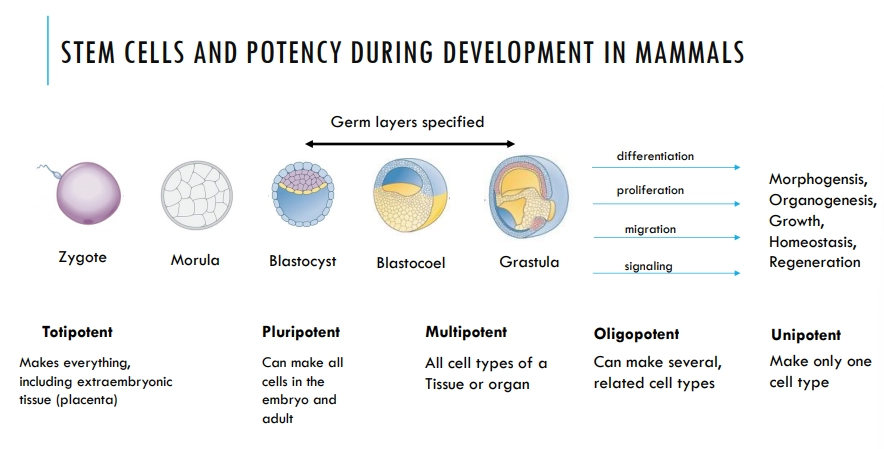
what are the different levels of stem cell potency during mammalian development?
stem cells become more determined and specific, with decreasing potency through development
the totipotent and pluripotent stem cells are lost and replaced by multipotent, organ-specific stem cells, that are retained throughout the rest of the organisms life
embryonic stem cells have three states of readiness to differentiate: naive → formative → primed
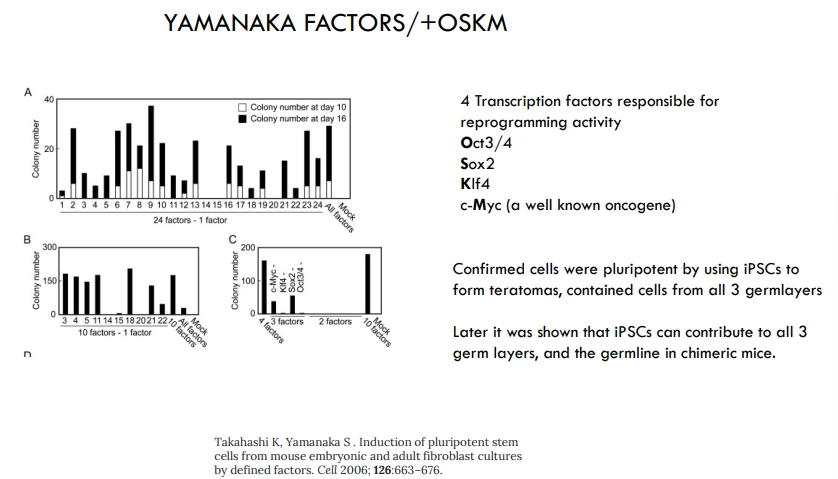
how can non-stem cells be reprogrammed to pluripotency?
4 Yamanaka TFs are responsible for reprogramming
if these are expressed together in any non-stem adult cell, it will become an induced pluripotent stem cell
these can form teratomas (small tumours containing cells from all three germ layers), as well as chimeric mice
what are the 4 major groups of plant pathogens?
ascomycetes and basidiomycetes fungi differ in their spore storage + release

what are the different lifestyles of plant pathogens?
saprotrophs aren’t strictly pathogens because they don’t kill the host, but many other pathogens possess a facultative saprotroph stage

what are the 9 ways plant pathogens can enter host tissue?
into tissues:
through stomata
through lenticells (gas exchange pores in potato skin/bark)
through hydratodes (water guttation points)
throud wounds
through lateral roots (at the break between the new root and the epidermis)
through nectarthodes (in flowers)
into cells:
using haustoria (particularly fungal pathogens) that extract nutrients from punctured epidermal cells
using appressoria that penetrate deeper into the tissue
using toxins to kill host cells
how can pathogens manipulate plant ‘behaviour’?
some extend plant cell life on green islands (even though surrounded by senescent cells)
some produce pseudoflowers to encourage spread by bees
some induce tall growth by releasing gibberelins to spread spores using the wind (‘foolish seedling’)

what are the different tools plant pathogens can use to manipulate their hosts?
phytohormones:
eg. gibberelins that induce plant elongation to release spores to the wind
eg. auxins/cytokinin to induce plant tumour growth (eg. following DNA transfer by Agrobacterium)
phytotoxins:
eg. coronatine which reopens stomata after pathogen recognition closes them
eg. syringolin A that suppresses stress hormone production at wounds
eg. rhizoxin, produced by an endosymbiont, that blocks microtubule formation (has a mutant tubulin itself)
small RNAs:
packaged in vesicles to be delivered into the cell
these small RNAs hybridise with host mRNA to degrade it/prevent translation
effector proteins:
pathogen-produced, secreted proteins that manipulate the host cell- including enzymes, inhibitors, TFs and opines
apoplastic effectors act outside the cell
cytonuclear effectors act inside the cell/nucleus
inhibitors + enzymes act to prevent immune recognition/degradation of the pathogen
cytonuclear TFs modify the expression of host genes eg. up-regulate sucrose export genes
opines are produced by Agrobacterium, which are N-rich and can’t be metabolised by the plant (so are a food source just for the bacterium)

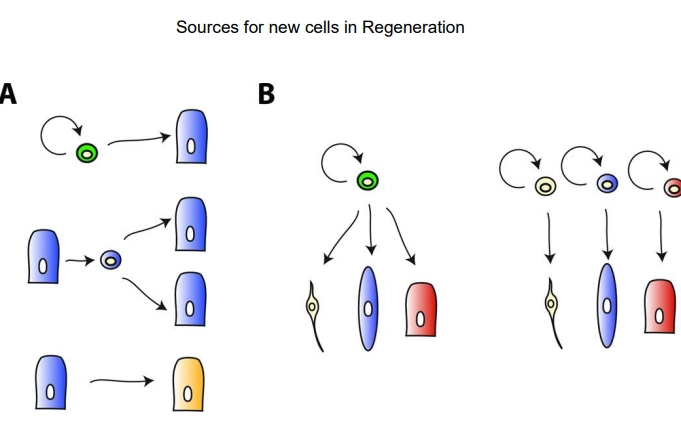
what are the sources for new cells in regeneration?
