Exam 2
1/131
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
132 Terms
Phase 1 Clinical Trials (1 year)
healthy human volunteers. Small short studies done in a few subject (<20)
test escalating single doses and multiple doses
pharmacodynamic and pharmacokinetic studies
safety and tolerance are the endpoints- does the drug mess with the person’s health
Phase 2 Clinical Trials (2 years)
several hundred patients with the target disease. Can involve multiple institutions
placebo controlled, double-blind study designs are the gold standard
Phase 2a: proof of concept. Do patients improve? Is the drug safe?
Phase 2b: dose response studies. Is it better? Is it safe? (nausea, headache, gastrointestinal side effects, liver function tests).
Phase 3 Clinical Trials (3-5 years)
Hundreds to thousands of patients with disease. Definitely involves multiple institutions.
Placebo controlled, double blind study designs are the gold standard
Must show clinical efficacy. This is a key point in drug development
Many drugs die at this point although the company has already invested millions of dollars
Concurrent work with Phase 3 Clinical Trials (3-5 years)
Drug interaction-studies in healthy volunteers
Long-term toxicology studies: chronic and reproductive. Is it teratogenic?
Manufacturing considerations
Preparing the market
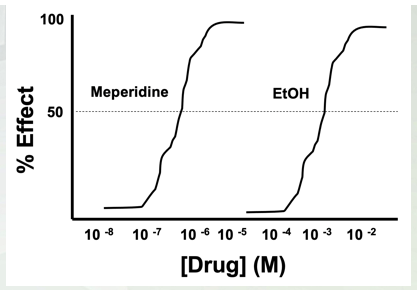
Alcohol Pharmacodynamics
GABA(A) Agonist
Increase intracellular Cl- (higher concentration outside then inside flow through GABA (A))
CNS depressant (increase of intracellular chlorine in the cell)
Very poor potency
Around 14g/EtOH/drink
Most drugs=mg-µg quantities
BAL of 0.1%=100mg/100ml of blood (legal=0.08)
Mempredine (opiate) high micromolar low mammolar. EtOH high millimolar concentration (10-4).
Alcohol Pharmacokinetics- Absorption
Absorbed into partial blood from stomach and intestines
Slow absorption into system
Long latency
Large 1st pass effect
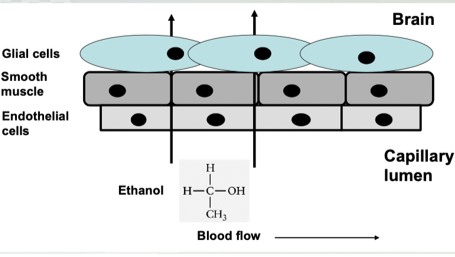
Alcohol Pharmacokinetics- Distribution
Small, uncharged molecule
Easily gets across BBB
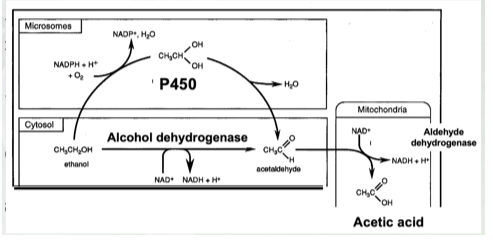
Alcohol Pharmacokinetics- Metabolism
Processed by 2 different liver enzymes:
ADH (alcohol dehydrogenase)
P450 (2E1)
ALDH
Know ethanol is produced by alcohol dehydrogenase and P450
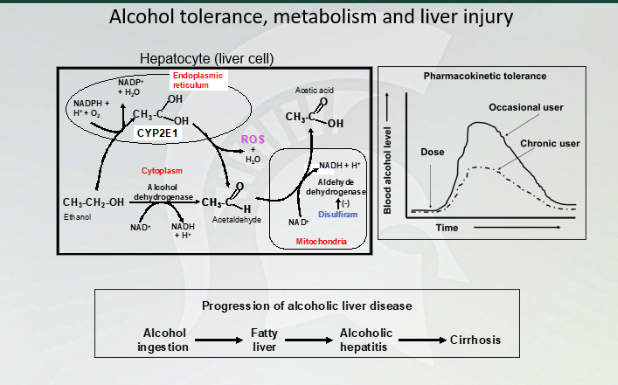
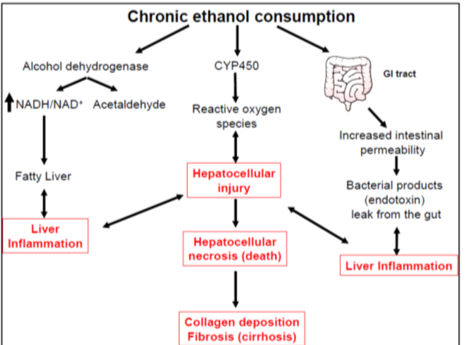
Alcohol tolerance, metabolism and liver injury
Breaking alcohol by P450 to ADH. to acetic acid. There is a different amount, chromic user will have a different latency and concentration in their blood than a person who doesn't drink. Fatty liver first then alcoholic hepatitis then leads to cirrhosis
rate of elimination
is constant per unit time: alcohol follows zero order elimination kinetics
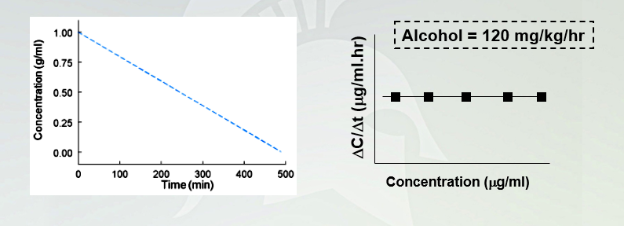
Alcohol: time vs. concentration
Plotting concentration in a function time, you will have a straight slope. Horizontal doesn't change because alcohol stays the same as a zero order kinetic drug.
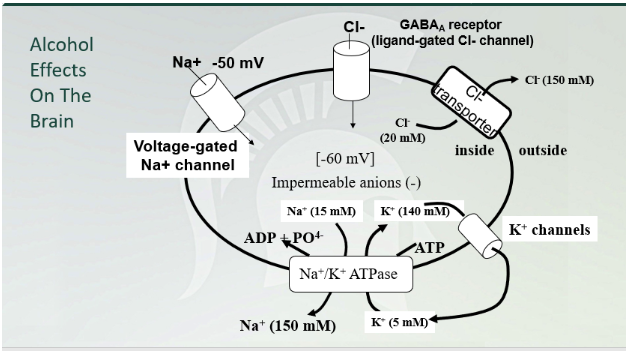
Alcohol Effects on the brain
GABA (A) chloride channel!!!
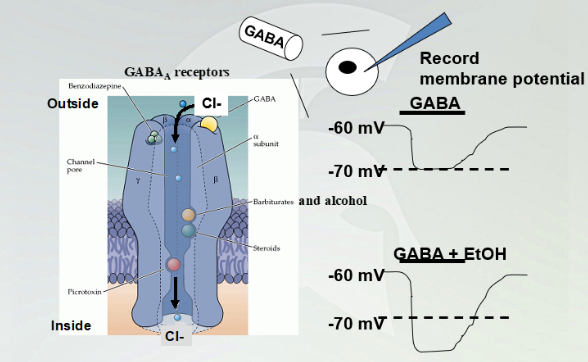
GABA(A) effect with alcohol
GABA(A) agonist, once it binds cause influx of chloride and have a hyperpolarized membrane (increases negative value). Adding etOH bind to a specific part and cause influx of chloride and hyperpolarize past the -70mV.

Ethanol Discriminating protocol experiment
Rats and mice can discriminate a reinforcing property of the EtOH as they prefer EtOH over sugar water.
A lever press, a drop comes out and they measure how many times they pick the lever and how each lever affects the mouse.
