Looks like no one added any tags here yet for you.
Types of Chemical Reactions
1. anabolic (requires energy)
2. catabolic (releases energy)
Enzymes
-catalyze metabolic reactions
-structure dictates function
-proteins and very occasionally RNAs (ribozymes)
-active sites are substrate-specific for enzymes
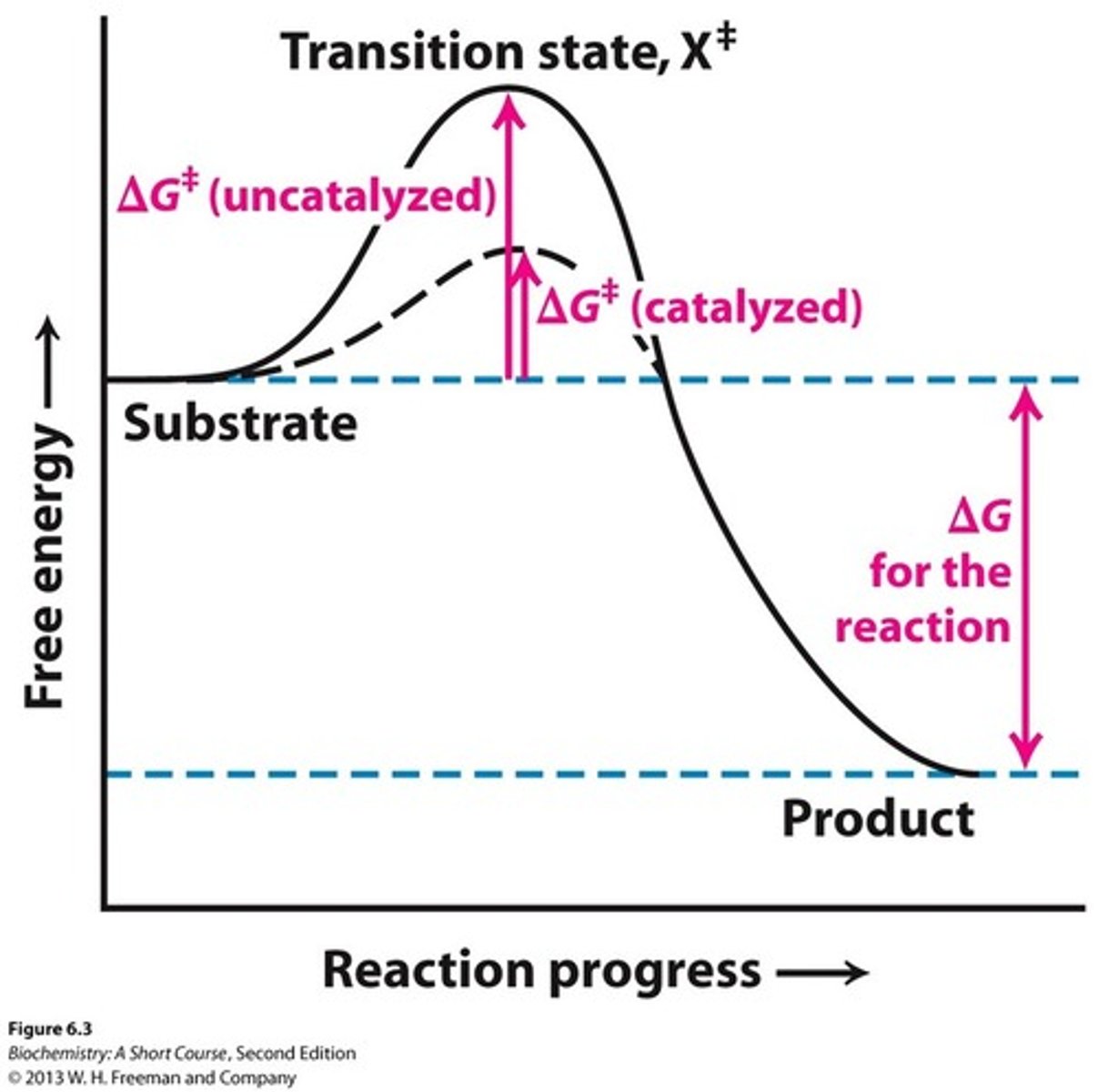
Function of Enzymes
decrease activation energy to allow reactions to happen more easily and quickly (make it so things that are reacting are closer to each other to react)
Enzyme Regulation
-allosteric regulation
1. can regulate availability of cofactors (ions, small molecules) and coenzymes (metals) which are necessary for enzyme function
2. competitive inhibitor (bonds to the active site)
3. non-competitive inhibitor (binds outside the active site and changes shape of active site)
What type of small molecules promote enzyme function?
cofactors and coenzymes
What type of small molecules block enzyme function?