mitosis of stem cells, dependent on the phyla/species, this may be at different levels of:
specialisation of the initial cell:
adult, undifferentiated cells are always self-renewing and can produce any cell type- in invertebrates/non-bilaterians these may be able to migrate from a different region to the wound site
specialised cells can undifferentiate, proliferate and redifferentiate to make more of themselves
specialised cells can undifferentiate, proliferate and trans-differentiate into a different cell type
levels of potency of the initial cell:
multipotent/pluripotent adult stem cell produces many cell types
oligopotent/unipotent, lineage-specific, committed stem cells produce one cell type or a few within a tissue
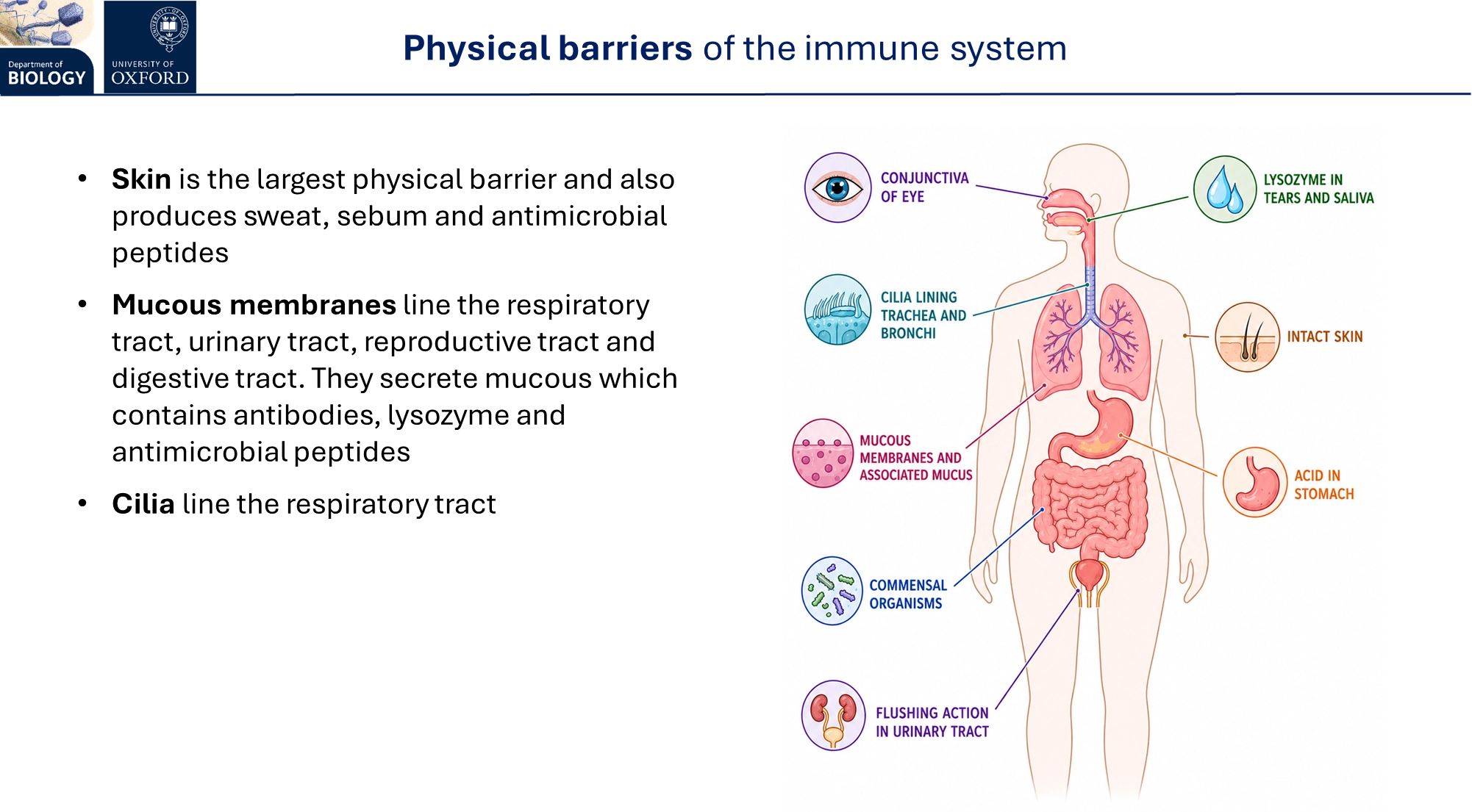
what are the physical barriers of the animal immune system?
sebum contains fatty acids that inhibit bacterial growth- together the sweat + sebum produce a slightly acidic, antimicrobial environment
stomach acid
commensal organisms in the digestive, respiratory and reproductive tracts compete with and create unfavourable environments for other microbes
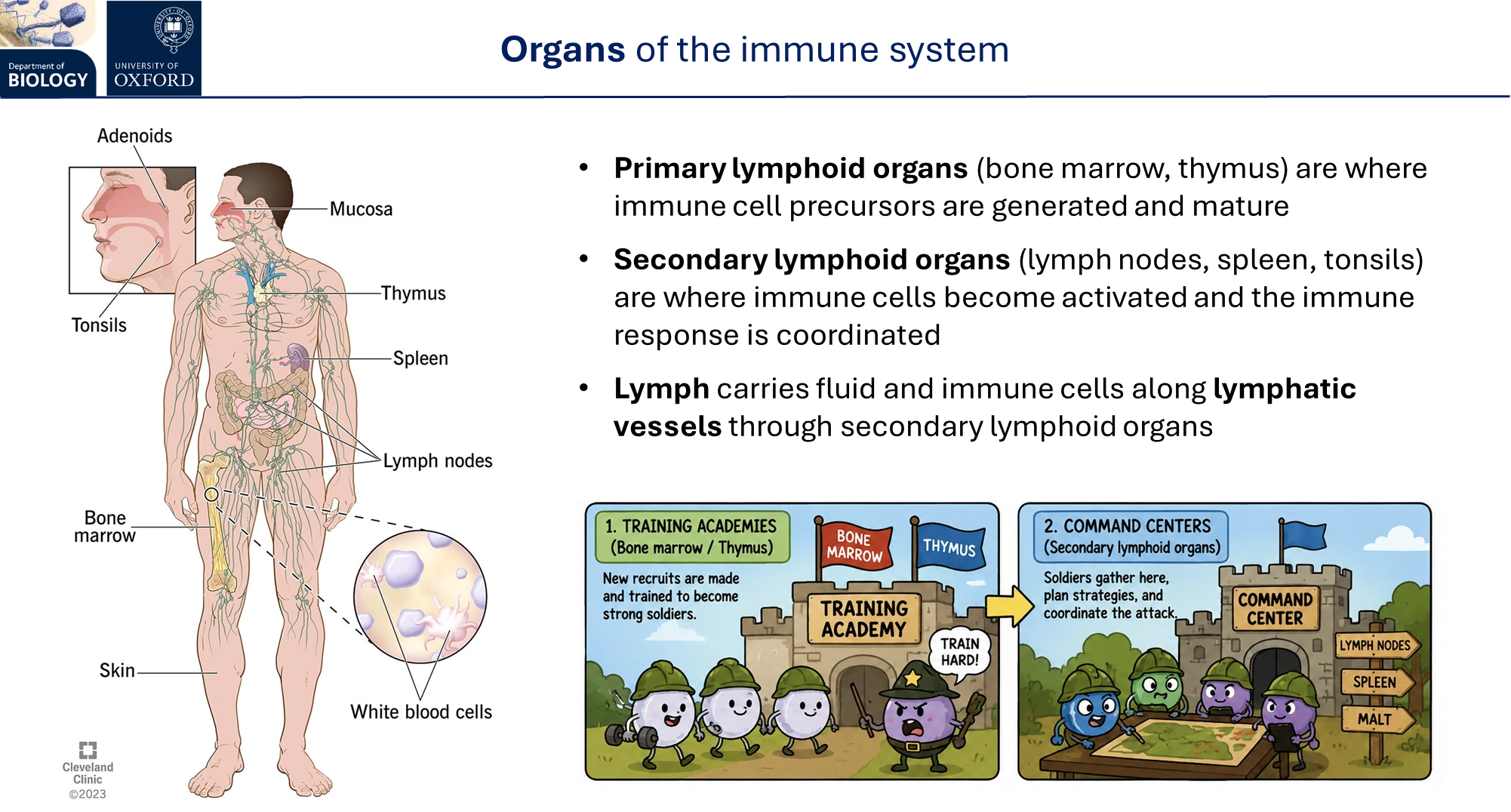
what are the organs of the immune system?