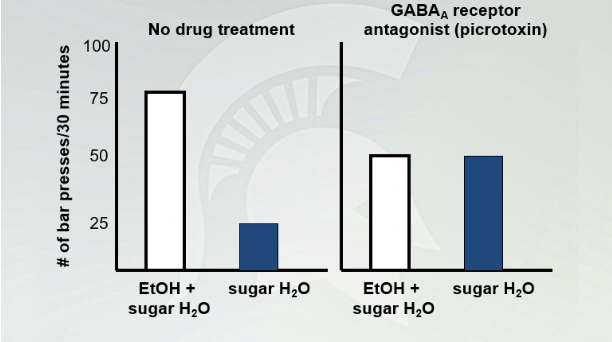
No drug treatment vs picrotoxin
Mouse press 25x on sugar. They prefer EtOH 3:1 to sugar water with the no drug treatment. We know they GABA(A) (picrotoxin) blocks GABA(A) receptor and wr see no difference with the 2. GABA A antagonist, the mouse does not tell the difference between etOh and sugar water since GABA A is blocked.
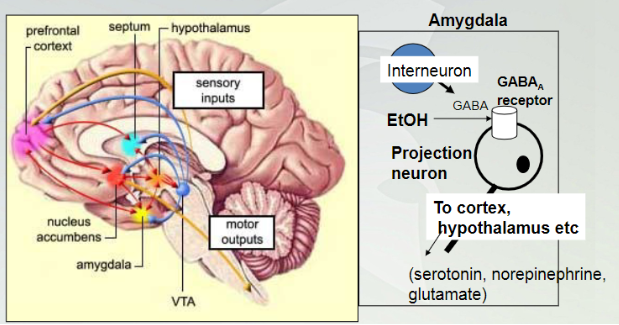
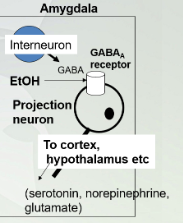
Alcohol effecting amygdala
The amygdala is the primary site of action for EtOH’s behavior and reinforcing (drug seeking) effects. These actions occur at lower doses of EtOH. High doses have more generalized effects on more diverse brain areas (motor function etc).
The amygdala is involved in perception of stressful conditions and responses to fear and stress
Lowest dose of alcohol affects the amygdala in our brian (starts here). Interneuron is a GABA and the receptor for this internourn is a projection neuron. Once GABA binds to receptors will cause decrease activity in that neuron.
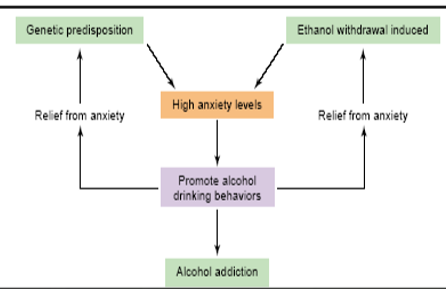
Alcohol and Anxiety
Amygdala involved in perception of stressful conditions and responses to fear/stress
Alcohol increases GABA activity and decreases output of downstream neurons leading to depressed fear response
High anxiety promotes alcohol drinking behavior, to become less stress, cause decrease in activity (calming feeling) but there is a population that have a genetic predisposition, once undergo anxiety it will reinforce itself and if you continue then you get frustrated and stopped and causes withdrawal which induce anxiety which makes you want to drink more which leads to addiction. The root is using alcohol to deal with anxiety and the GABA activity give you a depress fear response but does not help solve the stressful situation/
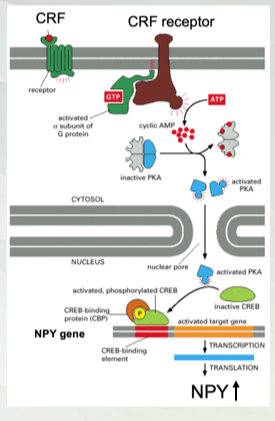
Molecular Pathway of Stress Relief
Neuropeptide Y (NPY)= anti-stress peptide
Corticotropin releasing factor (CRF)= pro-stress peptide
CRF binding to its receptor causes increase in NPY production (through activation of PKA and KREB)
CRF is released and bind to a GPCR receptor. Galpa binds CRF and activates and Galpha dissociate and active adenylyl cyclase to cyclic AMP then bind to PKA (inactive molecule) dissociate inhibitor then leave to inactivate PKA to nucleus and activates KREB and it will target to their target. Cyclic amp blinds to cyclic CBP. translate to NPY (increase inn system)
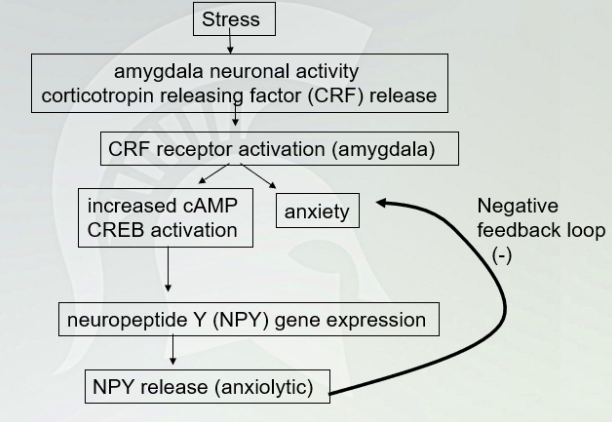
Stress Pathway (with negative feedback loop (-)
NPY shuts production of CRF during the negative feedback so it doesn't rapid increase anxiety.
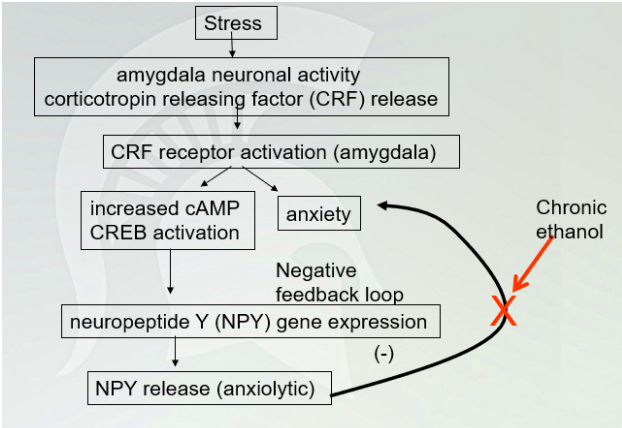
Stress Pathway (chronic ethanol)
Disrupting NPY when drinking (prevent NPY production)

Drug Tolerance (decreases in sensitivity to a drug after repeated administration)
Tolerance is a state of reduced drug sensitivity
Occurs as an adaptive response to the presence of a drug
Tolerance is an effort to maintain homeostasis
Removal of a drug disturbs the new homeostatic level (withdrawal)
Need more drug to produce the same effect.
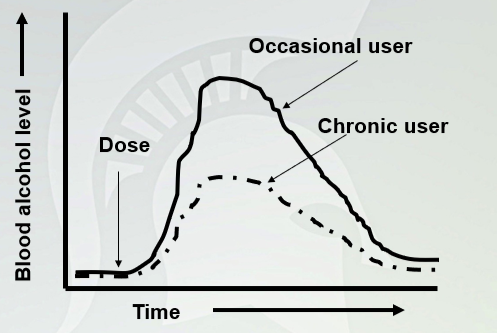
Pharmacokinetic Tolerance
Similar latency time between the two (it doesn't change absorption of the drug, but change metabolism. You will have less concentration in chronic user.