competitive inhibitors and non-competitive (allosteric) inhibitors
Energy Production
1. breakdown of large macromolecules into building blocks
-digestive process outside of cell
2. glycolysis, citric acid cycle, ETC convert the building blocks into energy
-occurs in cytoplasm and mitochondria
Energy Storage
-chemical energy
-ATP
-Glycogen (extra glucose storage in liver)
Glycolysis
-breakdown of glucose
-in: glucose (6 c) + 2 ATP
-out: 2 pyruvate (3c) + electrons (carried to ETC by NADH) +4 ATP (net is 2 ATP)
First Step Regulation of Glycolysis
1. hexokinase
-high affinity for glucose
-glycolysis can proceed with very low glucose concentrations
2. Liver hexokinase
-lower affinity for glucose
-insensitive to inhibition by products of glycolysis
-allows liver to aid in blood glucose regulation (liver take in glucose and convert to glycogen for storage)
What is the goal of glycolysis?
breakdown sugar into pyruvate to feed into the Citric Acid Cycle
Third Step Regulation of Glycolysis
-phosphofructokinase
-regulates overall reaction rate
-rate depends on inhibitors (ATP=inhibitor, ADP=activator)...high level of ATP means cell doesn't need to make more so glycolysis can slow down
Other Inhibitors of Glycolysis
-inhibited by oxygen and sometimes inhibited when fat can be used as an energy source
-pyruvate kinase (final enzyme before pyruvate) is inhibited by high levels of ATP and acetyl-coA (used in citric acid cycle)
Aerobic Metabolism
-pyruvate will proceed to the citric acid cycle in mitochondrial lumen if 02 is present
-34-36 ATP generated
Anaerobic Metabolism
-pyruvate will proceed to fermentation or lactic acid production if no oxygen present
