Pharmacokinetics
1/85
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
86 Terms
What is Pharmacokinetics?
The movement of a drug through the body (how it’s dispersed)
What is the therapeutic range/window?
The dosage range or blood plasma or serum concentration usually expected to achieve the desired therapeutic effect
What is the difference between Enteral and Parenteral administration of drugs?
Enteral (Enters the alimentary canal)
Oral
Suppository
Parenteral
IV,IM,SQ,Inhalation,sublingual, etc
What are the 4 stages of drug disposition (what happens to a drug after it enters the body)?
Absorption
Drug leaving the administration site and entering the bloodstream
Distribution
Metabolism
Excretion
Is there a difference in the absoprtion of a drug administered IV vs PO?
Yes, the IV administration has 100% absorption while the PO would be somewhere less than 100%
Absorption:
What is Bioavailability?
What is Bioavailability used for?
The % of the drug that enters systemic circulation compared to the 100% that enters with an IV injection
Bioavailability is used to determine the adjustment needed for an oral dose so that it can have the same effect as an IV dose
What is the first pass effect? Why is it important?
It’s when medication undergoes metabolism at a specific location in the body (Liver) before it can enter systemic circulation
The first-pass effect decreases the active drug's concentration upon reaching systemic circulation or its site of action when the drug is taken orally
What are the 4 different types/ways that a drug can be absorbed? Describe each, which is the most important?
Passive Diffusion
No E required
Driven by the concentration gradient
Most Important
Active Transport
Requires E
Uncommon
Facilitated Transport
Passive
Pinocytosis
Receptor-mediated endocytosis
What are the 2 subtypes of passive diffusion?
Aqueous Diffusion
When the drug passes through the aquoeus channels between cells
Lipid Diffusion
When the drug travels across the cell membrane
What 4 factors affect that can affect a drugs ability to be absorbed?
Size
Must be 150-200 MW
Solubility
Lipid vs Aquoeus
Polarity
Polar drugs are more water-soluble
Ionization
Ionized=more water soluble
Non-ionized = more lipid soluble
Acids are proton _____ while bases are proton ______
Donors, acceptors
Ionized drugs are more _____ soluble, while the non-ionized form is more _____ soluble
Water, Lipid
If the local pH<pKa which form of the drug predominates?
The protonated form (HA and BH+)
Acids
The environment is acidic
Acids are non-ionized in acids due to the inability of the acid to get rid of their protons (H+)
HA
Bases
Environment is acidic
Bases are IONIZED in ACIDS due to the acids donating them their proton (H+)
BH+
If the local pH>pKa which form of the drug predominates?
The De-protonated form (B + H+ and A- + H+)
Acids
The environment is acidic
Acids are ionized in basic environments due to the ability of the acid to get rid of its protons (H+)
A- (IONIZED)
Bases
Environment is acidic
Bases are Non-Ionized in Bases due to the acids not donating them their proton (H+)
B (Non-ionized)
This is because a higher pH (more basic conditions in the environment) favors the removal of protons from the drug molecule.
What happens if the pH=pKa?
50% of the drug is ionized and 50% of the drug is non-ionized
If you have a drug that is slightly acidic that needs to be cleared via the kidney/urine, how can you accomplish this?
Make the urine more basic, acids move towards more basic environments
Which variant of a drug diffuses better? Ionized/Non-Ionized? Why?
Non-ionized, it is lipophilic and can cross plasma-membranes better
If an Acidic drug is in an Acidic environment, which state will it be in, Ionized/Non-Ionized
Non-Ionized, likewise for a basic drug in a basic environment
T/F: Drugs diffuse to different areas of the body better than others
True, drugs can diffused into the heart, liver, and kidney better than fat
If a drug is bound to a protein in the blood can it perfuse into tissues?
No, that protein has trapped it in the blood
If a drug has a high protein binding, what is done to mitigate that so that the drug can reach its desired effect?
You have to up the amount of drug given to accommodate for the % of the drug that will get bound to the proteins
What happens to a drug with high-protein binding if the patient has hypoalbuminemia?
They would have increased active drug in their system
Changing the dose that would be required/potentially making a normal dose toxic
A drug is eliminated either by _______ or _______
Metabolism, Excretion
When a drug undergoes biotransformation by the body, what happens?
It’s structure is altered by enzymes and it becomes a metabolite
There are 2 main phases to drug metabolism, what are they?
Functionalization reactions (Phase I)
These reactions convert drugs into more polar metabolites
This provides a place for a conjugant to bind
Conjugation Reactions (Phase II)
Reactions that conjugate a drug/metabolite with an endogenous substrate to increase the water solubility of a drug
T/F: All drugs require functionalization reactions?
False, some already have binding sites for conjugates and therefore don’t require functionalization
Conjugation (Phase II) Reactions have 4 different variants, what are they? Briefly describe each
Glucuronidation
Catalyzes the transfer of glucuronic acid
Drug conjugate is secreted in bile and or urine
Acetylation
Performed by N-acetyltransferase (NAT)
They transfer acetyl groups to drug molecules
Sulfation
Sulfanotransferases
Catalyses drugs and endogenous substances
Glutathione conjugation
Glutathion-S-transferase
Not usually important for drug excretion
Can be important for detoxifying reactive metabollites
Most Functionalization (Phase I) reactions are performed by 1 enzyme, what is it?