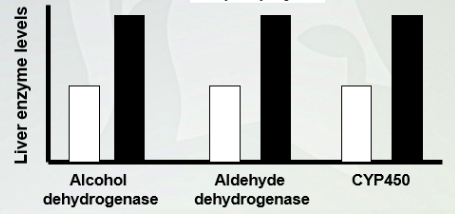
Mechanism of Pharmacokinetic tolerance
Have higher levels in the liver with chronic user of alcohol so they can metabolize alcohol quickly
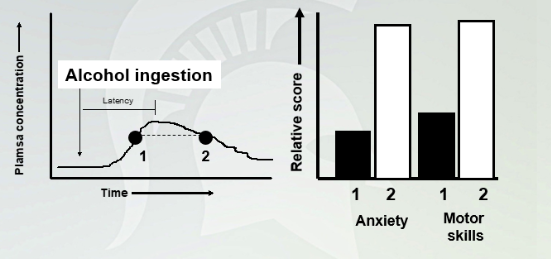
Acute Ethanol Tolerance
Ethanol tolerance occurs quickly (within hours after a single exposure) and is demonstrated by behavioral assessments at times when blood alcohol levels are rising and falling but are at equal levels.
At distent and intent when alcohol match. Have more anxiety when alcohol was being broken down. Anxiety is higher one the down turn.
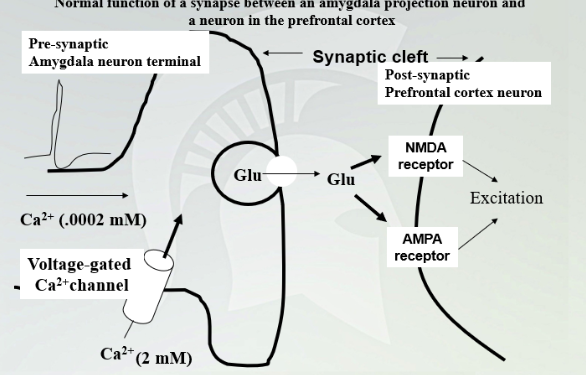
Normal function of a synapse between an amygdala projections neuron and a neuron in the PFC
Calcium is higher outside and once it comes in and glutamate release in synapse and NMDA and AMPA receptor targets.
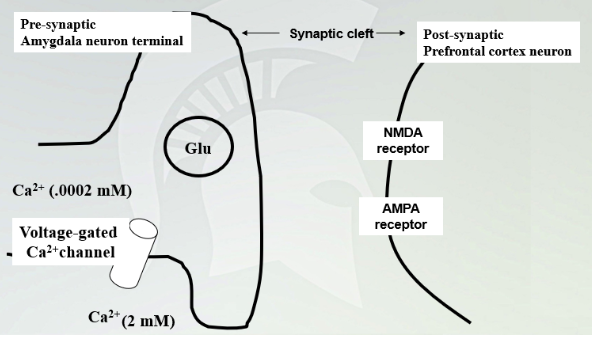
Function of a nerve terminal in the presence of ethanol
Hyperpolarizes the membrane and no release of glutamate due to high chloride levels. Without glutamate being released then you have an increased amount of glucose in the presynaptic neuron. Then it makes more glutamatergic vesicles and increases the number of receptors but only NMDA and AMPA stays the same. This is the response to ethanol in the system.
Stop administering ethanol, everything gets released. Have an influx of calcium and AP activates and a lot of glutamate release with a lot of receptors with a high excitatory response. (headaches, anxiety withdrawal) can also cause seizures
Alcohol withdrawal syndrome
Alcohol craving
Irritability, tremor, nausea
Sleep disturbances
High blood pressure, rapid heartbeat
Delirium tremors (DTs) in extreme cases
Agitation, confusion, hallucinations, nausea, diarrhea
Barbiturates/Benzodiazepines
All are retrieving the same symptoms like anxiety, sleep disorders, and seizures.
Genes and Alcoholism
Two different studies found 40-60% of genetic contributions; these studies compared alcoholism rates in fraternal twins related in separate foster homes with alcoholism rates in identical twins reared in separate foster homes. Alcoholism was 3-5 times more likely to show up in identical twin pairs than in fraternal twin pairs.
(last lecture): genetic predisposition sets you up to drink when you are stress which leads to addiction
Genetics of Alcoholism
Endophenotypes( a type of characteristic that have an origin that have multiple genes involved, combination of genes that contributes developing an alcohol addiction) link alcoholism in families
You have same genes as your parents and they develop alcohol dependence and addition and you have a genetic predisposition
Polymorphism (mutations below)
ADH
ALDH
D2 and D4 receptors
Gives rise that you may develop alcohol addiction based on genetics
GABAA subunits
𝞪2 and 4, 𝛃1 and 𝛄1
Mutations here, have been show rise in alcohol addiction
Alcohol Toxicity
L-T is cirrhosis of liver and terminal death.
Wernicke’s encephalopathy is a vitamin B1 (treated with) (thiamine) deficiency that occurs when cirrhosis
confusion , ataxia and visual disturbances are symptoms. Reversible with vitamin B1 administration
Alcohol can also have a direct cytotoxic effect on the brain: loss of brain tissue (Korsakoff’s psychosis), this requires large amounts of alcohol intake for long periods of time. Irreversible loss of neurons
Treatment for alcoholism
Disulfiram inhibits aldehyde dehydrogenase (block this enzyme so you have an increase of acetaldehyde) once you start giving this, people hate it because they get very sick.
Naltrexone for treatment of alcoholism
naltrexone= opioid receptor antagonist
Naltrexone is an opioid receptor antagonist that has been used as an adjunct to behavioral therapies. Naltrexone blocks opiate receptors that may be involved in the activation of dopamine neurons (stop reward system) in the VTA. This may reduce the reinforcing properties of alcohol. (extreme case) (beta-endorphin signals and shuts off gaba in vta, dopamine on, you have a reward system, having gaba on, shuts dopamine off and reward system is off)
acamprosate= NMDA receptor antagonist
Acamprostate blocks NMDA receptors. If these receptors are upregulated in alcoholics, inhibition of these receptors might relieve some of the anxiety associated with ethanol withdrawal and abstinence. (block withdrawal systems and long term treatment until NMDA receptors goes down)
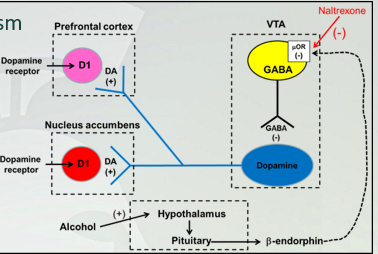
Alcohol dopamine pathway
VTA projection neuron into NA and PFC (dopamine neuron) but gaba took over. If gaba is activated, dopamine is inhibited (vice versa). When you are using alcohol, you will have one of the rxns, if there beta-endorphin, inhibits gaba and activates dopamine to get reward. Prolonged alcohol use releases endorphins.