-main goal is to recycle NAD+
-lactate dehydrogenase
-alcohol dehydrogenase (fermentation)
-2 ATP generated
If oxygen is present, where will pyruvate go?
Be changed into acetyl CoA and fed into Citric Acid Cycle
If oxygen is NOT present, where will pyruvate go?
either be fermented into ethanol or made into lactic acid
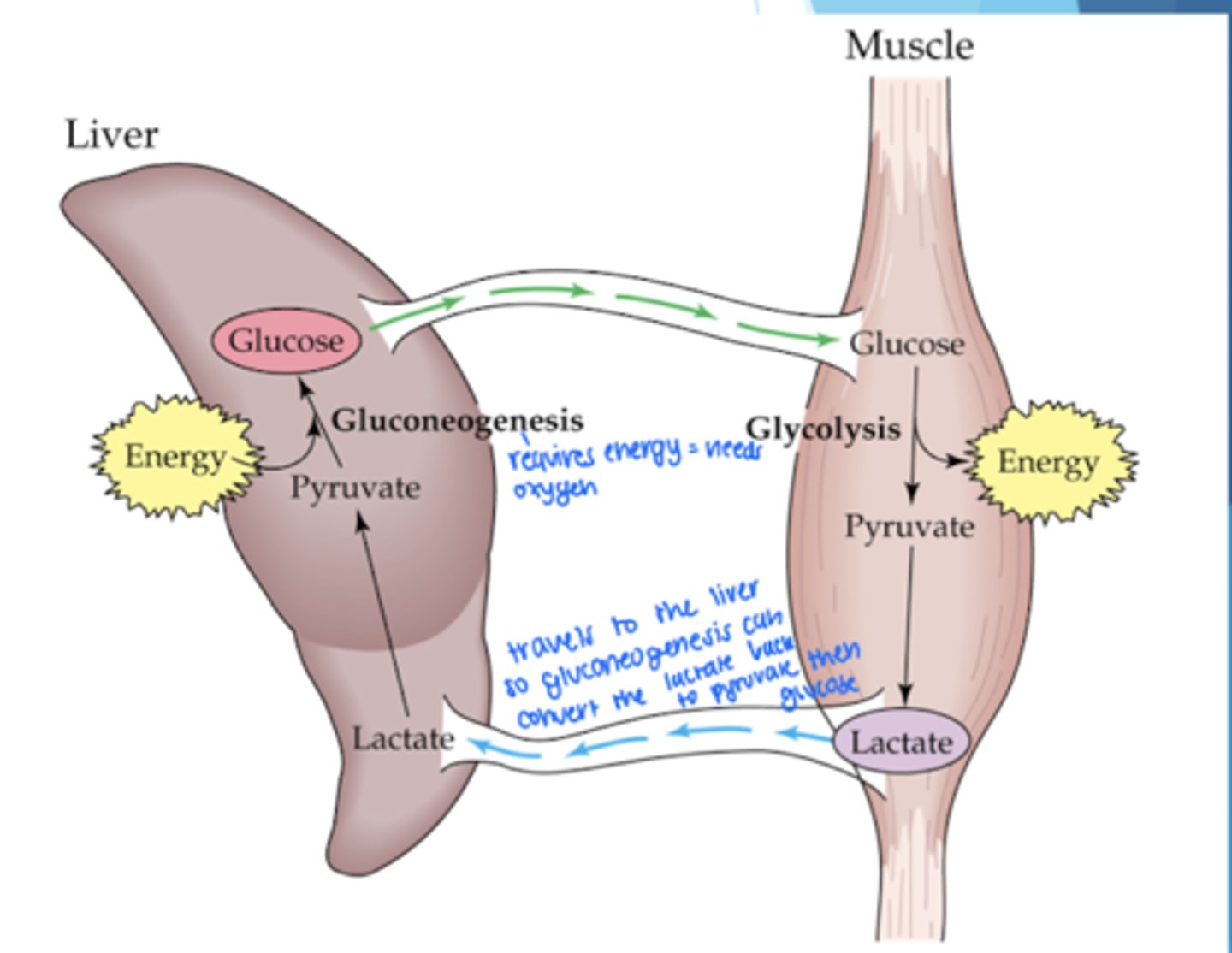
Lactic Acid Production in Muscles
-occurs with oxygen debt (using energy faster than producing)
-lactate circulates to liver and is converted to glucose to get ATP back to muscle cells
-requires energy which is often depleted during exercise (oxygen required to restore energy)
What is the main goal of both lactate dehydrogenase and alcohol dehydrogenase?
recycle electron carriers for more glycolysis
Citric Acid Cycle
-also known as krebs cycle
-occurs in lumen of mitochondria
-pyruvate is converted to acetyl-coA and that enters into the cycle
-process of breaking bonds to release electrons which then bind to NAD and FAD (go to ETC)
-products:
1. 2 GTP
2. CoA regenerated
3. NADH + FADH2 (go to ETC)
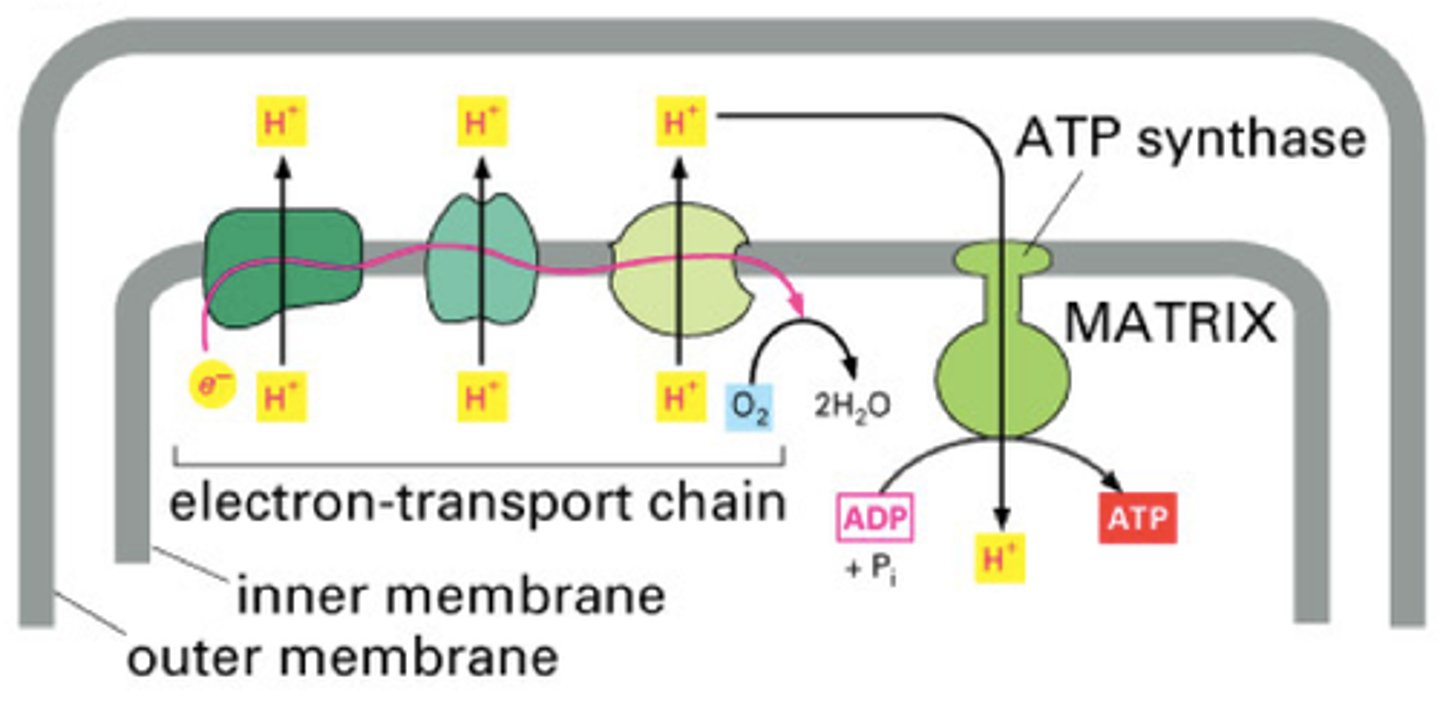
Electron Transport Chain
-oxidative phosphorylation
-occurs in inner mitochondrial membrane
-electrons are passed form one complex to the next, decreasing in energy over time
-energy helps to pump H+ ions across the membrane
-electrons are accepted by oxygen at the end and produce water
-generate 32 ATP
Gluconeogensis
-reverse synthesis of glucose from pyruvate or other intermediates
-some tissues rely heavily on glucose (brain and muscles)
Where does the majority of ATP come from in metabolism?