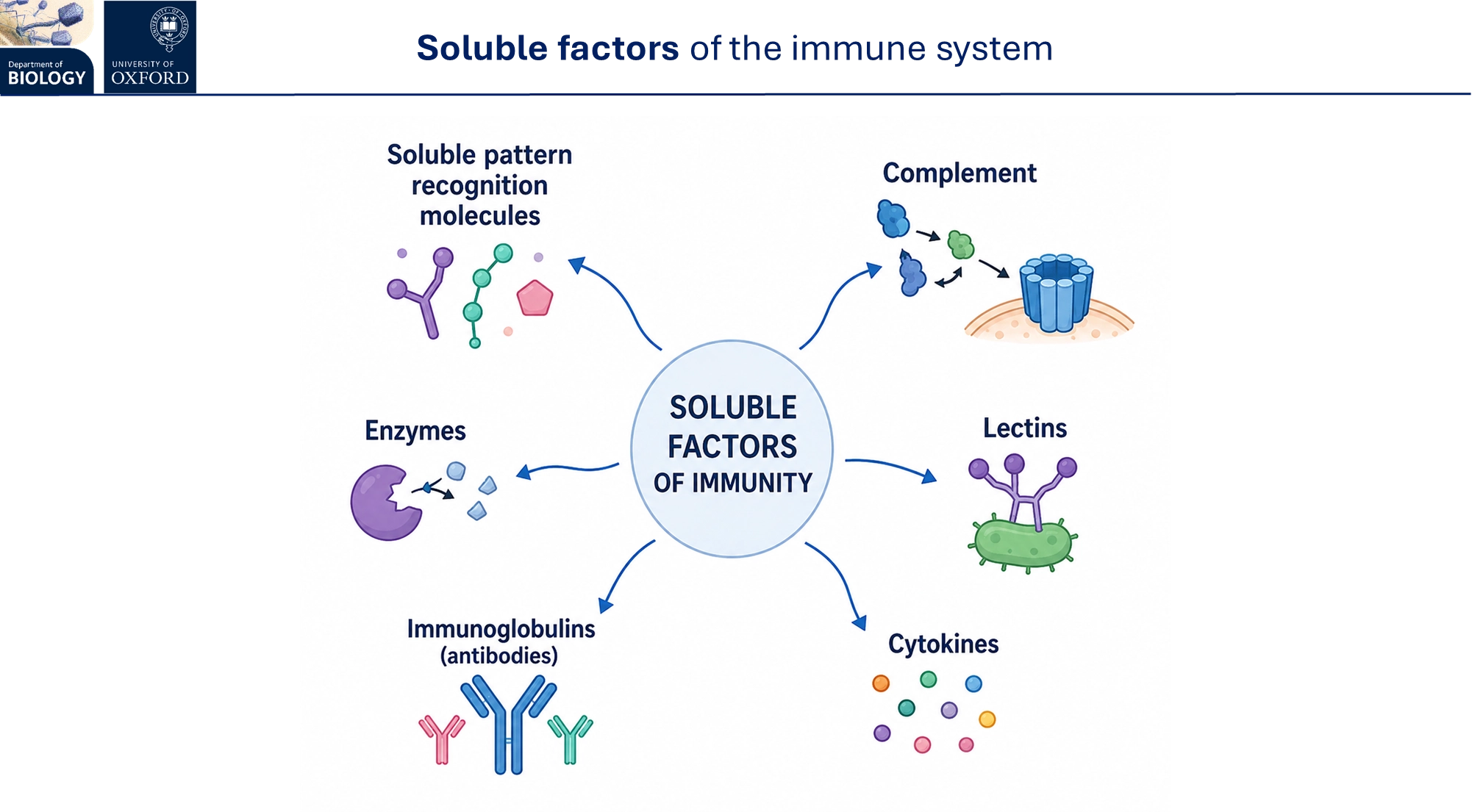
what are the soluble factors of the immune system?
the complement is a system of plasma proteins that complement the innate immune response, triggering microbial lysis and increased phagocytosis
lectins are proteins that bind to sugar on microbial surfaces to assist in recognition
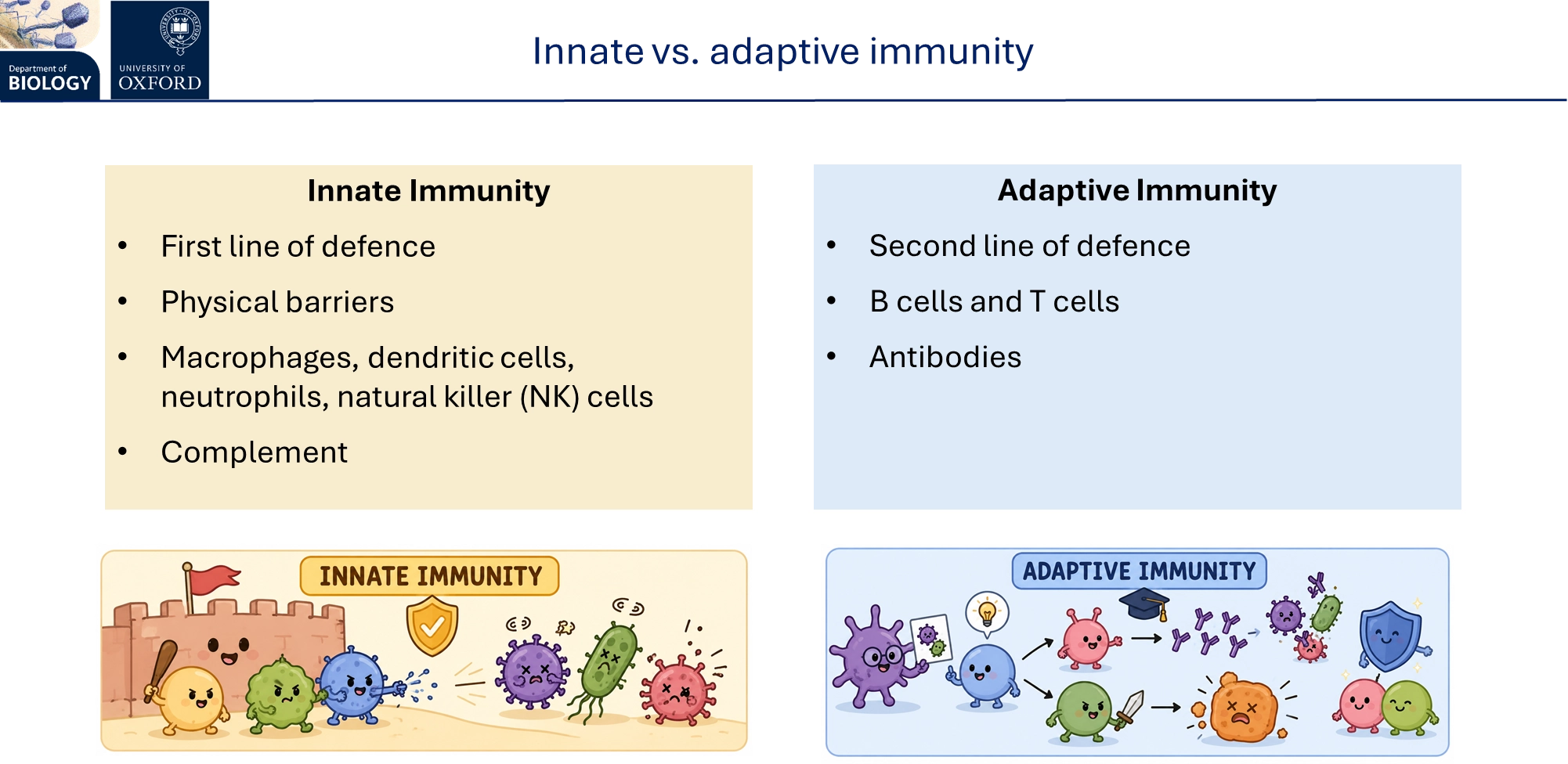
what is innate vs adaptive immunity? what stages are involved in each
innate immunity is fast, non-specific, broad and has some non-specific memory by epigenetic modification- all animals have this, it involves:
rapid detection
phagocytosis
triggering inflammation
clean up
activating adaptive immunity
adaptive immunity is slow, specific, narrow and has memory- evolved independently in the agnathans and gnathostomes, it involves:
antigen-specific recognition
targeted effector responses
long-term protection
adaptive immunity is made up of humoral (extracellular pathogens/toxins) and cell-mediated immunity (intracellular pathogens/abnormal cells)
how does the innate immune response detect a pathogen?