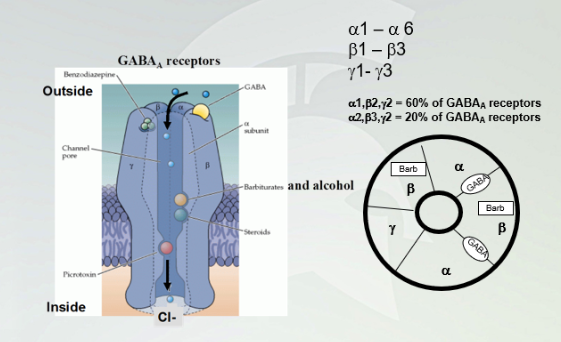
GABA(A) receptors
alpha1-alpha6
beta1-beta3
gamma1-gamma3
Penamere of the first stuff (know the percentages) Gaba binds between alpha and beta subunits. Baribiates bind with alcohol (ligand gated channels, allow influx of chlorine)
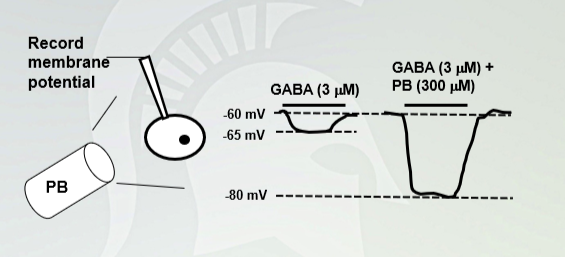
Pentobarbital (PB) increases
Gaba causes an increase in negativity, hyperpolarizes the cell, gaba with alcohol does the same thing. Gaba with barbiturates have a maximum hyperpolarize with an influx of chloride ions
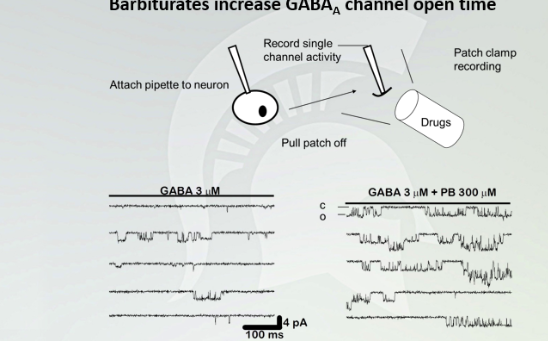
Barbiturates increase GABA(A) channel open time
Each of these recordings came from the electrode of the cell. The dips (wells) that is live recording of a hyperpolarization with GABA. this is how GABA bind to GABA (A) receptors. You see more drastic dips with barbiturates because there is more hyperpolarization and it occurs for a longer period of time
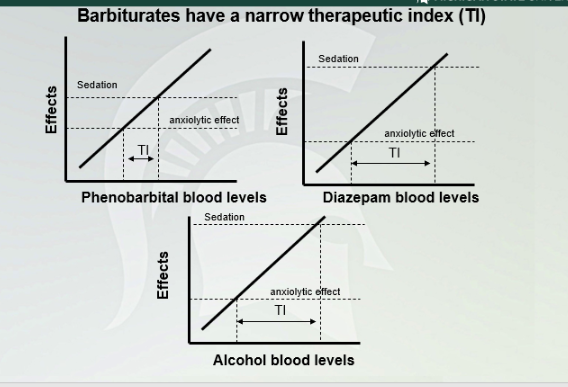
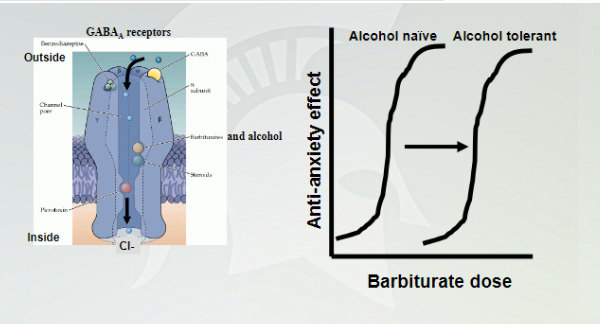
Cross Tolerance between barbiturates and alcohol
Alcohol tolerant need a lot more of barbiturates to get the anxiolytic effect. Alcohol naive needs less barbiturate since they are both working on the same receptor
Benzodiazepines
Xanax has better TI, and doesn't produce a narcotic effect. They decrease anxiety
Chlordiazepoxide (Librium) was synthesized in the 1950’s as a sedative-hypnotic and anticonvulsant the most prescribed drugs in the world through the 1970’s-1980’s
Atypical benzodiazepines
“Atypical” benzodiazepine receptor agonists such as zolpidem (ambien) and eszopiclone (Lunesta) have been developed and these are used as hypnotic drugs to treat sleep disorders
Benzodiazepines (alprazolam) are used to treat sleep disorders, epilepsy and anxiety. They are also used as sedatives and anesthetics for some surgical procedures (conscious sedation). They produce short lived sedation and amnesia
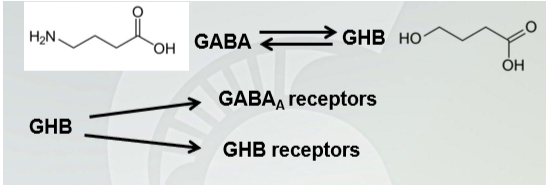
𝛄-hydroxybutyrate (GHB) (GABA metabolism)
GHB is anxiolytic similar to alcohol and benzodiazepines (it basically is taking a lot more alcohol than just drinking alcohol by itself)
Short half life (½ hour) (short pathway)
Synergistic effects with alcohol and produces amnesia at high doses.
Propofol (Michael Jackson)
Drug of choice for induction of anesthesia and for short term surgical anesthesia. It is a powerful sedative and amnesia inducing drug.
Very safe and short acting and there is reduced “hangover” or drowsiness after use
Tests for innate anxiety
Studies of the anxiolytic properties of a drug are difficult. Good predictive tests are important because it is desirable to separate the various effects of benzodiazepines in order to produce drugs that will produce only one type of effect. Two types of anxiety or fear responses: innate anxiety and conditioned anxiety.
Innate anxiety: is shared by all (fear of open spaces in rodents). Conditioned anxiety is learned.
Tests for these types of anxiety conditions can be used to predict anxiolytic properties of drugs like the benzodiazepines
Elevated plus maze and novelty suppressed feeding are tests of innate anxiety in rats and mice
Benzodiazepines are anti-anxiety drugs
Decrease the latency to feeding in the novelty suppressed feeding assay
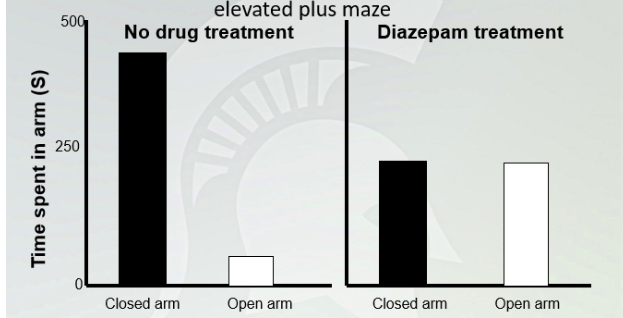
Anxiolytic effects of diazepam in the elevated plus maze
No treatment, mouse stays mostly in closed arms because it is scared. Adding a drugs, it will spend equal time of going in and outside the arm
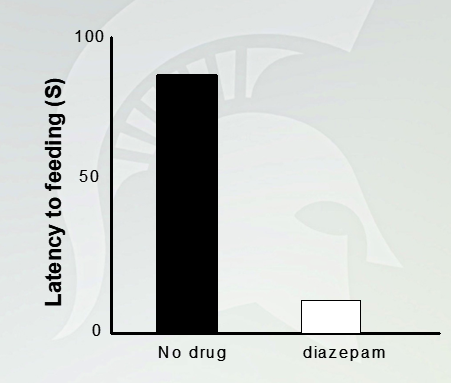
Novelty Suppressed Feeding
A minute and half to get food. With drugs takes 10-20 seconds to get food

GABA(A) Receptors
BDZs bind to between alpha and beta subunits , gaba bind to alpha and beta
Sedation vs Anxiolysis
The amygdala plays a central role in neuronal circuits responsible for anxiety
a2b2g2 subunits found in highest concentration in the amygdala
a1 subunits found in other brain areas
There are efforts to develop benzodiazepines like drugs that are selective for a2 subunit containing
GABAA receptors. These would reduce anxiety without causing drowsiness. This would be a benefit to the patient.