Cytochrome P450
If a drug increased CYP P450 #s/activity what affect would that have on the body?
it would increase the enzymes overall activity and therefore cause faster drug metabolism
Which animal has less Glucuronidase activity? What effect does this have on them?
Cats
It causes them to be more prone to drugs that are metabolized by glucuronidation
Which animal completely lacks NAT genes?
Dogs (Not A Terrier=Dogs don’t have the NAT gene)
T/F: Dogs can acetylate as a form of drug metabolism
False, they lack NAT genes
What is Enterohepatic Circulation? Where does it fit within drug metabolism?
It is when conjugated lipophilic metabolites are excreted in bile (Glucuronidation?) and then get reabsorbed in the intestines
It is something to consider as bacteria can de-conjugate the drug back into its active form when its in the intestines, essentially allowing for a drug to be absorbed twice
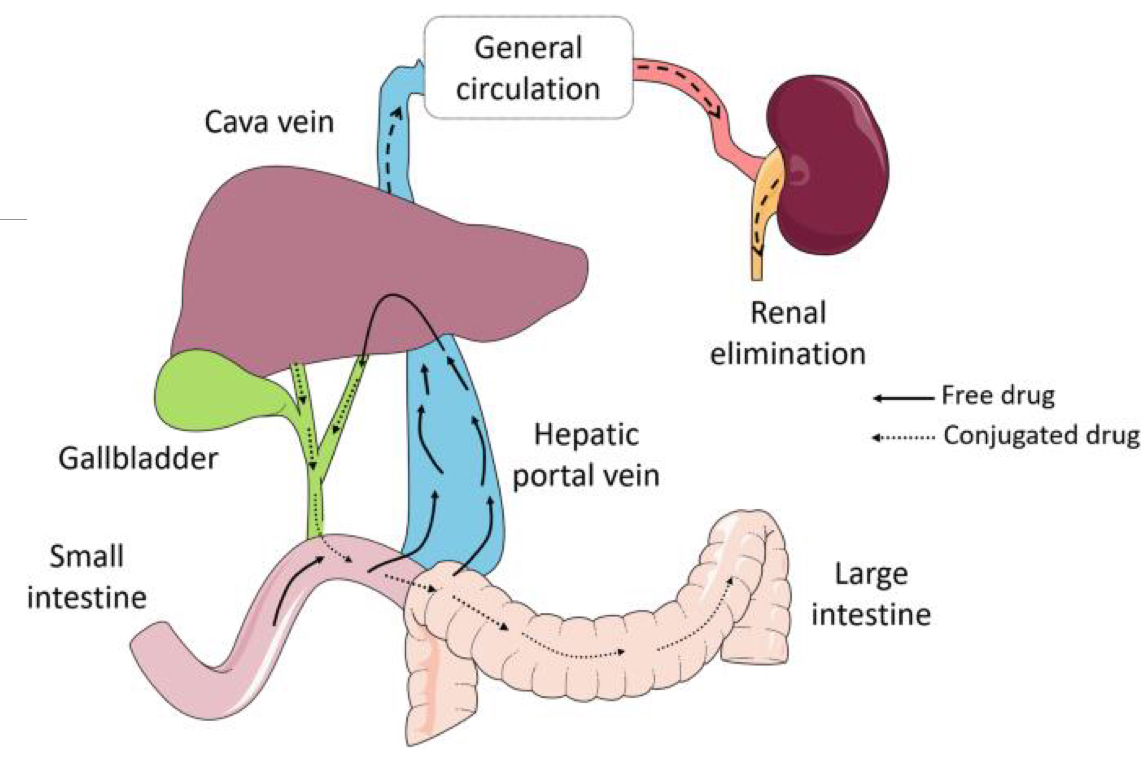
Where are drug transporters (transport proteins that only transport drugs) found? Are they important?
BBB
Intestines
Renal Epithelial Cells
They are very important, they pump drugs out of important structure back into an area (circulation, lumen, urine) so that they can be excreted
________ excretion is the major route of drug excretion
Urinary
In the kidneys _______ substances/drugs are excreted and ________ substances/drugs are reasborbed
Hydrophilic
Lipophilic
Why are many lipophilic drugs biotransformed in the liver into hydrophilic drugs before they are sent to the kidney?
Because hydrophilic drugs are easier to excrete in urine
Rate of renal excretion = rate of filtration + rate of secretion - rate of _________
Reabsorption
Glomerular Filtration….
Has size limitations due to what structure?
T/F: Protein-bound drugs are filtered here
Is a ______ (active/passive )process
Capillary endothelium
False, only free drugs are filtered I the glomerulus
Passive, caused by the high BP
In the kidney, Tubular secretion in the proximal tubules plays an important role, it is an _____ transport that is responsible for transporting what?