rapid detection of harmful non-self, using both:
highly-conserved pathogen-associated molecular patterns (PAMPs), not antigens, are recognised (eg. flagellin, chitin, peptidoglycan) as non-self by pattern recognition receptors (PRRs, eg. toll-like and NOD-like)
animals without an adaptive immune system, eg. sea urchins, have very diverse PRRs to make up for it
stressed, damaged, or dying cells release danger signals called damage-associated molecular patterns (DAMPs)
how does the innate immune response trigger phagocytosis?
phagocytosis (pseudopodia engulf pathogen in a phagosome, fuses with lysosome into a phagolysosome for digestion, requiring ATP)
this is primarily carried out by neutrophils, macrophages (+ immature monocytes) and dendritic cells
this is facilitated by opsonization, coating the pathogen using antibodies or complement proteins for phagocyte recognition by receptors
however, some pathogens can survive phagocytosis by escaping out of phagosomes, preventing phagolysosome formation, or withstanding oxidative stress
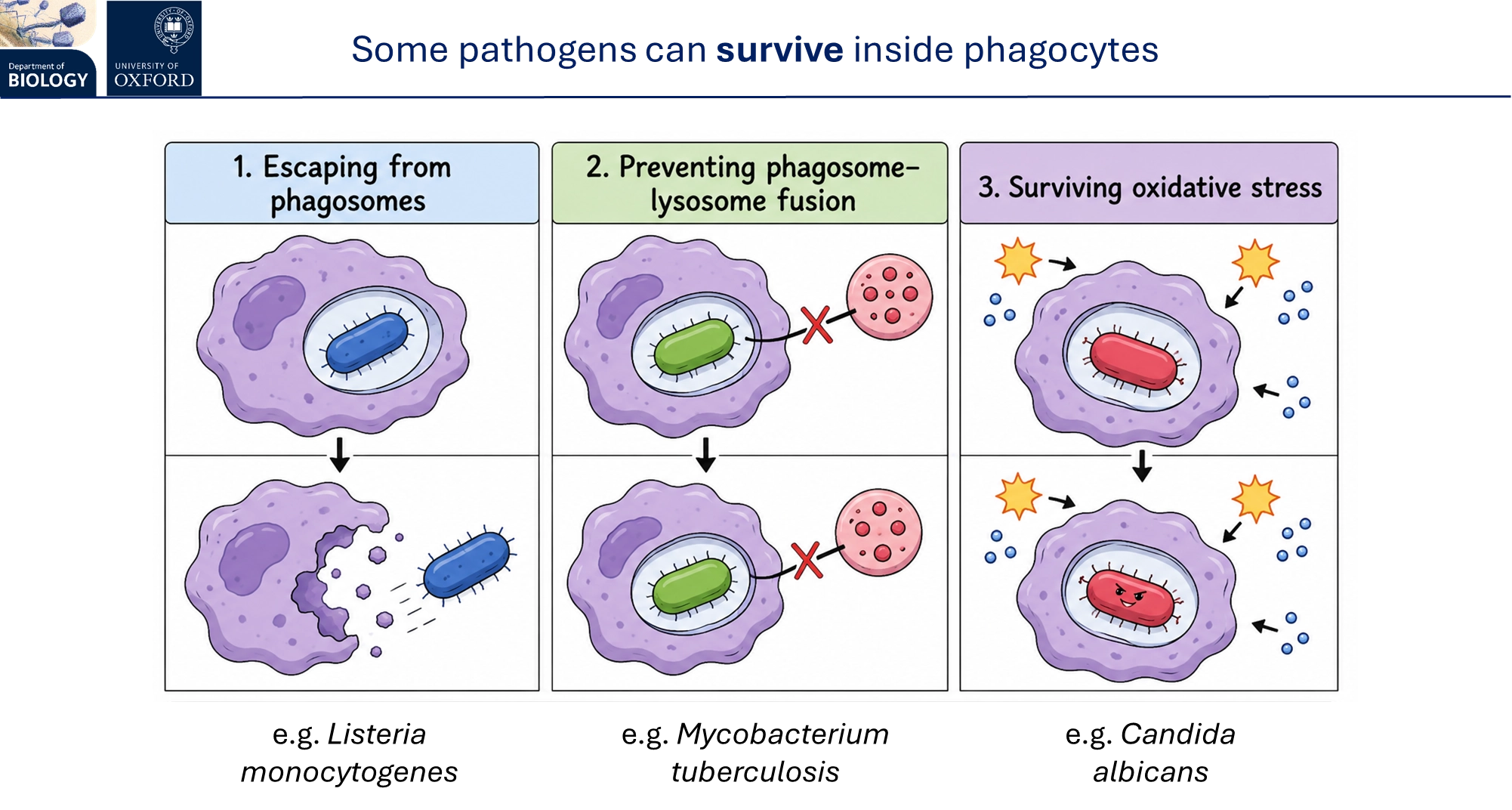
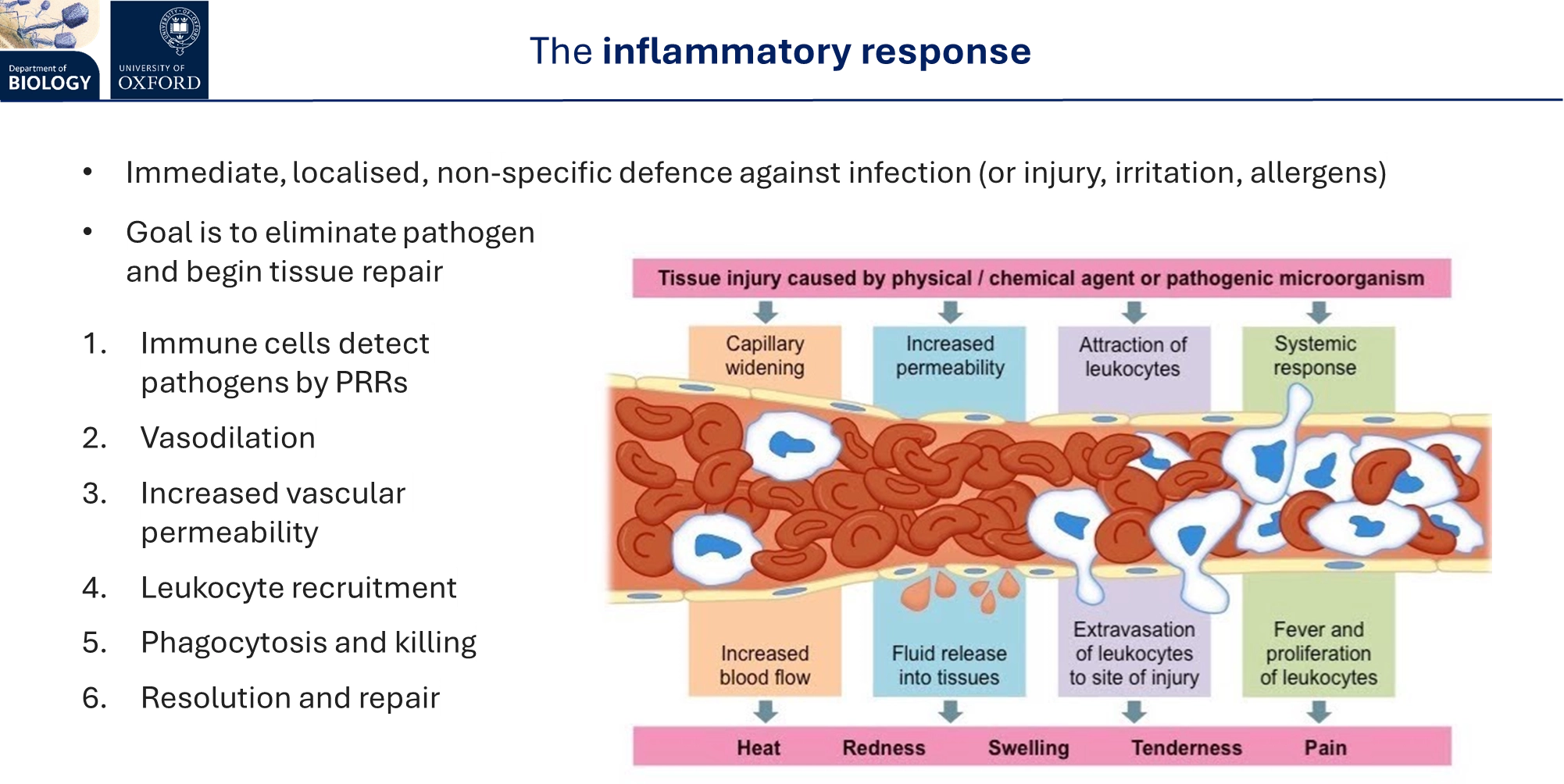
how does the innate immune response trigger inflammation?
following activation by PAMPs and DAMPs, the innate immune cells release inflammatory mediators:
cytokine and chemokine release by innate cells eg. macrophages
histamine release by mast cells
complement release
this triggers the inflammatory response, an immediate, localised, non-specific defence:
vasodilation (leads to heat)
increased vascular permeability (leads to redness + swelling)
WBC recruitment
phagocytosis
resolution + repair
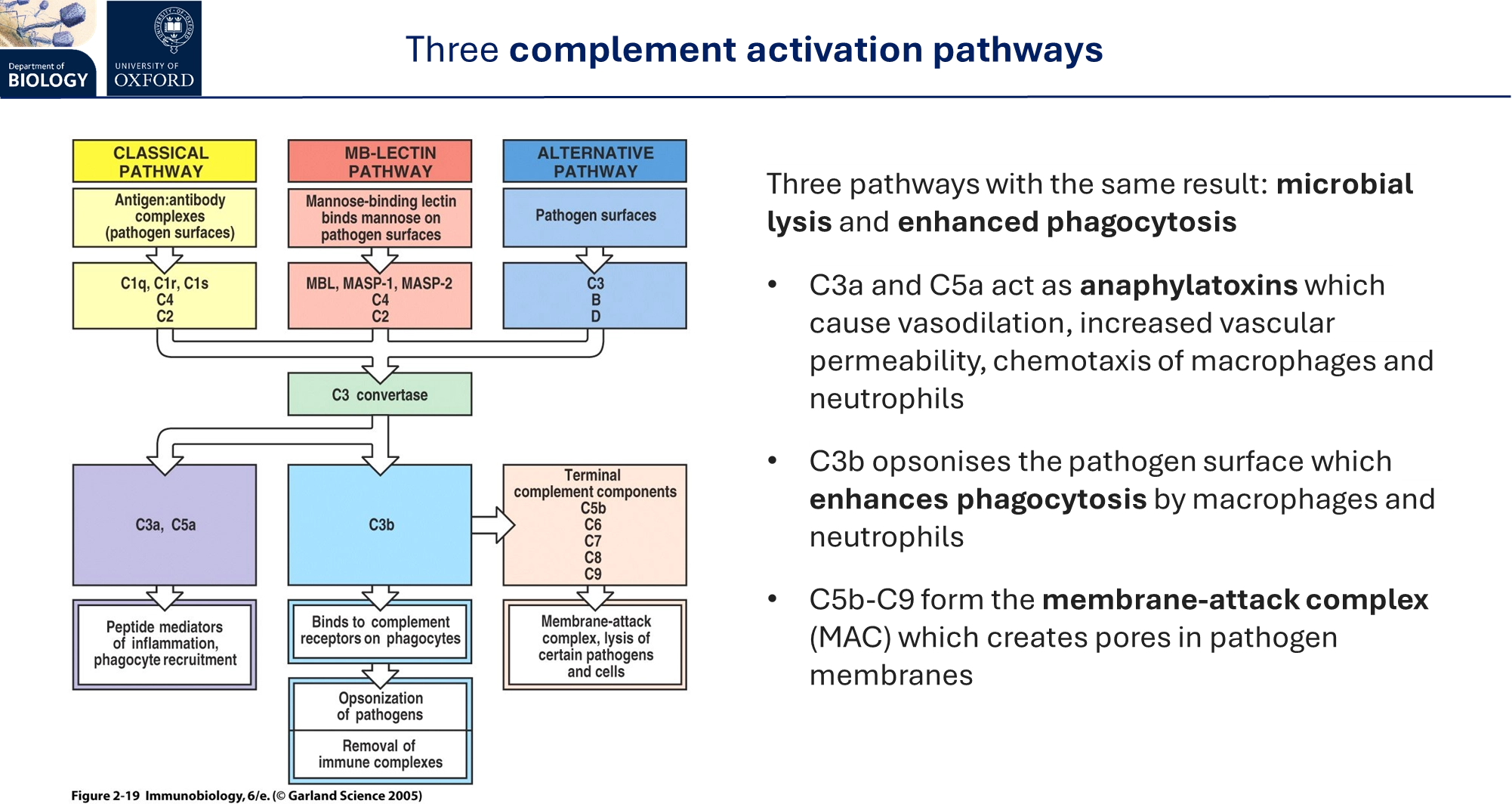
what is the complement in the animal immune system?