Red and yellow, a lot of expressive of the subunit, blue and purple less expressive
Anterior amygdala see a lot of alpha 1
Anxiolysis alpha 2- specific to hit anxiety and avoid areas that causes seizures and anxiety
Endocannabinoid System
CB1: in CNS
CB2: in PNS
Where different receptors are located and expressed (dots showed in image) (don’t memorize the list, know it in molecular level)
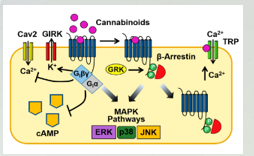
Two types of cannabinoid receptor
Cannabinoid (CB) receptors are G-protein coupled receptors.
CB1 receptors are neuronal receptors (in CNS)
CB2 receptors are expressed by immune cell and microglial cells (macrophage type cells in the brain)
Both are coupled gcouple i and inhibit adenylyl cyclase and cyclic amp. Another layer of control is we increase a flow of K+ through gert channels and blocks calcium channels.
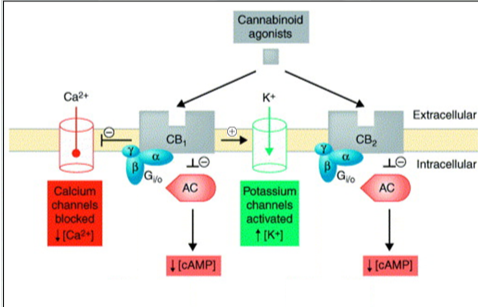
Signaling Pathways linked to cannabinoid receptors
-yellow bar is the membrane. Both receptors are embedded in the membrane. GalphaI we have a decrease in cyclic amp. K+ channels (activates) and ca2+(inhibits_
Endocannabinoids
AEA–Agonists at CB1 and CB2 receptors (High affinity for CB1)
2-AG–Agonist at CB1 & CB2
Come from arachidonic acid and forms anandamide, AEA and 2-Ag( don't know structures)
Both agonists for CB1 and CB2
Exogenous Cannabinoids
Rimonabant CB1 inverse agonist (2006 marketed by Sanofi)
All these signals in our bodies based on how much we ingest. Not tested
Planted-Derived Cannabinoid Compounds
Cannabinoids are the group of more than 60 plant-derived compounds from Cannabis sativa
The primary psychoactive congener in marijuana is 9-tetrahydrocannabinol or 9-THC
Other plant derived cannabinoids are CBD little psychoactivity (cannabidiol) and CBN (cannabinol)
Synthetic cannabinoids (K2, Spice)
In the 70s is when they started to synthesize . spice- used as a spice on different herbs and can be smoked to get the effect (the two on the left) all can be K2. all produced in the lab by CB1 and have high potency and efficacy than THC.
Spice molecule relates to opiate receptor
Can be used a surgical molecule for THC
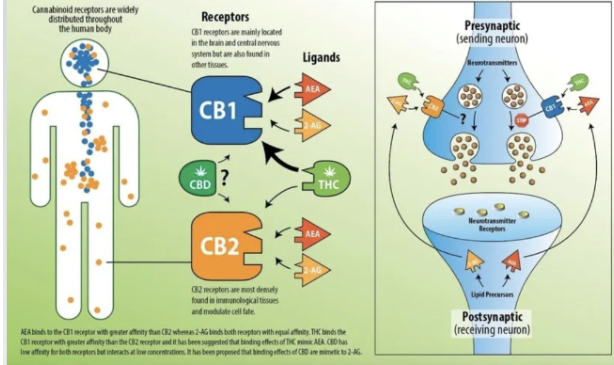
CB1 VS CB2
Circles represent expression
Orange and yellow triangles are endogenous receptors and bind to CB1 and have high potency in CB1.
AEA and 2AG have same level of potency in CB2
THC has higher potency in CB1
THC don;t have good potency in CB2
Receptors are presynaptic terminal, signals CB1and it prevents release of NTS in that nerve. Inhibition of ca2+ pulse because NT can’t be release.
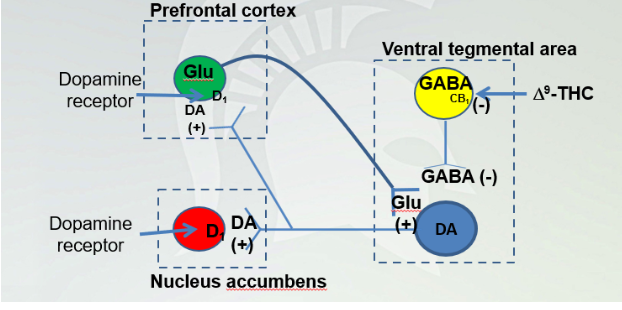
Site of 9 - THC in the reward pathway
Ingesting THC creates a high because we are signaling dopamine. In VTA, GABA and projection neuron of DA from nucleus accumbens and it synapses back to DA.
THC prevents binding CB1 and inactivating GABA and allows DA to be turned on and it turns on DA and glutamate neurons. And continue to open DA and circuit will be active which leads to addiction when glutamate is activated
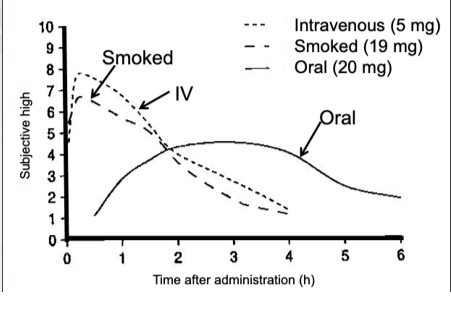
Pharmacokinetics: Routes of Administration
Ingest by injection, smoke, or eating.
Level of euphoria experienced from ingesting weed. Intravenous or smoked has the shortest latency and highest blood concentration. Slowest way is through eating it and it has to go through ADME.
Smoking and IV has shortest latency and oral has the longest latency
Don’t have the same withdrawal effects like you would with opiates and alcohol.
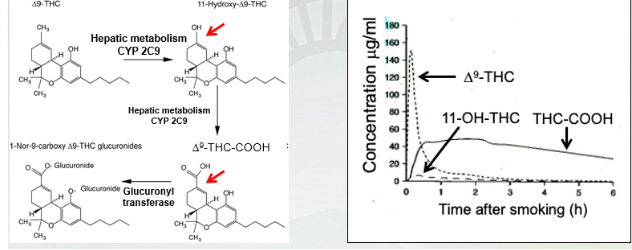
Pharmacokinetics: Metabolism of 9-THC
Blood, urine and saliva tests can detect 9 -THC and its metabolites for as long as 1 month after the last use in heavy marijuana users. 5ng/mL is the intoxication threshold in Colorado.
2 steps for 1st pass and carry by CYP2C9
2nd pass of THC by glucuronyl transferase metabolism
9 -THC is psychoactive molecule and activates in largest molecule and shortest latency when smokes and quickie metabolize by 11-OH-THC (less signaling since it breaks down so quickly) and THC-COOH (most signaling in the body and sticks the longest). Little dash ends 1st pass of metabolism. In circulation for the longest time.
9 -THC has highest potency
Physiologic Responses to 9 THC Activity In the Brain know physiological response
VTA-NAcc: Reward Pathway
Ventral tegmental area (VTA) Nucleus accumbens: reward pathway (mesolimbic dopamine system).