Active
Specific, Ionized drugs
Tubular reabsorption can be active/passive; which is more important?
Passive
T/F: Urine pH affects
There are 3 main parameters for Pharmacokinetics, what are they?
Volume of distribution
Clearance
Half-life
What is Volume of Distribution (Vd)?
It's the fluid volume that a drug needs to be dissolved in to achieve the concentration observed in plasma
What does a poor Vd indicate about a drug? What is one example of something that could cause this?
Low Vd = drug is bad at entering tissues
Protein binding
Drugs with a high rate of protein binding have a poor Vd
T/F: A drug with a high Vd perfuses all across the body well
False, it will distribute widely but not necessarily everywhere
What is the most important cause of altered excretion for drugs that are normally excreted renally?
Renal Failure
If a patient is in renal failure and is receiving a drug that is normally excreted in the urine, what needs to happen to the dosing?
It needs to be decreased to avoid toxicity
What is the difference between 1st order kinetics and Zero-order kinetics?
1st
Most common
A constant fraction of drugs is eliminated per unit time
Zero
Less common
A constant amount of a drug is eliminated per unit time
Can occur due to the metabolism/transport of a drug can be saturated
Drug clearance is dependent on drug concentration
Clearance of drugs is primarily done by what 2 organs?
Kidney and Liver
What is the Half-Life of a drug? Why is it important?
It is the time it takes for the drug concentration in the plasma to decrease by 50%
It’s important because it reflects the rate of elimination
The faster the clearance of a drug is the ______ the half-life
Shorter
The higher the Vd of a drug is the ______ the half-life
Longer
The pharmacokinetics of a drug are determined by the ___ and ___, but the ___ is what we observe clinically (t-1/2, Vd, Cl)
Cl (Clearance)
Vd
t1/2
Define Steady State, describe its importance
When the amount of drug going in matches the amount of drug being excreted
It is at this point that the plasma concentration of the drug will remain constant
It takes _ half-lives to reach a steady state, irrespective (not taking into account) of half-life
5
The mean steady state concentration is dependent on the ___ and ______ of the drug
Dose
Clearance
T/F: Changing the dose or dosing rates changes the steady state concentration and the time to steady state
False, it doesn’t affect time to steady state, that is only affected by the length of the half-life
The steady state concentration is proportion to the ________ and __________ of the drug
Clearance, Dosage/Dosing Interval
How many half-lives does it take to reach steady state?
5
Drugs that accumulate have a higher steady state than a single dose. Prevent this by giving at intervals less than t1/2
t1/2 is a function of _______ and __
Clearance, Vd
The greater the Clearance, the _______ the t1/2
Shorter
The greater the Vd, the _______ the t1/2
Longer
The duration of a drug is directly tied to its ____
t1/2
Define Steady State
The time at which the amount of drug going in matches the amount of drug being excreted.
How many half-lives does it take to excrete 95% of a drug?
5 (50→75→87.5→93.75→95)
If you wanted the drug you are going to give to have the least amount of fluctuations, how should you administer it?
IV (oral causes too much fluctuations)
Which is not a factor that controls/influences fluctuations in plasma concentrations?
Steady State
How long does it take to eliminate a drug from the body?
5 half lives
If you are using an ____ loading dose remember to adjust for bioavailability!
Oral
When you repeat dosing regimens, what is the main factor that affects the time at which you dose?
The t1/2 (half-life), how often you repeat a dose, depends on how quickly that drug is metabolized/excreted from the body
For safe drugs with short half-lives (<12hrs), drugs may be administered at interval _ t1/2, why is this?
Drugs may be administered at intervals > t1/2, because the fluctuations are not dangerous
Drugs with a narrow therapeutic range the dosing interval should be _ than the half-life to minimize fluctuations
<<< (much less), this is don’t to make sure the fluctuations stay within the therapeutic range so they have their intended effect
What is Therapeutic Drug Monitoring?
The measurement of plasma/serum drug concentration to determine if the dose is correct
If you are worried about toxicity in steady state, when should you sample during TDM?
When the drug is at it’s peak concentration (Cmax)
If you are worried about a lack of effect for a drug in steady state, when should you sample during TDM?
Cmin just before the next dose is given
How are withdrawal times determined?
They are determined from measurement of drug residue in tissues
T/F: Withdrawal times can be calculated and estimated in the event that you can’t get access to tissue
False, it always has to be measured from the tissue
When calculating the safety factor for the withdrawal time of a drug, it should be set at ___-____ times more than the “safe dosage“
100-1000
In regard to withdrawal times, when calculating the consumption factor (allowable concentration of a drug consumed) it is estimated on the basis of x amount ingested _____ (timeframe)
Daily
T/F: Giving a drug off-label, changing the intended administration site, intended frequency of administration and or intended species makes the calculated withdrawal time null and void
True
Drug residues are most often the result of ________ drugs
Injectable
What is drug residue?
Drugs left in the animals product or the animal itself at the time of consumption
T/F: If the t1/2 of a drug changes (change to clearance/Vd), then the withdrawal time of the drug also changes
True
What is gFARAD
Global food animal residue avoidance databank