a system of plasma proteins that complement the innate immune response
they have three activation pathways:
classical pathway (requires adaptive immunity)
lectin pathway
alternative pathway
each of these pathways triggers microbial lysis by forming a membrane-attack complex, and increased phagocytosis by opsonisation and triggering inflammation
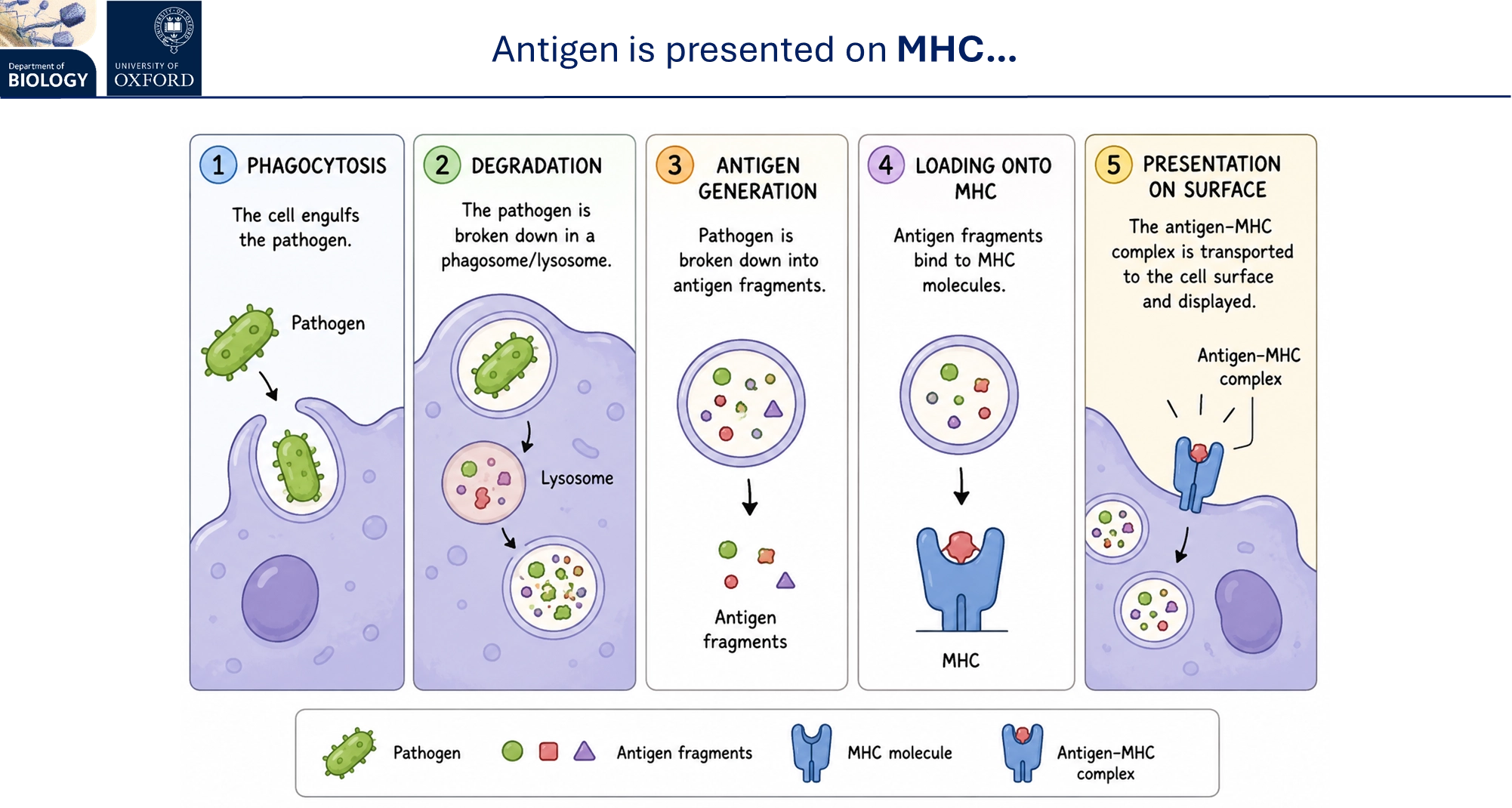
how does the innate immune response activate adaptive immunity?
following phagocytosis in antigen-presenting innate cells (dendritic, macrophage and B cells), peptides from the broken-down antigens of the pathogen are presented on the major histocompatibility complex (MHC) on the cell surface
these are recognised by T cells as being self or non-self, using T cell receptors
innate cells also produce cytokines to activate different cells
many pathogens can downregulate the MHC to avoid recognition
in response, the host evolved natural killer cells that recognise when a cell is under stress, and its MHC is missing, to activate apoptosis of the cell (by releasing perforins + granzymes)
how does the innate immune response clean up debris?
by phagocytosis of debris + cell fragments
by efferocytosis of aptotic cells (similar but produces anti-inflammatory cytokines)
macrophages hence have a pro-inflammatory and an anti-inflammatory phenotype dependent on the infection stage, changing which cytokines they produce
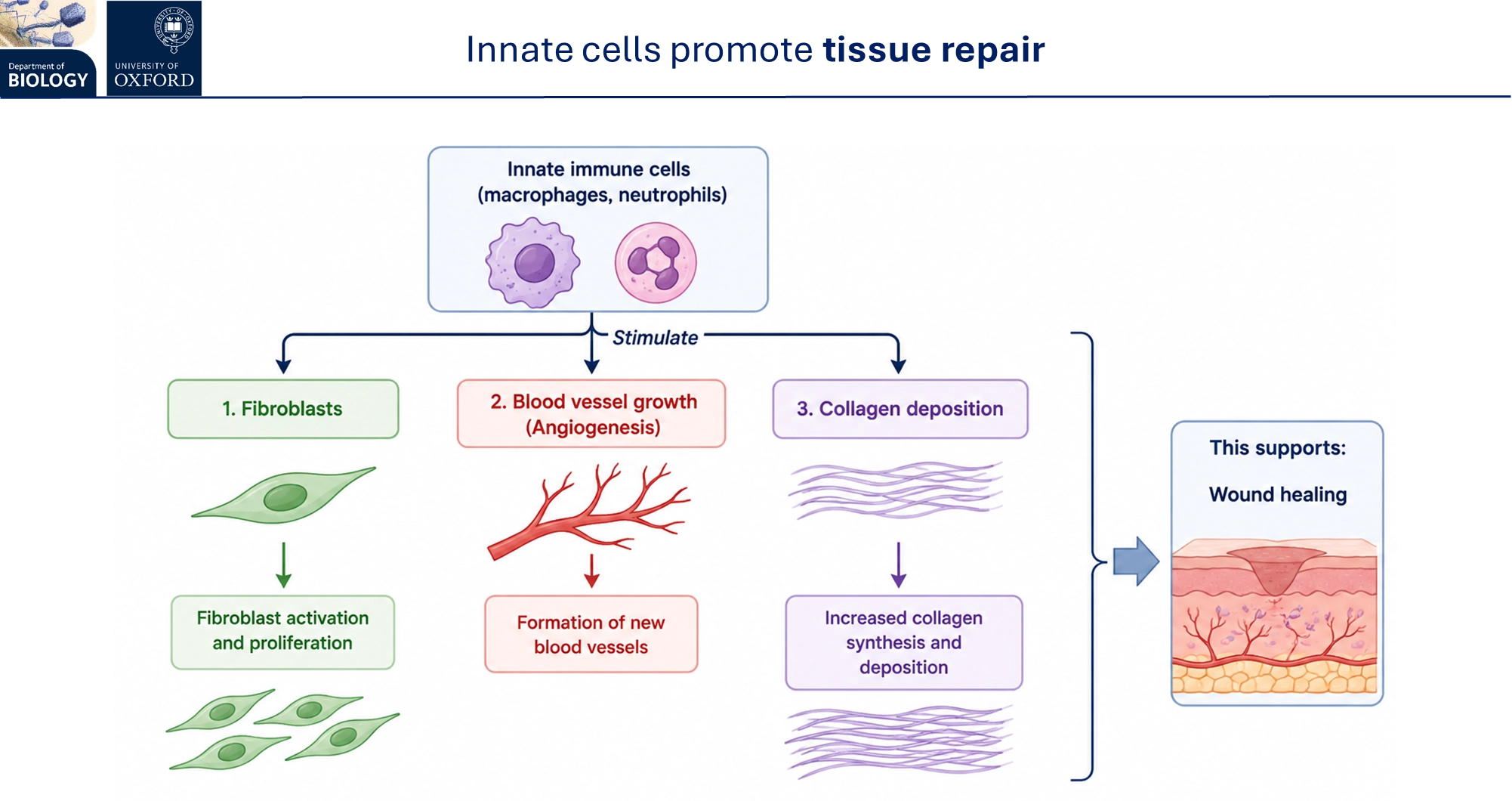
innate cells promote tissue repair by triggering fibroblast proliferation, formation of new blood vessels and increased collagen synthesis + deposition
how does the adaptive immune response recognise specific antigens?