NTS: Nausea relief
Nucleus tractus solitarius- afferent input from the GI tract. Nausea and vomiting
Hypothalamus: Appetite stimulation
Hippocampus: memory
Amygdala: anxiety and wakefulness
Nucleus of meynert-cerebral cortex:cognition
Periaqueductal gray: Pain Pathway
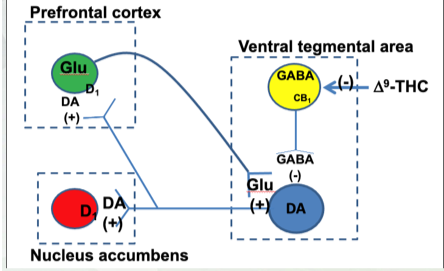
9-THC in the Reward Pathway
Agonist at CB receptors
Turns off-lost of 9 -THC in circulation
On (from above)
Concentration goes up with potency
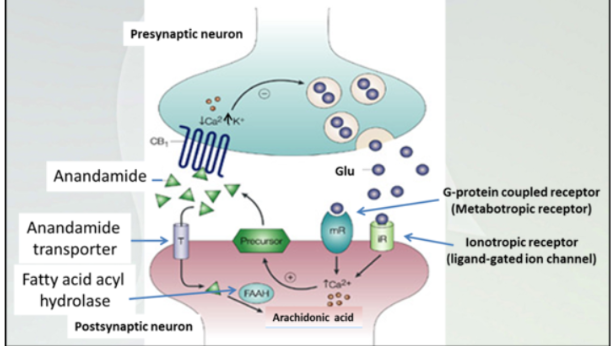
Endocannabinoids are retrograde neurotransmitters
Right half- signaling on presynaptic membrane and how release of NT happens.
9 -THC Suppresses Chemotherapy Nausea and Vomiting
9-THC is a powerful antiemetic drug. Blocks chemotherapy induced nausea and vomiting. Chemotherapy drugs damage rapidly dividing cells including healthy cells including healthy cells in the GI mucosa.
Enterochromaffin cells in the GI mucosa are damaged by chemo drugs and these cells release 5-HT which stimulates vagal nerve fibers which terminate in the nucleus tractus solitarius (NTS) in the brain stem.
NTS stimulates nerves in the chemoreceptor trigger zone (CTZ) which stimulates neurons in the vomiting center to cause vomiting.
THC acts at inhibitory CB1 receptors on the vagal nerve terminal to inhibit glutamate release. This inhibits the vagal signal causing vomiting
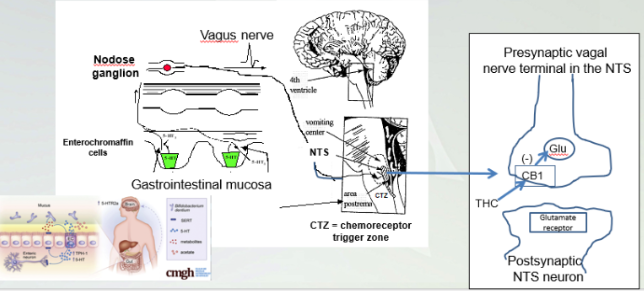
Presynaptic Vagal Nerve Terminal in the NTS
Brain stem, NTS connecting to CTZ where stimulus for nausea originates and goes through the body for vomiting. Dominated serotonin receptors in enterochromaffin cell
THC signals through CB1 prevents nausea and vomiting.
NTS contains the vomiter cell and stimulate cells in stomach and NTS stimulates CTZ that cause vomit and nausea. THC binds CB1 prevents glutamate which reduce vomit and nausea
Hypothalamic Regulation of Feeding Behavior
Cannabinoids act in the hypothalamus to stimulate appetite.
The arcuate and lateral hypothalamic nuclei of the hypothalamus control appetite and feeding behavior
Activation of the lateral hypothalamus(LH) stimulates appetite and feeding. Inhibition of the LH suppresses appetite and feeding
VTA with cannabis
3rd ventricle (dotted red)- arcuate nucleus. When you eat, stomach signals to brain and you are not hunger, arcuate nucleus will allow you to not eat. Having THC, inhibits from arcuate nucleus and it tell us to eat (munchies ) even when you are not hungry
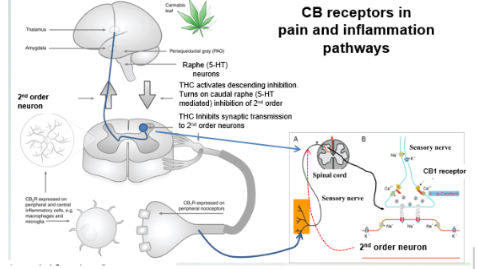
CB receptors in pain and inflammation pathways
Macrophages are first responder inflammatory cells
Microglial are nervous system macrophages
Inflammatory substances released from microglia and macrophages sensitize sensory nerve endings
THC acting on CB2 receptors inhibits macrophages and microglia
9-THC stimulates neurons in the PAG which then stimulates 5-HT neurons in the raphe nucleus. These 5-HT neurons sends axons down the spinal cord to the dorsal horn where peripheral sensory neurons synapse with 2nd order sensory neurons. The 2nd order sensory neurons project up the spinal cord to the thalamus carrying pain signals.
The nerve terminals of peripheral sensory nerves in the dorsal spinal cord also have CB1 receptors. 9-THC acts on these receptors causing inhibition of Ca2+ channels and this prevents neurotransmitter release. Therefore, there is no signal coming from the peripheral sensory neurons to the 2nd order neuron. CB1 receptors are also on the peripheral nerve terminals (in tissues) which detect painful stimuli . 9-THC acts on these nerve terminals to inhibit them as well
CB2 receptors are expressed by immune cells such as macrophages and microglia. These cells become active at sites of tissue injury and they release substances that sensitize sensory nerve endings. This is part of the pain response at sites of tissue injury and inflammation
9-THC acting at CB2 receptors inhibits macrophages and microglia from releasing inflammatory substances. This is part of the analgesic effects of 9-THC.
Brain to spinal cord (1st order) and we have a sensory nerve, the red nerve (2nd order neuron). We get stimulus from sensory nerve (pain) into spinal column for sensory neuron on to 2nd oder neuron and will communicate to brain we have pain.
Synapses onto 2nd order neuron, CB1 has its receptor in presynaptic bever, THC on bard prevents the transmission (ex:glutamate)) and block this pathway to the peripheral.
2: end of sensory nerve, these dendirtes of CB1, THC can signal there and shutting off pain closer to sensory and 2nd order neuron.
CB2 receptors (periphery) micorgailia- inflammaoty response. THC acting on CB2 will blunt that to not feel pain and blocks inflammatory response.
Singaling through CB2 will desynthrsize this
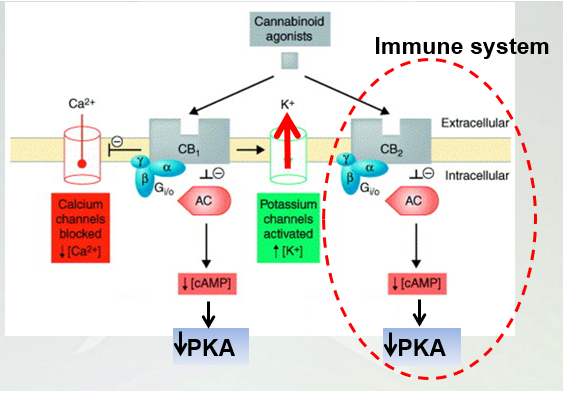
Signaling pathways linked to cannabinoid receptors
Both GCPR once an agonist, the alpha will dissociate and g alpha i inhibits the adenyl cyclase and cAMP. CB1 activates potassium and inhibits calcium.
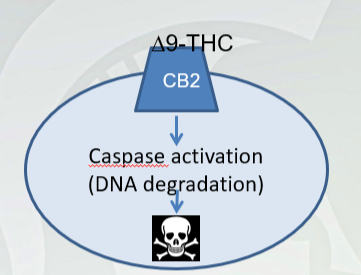
9-THC induced programmed cell death (apoptosis) in T-cells of the immune system (first thing)
T-cells facilitate immune system activation
T-helper cells, Natural Killer cells, memory cells, B-cells)
Due to CB2 signaling at THC (caused above)
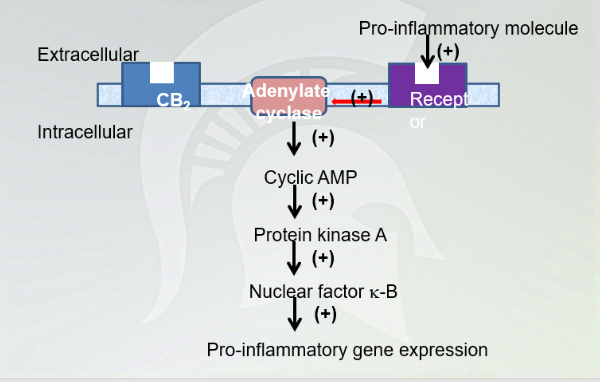
Cannabinoid Pathway
Inhibition cAMP and decrease signaling cascade and pro-inflammatory gene expression. Decrease immune response
Crohn’s Disease
Chronic inflammation of the last part of the small intestine (ileum) and first part of the colon.