Electron Transport Chain!
Glucose Storage
-excess glucose is stored as glycogen in the liver (separate stores in muscle and brain)
-glucagon (the reverse of insulin) mediates the release of glycogen back into glucose
Fate of Glycose 6-phospate
-intermediary of glycogen
1. glycolysis (muscles, brain)
2. gluconeogenesis (liver)
3. pentose phosphate pathway (generate ribose and NADPH)
Fats and Energy Production
1. contribute to acetyl CoA synthesis (necessary for citric acid cycle) "fatty acid oxidation"
2. liver: gluconeogenesis from glycerol in fats
Proteins and Energy Production
amino acids can contribute carbons to citric acid cycle if needed
Lipid Metabolism
-production of Acetyl CoA
-occurs in mitochondria
-Carnitine acts as a carrier for fatty acids across the mitochondrial membrane
-partially controlled by cAMP levels (need high cAMP to function, get cAMP when glucose levels are low)
Fatty Acid Oxidation
-good at producing energy but not as fast as using glucose
-for every 2 C in fatty acid chain, 1 acetyl CoA is generated
-FADH2 (2 ATP) and NADH (3 ATP) generated
-ex: 14 C would give 7 cycles= 7 FADH2 (14 ATP) and 7 NADH (21 ATP)
Diabetic Ketoacidosis
-occurs when body isn't making insulin or isn't using glucose properly
-insulin is required for glucose uptake in cells, insufficient insulin results in metabolic stress
-can cause: nausea, vomiting, swelling of brain, coma, death, acidosis, hyperglycemia
Pathophysiology of Ketoacidosis
-lack of insulin= no glucose metabolism
-glucagon is also in excess > gluconeogenesis (the liver is metabolizing more glucose, thinking the body doesn't have enough)
-cell uses triglycerides, amino acids for energy
-glucagon stimulates conversion of fatty acids to ketones (acetone is exhaled = sweet breath)
Amino Acid Degredation
releases nitrogen which is then carried away in urea cycle
Essential Amino Acids
-cell can't synthesize all amino acids
-essential ones must be consumed dietarily
-in times of stress, we must also consume conditional amino acids
Transaminiation
-how non essential amino acids are produced
-transfer of amine groups from one molecule to another, substituting for a ketone
-requires a variety of coenzymes to occur (ex: folic acid)
Urea Cycle
-amino acid degradation
-produces urea from ammonia (a way to get rid of excess nitrogen we don't need)
-required to clear ammonia from blood (elevated NH3 in blood indicates Kidney and liver damage)
-"coupled" with TCA cycle
CoQ Deficiency
-lack of coenzyme Q synthesis
-loss of electron shuttle in ETC (so can't make all the ATP)
-result: skeletal muscle breakdown
-can be treated with CoQ or riboflavin supplements
Chronic Lactic Acidosis (CLA)
-genetic deficiency in ETC subunits
-build-up of lactic acid in tissues due to lack of respiration
-result: tissue acidification
-pH can be regulated by IV fluid
-also linked to metformin use (drug that alters metabolism)
what happens if you have increased blood glucose levels in a healthy patient?
-pancreas releases insulin
-cells pull glucose out of blood
-blood sugar decreases
What happens if you have decreased blood glucose levels in a healthy patient?
-pancreas releases glucagon
-glucagon stimulates breakdown of glycogen into glucose
-glucose gets into our blood to be used
-blood sugar increases
What do fats contribute to energy production?
contribute to Acetyl CoA synthesis (oxidation of fatty acids)
what do proteins contribute to energy production?
amino acids can contribute Carbons to Citric Acid Cycle if needed (last ditch effort)