each T cell has an identical set of T cell surface receptors (alpha/beta chains)
these only recognise antigens that have been broken down and presented as peptides on the MHC of antigen-presenting cells
TCR specificity/affinity is fixed
cytotoxic (CD8+ co-receptor) T killer cells directly kill infected/abnromal host cells, by recognising MHC class I (on all nucleated cells)
T helper (CD4+ co-receptor) cells coordinate the immune response, by recognising MHC class II (only on specialised APCs)
each B cell has an identical set of B cell surface receptors resembling the antibodies they produce (light/heavy chains)
these bind directly to native antigens (not using MHCs), but need T cell-produced cytokine signals for activation
BCR specificity/affinity can increase in strength over time
these both recognise variable pathogen-specific antigens, not PAMPs
T and B cell activation triggers clonal expansion, producing many effector cells and a fe memory cells
how does immune tolerance work?
the immune system has to be tolerant to the host’s own antigens (otherwise causing autoimmunity) and to innocuous antigens (otherwise causing allergies)
in normal funciton, if antigens are encountered without danger signals, in non-inflammatory conditions, innate cells promote tolerance over an immune response
central tolerance
in the thymus, self-reactive T cells (bind too strongly to MHC) are eliminated by apoptosis
in the bone marrow, self-reactive B cells are eliminated by apoptosis/anergy (neglect), or edited to change their receptor specificity
peripheral tolerance (if central tolerance isn’t completely successful)
after the T cells mature, excess stimulation by a self-antigen induces apoptosis, and regulatory T cells suppress the self-immune response
after the B cells mature, excess stimulation by a self-antigen induces apoptosis/anergy, and regulatory T cells suppress the self-immune response
this is why cell grafts from other organisms are normally rejected, unless MHCs are matched or autologous transplant (using patient’s own modified stem cells) is used
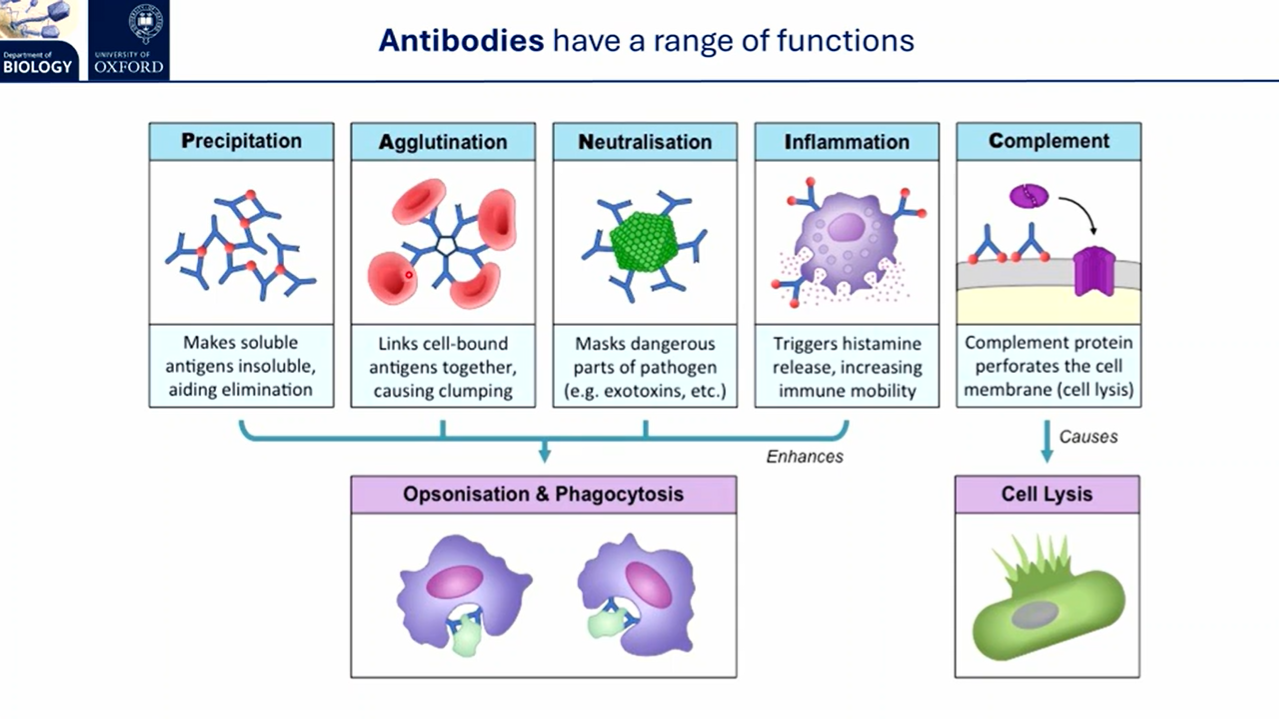
how does the adaptive immune response trigger targeted effector responses?
following antigen recognition and clonal expansion:
effector plasma (B) cells secrete large amounts of antibody and cytokines
antibodies precipitate soluble antigens, link cell-bound antigens together to cause clumping, mask toxins, trigger histamine release for inflammation, and target natural killer cells to infected cells in cellular cytotoxicity
effector T helper (CD4+ co-receptor) cells produce interleukins (cytokines) to coordinate the immune response and activate B cells
effector T killer (CD8+ co-receptor) cells release cytotoxic granules (perforin + granzymes) to trigger apoptosis
how does the adaptive immune response trigger long-term protection?