Caused by inappropriate immune response to normal bacteria living in the gut (microbiota)
Can be hereditary
Chronic diarrhea, intestinal bleeding, abdominal pain, weight loss, fatigue
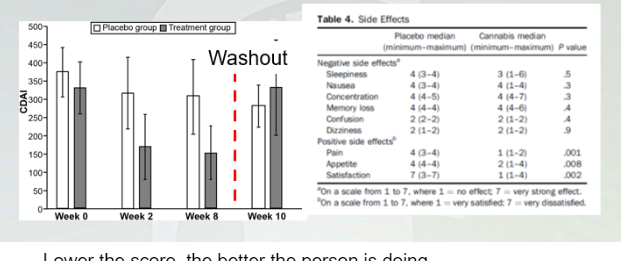
Smoking cannabis improves Crohn’s disease activity index (CDAI)
Lower the score, the better the person is doing.
Weekly treatments and then they did not get anything and got treatment again.
The drop in CBAI in people getting THC. THIs Improves condition with chrons.
(-) 1(no effect) 7(very effective)
(+) 1 (stasified) 7(not statsifed)
Effect shows in positive and got less pain, appetite
Cannabidiol enriched marijuana for treatment of childhood epilepsy \
19 patients and ages between 2-16
Seizures before CBD, people had over 100 seizures to 8-10 a day.
Reduction of seizures once they receive CBD treatment.
3/19 did not get the effect of CBD helped. 25-80% decrease of seizures in the 16 patients.
Cannabidiol in Dravet’s Syndrome
(placebo controlled/double blind)
40% difference in cannabidiol and placebo. CBD helps this condition.
Side effects: Sommolence, lethargy, decreased appetite, vomiting
Multiple sclerosis
Multiple sclerosis is an autoimmune disease in which the immune system becomes dysregulated.
Autoimmunity causes demyelination of motor axons and inflammation.
MS symptoms includes fatigue, muscle weakness, numbness in arms and legs, muscle spasticity and difficulty walking.
Anti-inflammatory effects of 9-THC/cannabidiol relieve symptoms.
Sativex (9-THC/CBD; oral mucosal spray)
People taking sativex vs placebo reported less muscle spasms . more on sativex, less muscle spasms that happens with MS.
Anandamide release from CA1 neurons…
Anandamide release from CA1 neuron inhibits GABA interneurons
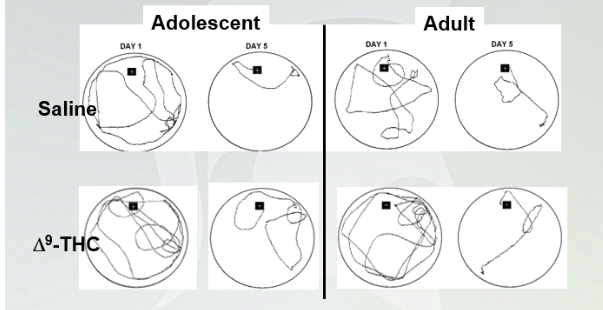
9-THC impairs spatial learning and memory in mice
Black square- stage
White liquid and the mouse can stand on platform. Researchers removes platform and the Mouse going to swim to find platform.
Day 5: they swim around but know where the platform is.
With THC, they have a harder time finding it. Both groups found a platform on day 5 then day 1.
Not statistically significant but for adolescents it is.
9-THC suppresses Eeg Gamma oscillations (fastest brain waves)
Associated with learning and memory formation.
Couldn't control the users that were taking on like alcohol.
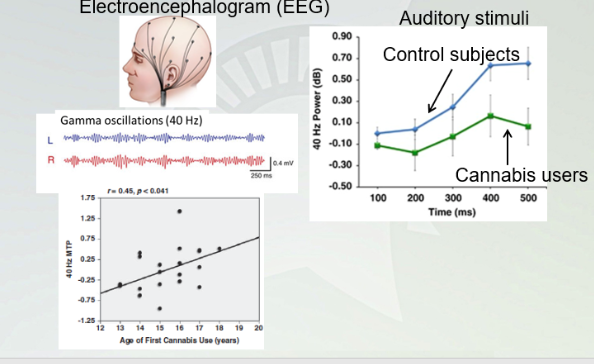
Electroencephalogram (EEG)
Study shows when you have people treated with cannabis and control, GAMMA oscillations decrease with people with cannabis. This has a larger effect with the younger the people go. Younger you go down, the worse you are for your brain to handle.
THC can be a problem with young brain developing for learning and memory
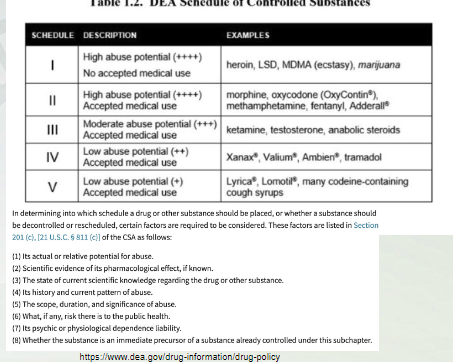
Medical Marijuana- Drug Scheduling
Schedule 1-5 determined by:
Abuse potential
Medial use
Criteria for drug to be a medicine
Chemistry must be known and reproducible
Adequate safety studies
Well-controlled studies proving efficacy
Accepted by qualified experts
Evidence widely available
Tetrahydrocannabinol and marijuana are Sch1
Dronabinol Sch3
Trail 1- highest abuse, no medical use
2: high abuse, some accepted medical use
Cannabinoid Pharmaceuticals- Dronabinol (Marinol)
Synthetic 9-THC in sesame oil (droplet)
Indications: attenuation of nausea associated with cancer chemotherapeutics and appetite stimulation
Oral administration
Marketed in US
Cannabinoid Pharmaceuticals- Nabilone (Cesamet)
Synthetic analog of 9-THC (capsule)
Indications; attenuation of nausea associated with cancer chemotherapeutics and appetite stimulation
Oral administration
Marketed in Canada
oral administration
– results in slow onset
– lower bioavailability than inhalation – difficulty with compliance if severe vomiting
• neither approved drug has found much utility because of the narrow window between effective dose and unwanted side effects
Cannabinoid Pharmaceuticals-Rimonabant (Acomplia in EU, Zimulti in US)
CB1 receptor antagonist (synthesized compound)
Indications: adjuvant to diet and exercise for treatment of obesity
Oral administration
FDA advisors rejected approval because of suicidal tendencies; causes powerful nausea
Discontinued in EU in late 2008)
Cannabinoid Pharmaceuticals- Sativex
Combination of THC and CBD
Anti-spasticity drug for MS patients
Efficacious for preventing muscle spasms, pain
Marketed in Canada, Europe, S.Pacific
First therapeutic cannabinoid available in a form other than capsule (oromucosal spray)
Other indications for Sativex include neuropathic pain that may or may not be associated with MS or cancer
CNS side effects are lowered relative to a “9-THC only” preparation

Opium Compounds
Endogenous that are produced in the body. Delta, kappa, mu or the three groups that are important
Peptide ligands bind (dynorphin and endrophorin)
Heroin- isolated from poppy and tweak it through chem.
Fentanyl no origin from poppy, just synthesized from lab
3 classes of endogenous opioid peptides
1) Endorphin
2) Dynorphin
3) enkephalin
Endogenous opioid peptides
Enkephalins and dynorphin
Widely distributed in the brain (nucleus accumbens, amygdala, cortex, hypothalamus)
Found in the high concentrations in the PAG (periaqueductal gray (central gray))and nerve fibers project to the raphe nucleus and descending pain inhibitory pathways.
Enkephalins activate and opioid receptors. Dynorphin activates on receptors.
Endogenous opioid peptides
beta-endorphin
Restricted largely to the pituitary gland
Acts as a circulating hormone rather than a neurotransmitter. Typically released in response to stress. Comes from the same precursor as adrenocorticotropic hormone (ACTH) that stimulates adrenal gland to release steroids like cortisol, a marker for stress.
beta -endorphin activates , and opioid receptors
Cellular localization of opiate receptors
expressed at different positions;
mu and delta at soma
mu, delta, and gamma at terminals
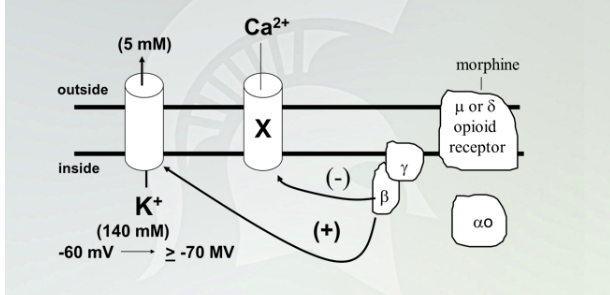
direct coupling of G-protein beta-gamma subunits to ion channels
Opioid receptors are GPCR, and couple to GCPRi
Once receptor bound to morphine, disassociates receptor, activates potassium and inhibit calcium
GCPR connected to Galphas and increase downstream through PKA.
Galpha i inhibit AC and decrease downstream
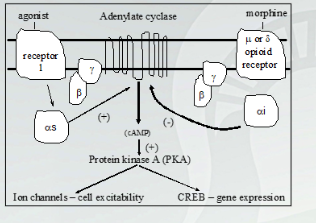
Opioid Receptors In the Brain
All 3 forms of opioid receptor expressed in most areas of the brain
Signaling at each receptor type involved in analgesia and has individual effects on the body.
Exception of PFC, does not have kappa or the medulla
Mu receptor main with euphoria effects
Site of Opiate Action in The Brain
Effects of opiates:
Euphoria
sedation , sleepiness
Reduced body temperature
Reduced BP and heart rate
Miosis (pin point pupil)
Reduced GI motility (constipation)
Analgesia
Effects of opiate withdrawal
Craving for opiates-opioids
Irritability, restlessness
Sweating (inc. body temperature)
Inc. blood pressure and heart rate
Mydriasis (large pupil)
Inc. GI motility (diarrhea)
Inc. sensitivity to pain (hyperalgesia)
withdrawal is a sign of physical dependence but not addiction
People develop a dependence prior to addiction.
Pharmacokinetics in opium
absorption:
Variable routes of administration
distribution
Widely, including brain
Metabolism
Large 1st pass (20-40% remaining)
Glucuronidation (M3G=80%, M6G=10%)
M6G is psychoactive, not m3G
elimination
Kidneys (urine)
Morphine less lipophilic compared to heroin and fentanyl
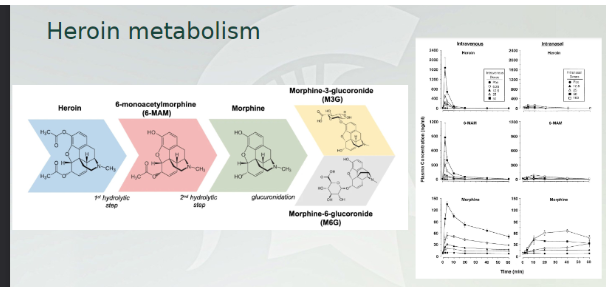
Heroin metabolism
Morphine is the main agonist. Many of these compounds are broken down into morphine through the brain.
This graph: IV vs IN. entire duration is an hour, huge spikes in all three and rapid breakdown. Very little in IN. broken down very quickly.
Avoid short duration of rapid breakdown
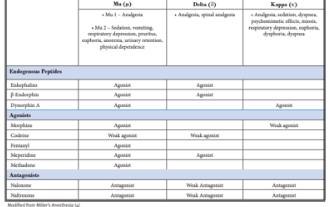
Pharmacodynamics for opium
Table show 3 different classes of opiates. Know this table know when they are agonist and antagonist. 2 subtypes of Mu (Mu1- analgesia, pain relief all the others Mu2- euphoria and sedation)
Structures: be familiar all of these are built off from original morphine molecule
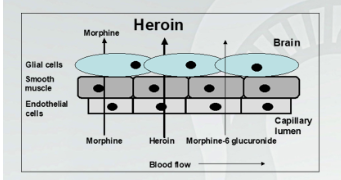
Heroin:Distinct Characteristics from Morphine
Heroin is not an opioid receptor agonist. It must be deacetylated in the brain to produce 6-monoacetylmorphine (6-MAM, this is an agonist) and then be deacetylated further to produce morphine.
This increases the duration of effect of heroin to about 5 hours. Heroin is more potent on a dose per body. Weight basis but is equi- effective at producing analgesia or other effects. You just need more morphine.
Heroin was originally produced in an effort to get around the undesirable effects of morphine use.
(Addiction, respiratory depression etc), However, heroin produces all of the effects of morphine only better.
There are no therapeutic uses for heroin, its clinical interest is based on addiction. Heroin is a particularly troublesome drug because of its pharmacokinetic properties (rapid penetration of BBB). in addition, in recent times (since the 1990;s), illegal production of heroin has improved in terms of quality control.
In the 70’s when heroin addiction was trendy, street packages of heroin contained only about 4-8%
heroin: i.v. injections were essential
Now, the heroin content is often more than 80%. It is not necessary to use i.v. administration to get high.
Smoking or snorting heroin can now produce the same effect. This lowers the threshold for heroin use (no need fo needles or concerns about hepatitis, HIV)
Codeine, Oxycodone, and Hydrocodone
All work as analgesics
Converted to morphine after administration
Comparing parental (other than oral) v oral. These meds tend to be used as an oral application.
Fentanyl
High potency opioid (specifically Mu receptor)
Variation from morphine structure
Methadone
Synthesized as analgesic
Used to treat opioid use disorder
Drug craving reduced with time
Cross tolerance with morphine and heroin
Methadone is an analgesic but it is largely used to treat withdrawal symptoms and morphine/heroin addictions. It is orally active and has a half-life of many hours. With repeated dosing, a stable blood level of methadone can be achieved.-
Tolerance also develops to the sedative effects of methadone, so it is possible for subjects to function (geta job) while on methadone maintenance therapy.
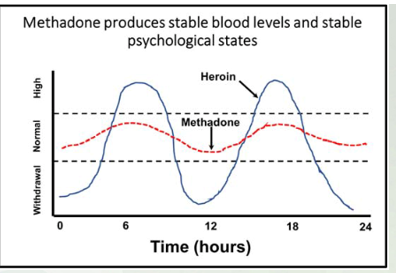
Methadone produces stable blood levels and stable psychological states
Below level- withdrawal, middle, use to having this drug, high is the euphoric response. As it gets broken down, you feel normal to leads to craving and take. Reduces cravings and stay withdrawal symptoms, you don’t get the euphoric response
Doesn't give you the high and avoids the low
Need more morphine if you want something stronger