the memory cells produced in clonal expansion respond faster, stronger and more specifically upon re-exposure to the same antigen
the secondary response has a shorter/no lag phase, because the memory cells are already in place and don’t need to wait for recognition, activation and clonal expansion
this is exploited in vaccines (most are B-cell inducing)
why are plants more at risk of pathogens and in what ways do they survive them?
plants are at risk because they:
are sessile
don’t have adaptive immunity by somatic recombination of immunoglobulin genes
no mobile immune cells/lymphocytes
however, though adaptive immunity isn’t present at an individual level, it is present at the population level due to non-fixed R gene diversity

what pre-existing barriers do plants have against pathogens?
bark
cuticle + cell wall
elevated stomata
chemical barriers called phytoanticipins
resistance against phytotoxins
the target proteins of the pathogen may be missing in the plant
the plant could not provide essential nutrients desired by the pathogen/ it could be unrecognisable
what induced responses do plants have against pathogens?
organ-specific:
stomatal closure after sensing pathogens (pre-invasive)
outgrowths of xylem parenchyma into the xylem vessels to block them and prevent spread
partial/whole leaf abscission
cork formation (made of suberin, a waxy hydrophobic layer) that is difficult for pathogens to penetrate
cellular:
release of reactive oxygen species (ROS, eg. hydrogen peroxide) in an oxidative burst in cytoplasm/apoplast- to cross-link the cell wall glycans, in/activate proteins, damage the pathogen directly and signal to other cells
hypersensitive response at site of infection- rapid programmed cell death (not apoptosis)
extracellular traps using secreted DNA
extracellular callose deposition for focal cell wall strengthening at the infection site
accumulation of pathogenesis-related (PR) proteins in apoplast + vacuole
accumulation of anti-oxidative phytoalexins (antimicrobials), dependent on genus
how do plants have temporal/spatial immune responses?
temporal:
immediate responses- protein phosphorylation, oxidative burst, ion fluxes (calcium import)
hours later- transcriptional reprogramming (due to TF phosphorylation), hypersensitive response, salicylic acid stress hormone biosynthesis
days later- callose deposition, PR protein accumulation, phytoalexin production
spatial:
local responses- highest strength, hypersensitive response, immune response in surrounding tissue
systemic responses- induced systemic acquired resistance (SAR), broad range, eg. productoon of salicylic acid stress hormone
how do plants recognise pathogens?
plants can recognise pathogen-associated molecular patterns (PAMPs, e.g., sequences in flagellin, peptidoglycan, chitin) using pattern recognition receptors (PRRs) in the plasma membrane- this activates PAMP-triggered immunity (PTI)
however specialised pathogens have effectors, coded by Avr genes, that block PTI signalling within the cell, causing effector-triggered susceptibility (ETS) to the pathogen
some plants can recognise these effectors using corresponding nod-like receptors (NLRs), coded by R genes, to cause stronger effector-triggered immunity (ETI), including the hypersensitive response (more severe, cell death)- this makes them resistant
NLRs can bind directly to the effector or indirectly, using an intermediate host target protein- this means R genes can recognise multiple pathogens
both of these are universal immune responses triggered by any pathogen, and active against all of them
what are the structures of PRRs and NLRs in plants?
transmembrane pattern recognition receptors (PRRs):
multiple motifs, but often use apoplastic leucine-rich repeats as a protein binding platform
often have a cytosolic kinase subunit- these recognise PAMP sequences to activate PAMP-triggered immunity (PTI)
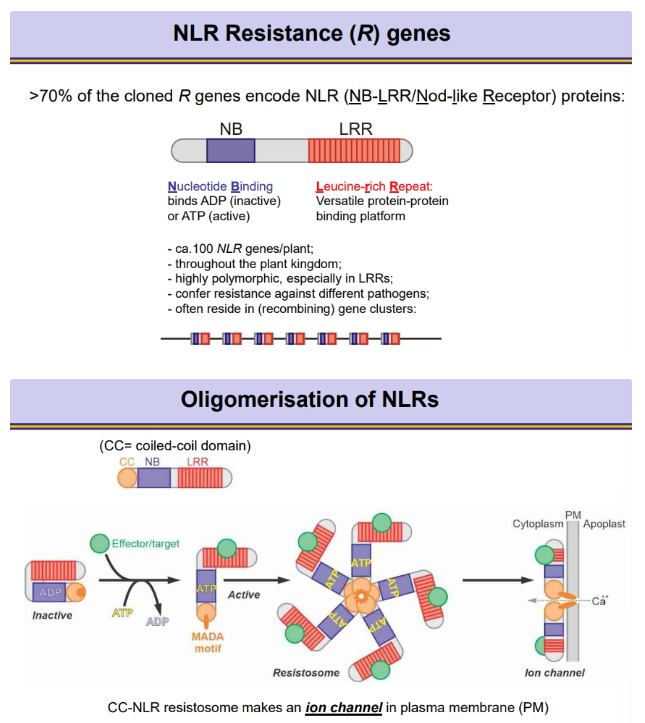
nod-like receptors (NLRs):
always have a nucleotide binding site, bound to ADP when inactive, but exchanged for ATP when active, following the recognition event
always have a leucine-rich repeat as a protein binding platform
most NLRs have an N-terminal coiled-coil domain- when in the active ATP-bound form, multiple subunits oligomerise into a resistosome, using this domain to form an ion channel through the membrane for calcium import (for programmed cell death)
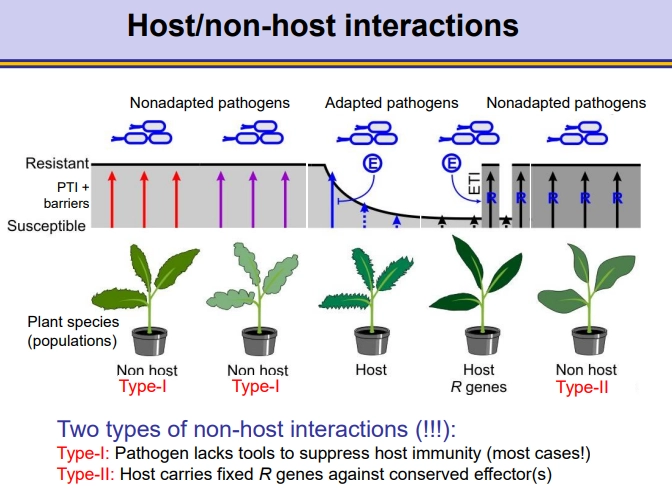
what are the different kinds of host/non-host interactions in plant immunity?
in non-host interactions, the pathogen doesn’t affect the plant, either because:
type I interaction- the pathogen is non-adapted to the plant and can’t suppress the plant’s PAMP-triggered immunity (PTI)
type II interaction- the pathogen can suppress the PTI response using effectors (produced by Avr genes) but the plant carries fixed corresponding R genes to activate effector-triggered immunity (ETI)
in host interactions, the pathogen affects the plant, because:
the pathogen is adapted to suppress the plant’s PTI response (compatible host)
only a select few of these plants will have the correct corresponding R genes for the effectors of the pathogen, so they can activate the ETI response and not be affected