(pt 2) exam #1 - intro to molecular diagnostics (cls 605)
1/88
Earn XP
Description and Tags
PCR + qPCR
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
89 Terms
polymerase chain reaction (PCR)
in vitro DNA replication
A way of selectively replicating a particular segment of DNA in a complex mixture of DNA
The segment to be copied is identified for the DNA polymerase by primers
useful in research, making specific proteins, infectious disease, genetic diseases, forensics
basic ingredients for PCR
Heat stable DNA polymerase (Taq polymerase)
DNA nucleotides: DNA building blocks
Primers: short ssDNA
Template DNA: DNA to be copied
Reaction buffer--contains magnesium ions (cofactor for enzymes)
components of standard PCR reaction mix
0.25 mM each primer
25 mM each dATP, dCTP, dGTP, dTTP
50 mM KCl
10 mM Tris, pH 8.4 (buffer)
1.5 mM MgCl2
2.5 units polymerase
102-105 copies of template
50 uL reaction vol total
PCR master mix
mixture of all the ingredients of PCR minus the template
Benefits: more efficient, less room for error
Multiply all the amounts of each ingredient by the number of reactions you are going to run (plus extra)
Distribute to each individual reaction tube before adding template
(PCR ingredients) Taq polymerase
isolated from hot water bacterium Thermus aquaticus

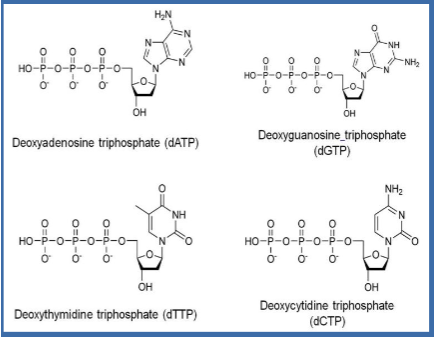
(PCR ingredients) deoxynucleotides
dATP
dTTP
dGTP
dCTP
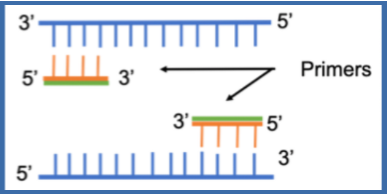
(PCR ingredients) primers
Synthetic single stranded DNA between 18 and 50 nucleotides long (oligonucleotides)
Necessary because:
Need to define DNA sequence to be copied
DNA pol needs 3' OH to add nucleotide to
types of primers for PCR
forward and reverse
Forward primer (5’→3’) = binds to antisense strand (3’→5’)
Reverse primer (3→5’) = binds to the sense/coding strand (5’→3’)
Flank sequence of interest
Each primer is complementary to a different strand
(PCR ingredients) buffer
Tris-HCl: maintains appropriate pH
Salts:
KCl--stabilize DNA, promote primer annealing
MgCl2--Mg is a cofactor for Taq pol
Other stabilizers and enhancers: DMSO, BSA
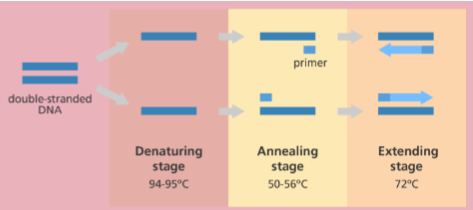
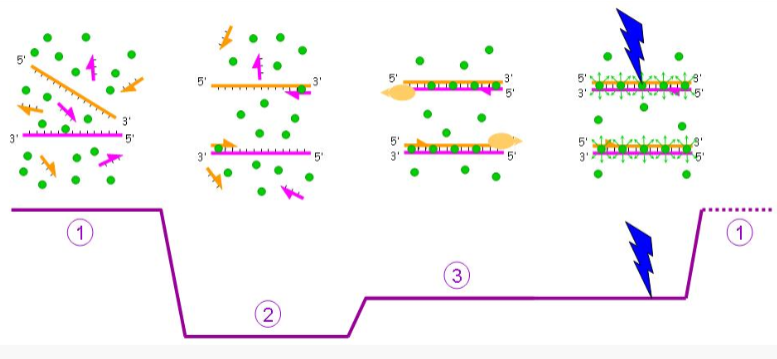
PCR cycle/steps (general)
Step 1: denaturation (~94 deg C)
dsDNA becomes single stranded
Step 2: annealing (depends on primers, 54-60 C)
Primers bind to template
Step 3: extension (depends on pol, 72 C)
Polymerase synthesizes complementary strand
Repeated for 30-40 cycles
exponential amplification
qualitative vs quantitative PCR
Qualitative: electrophoresis and staining
answers if there is DNA or not
Quantitative: qPCR (aka real-time PCR)
Can determine viral load
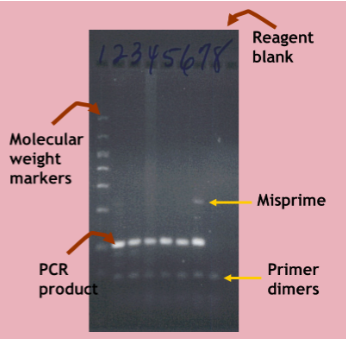
interpreting PCR results
The PCR product should be of the expected size (or melting point)
No product should be present in the reagent blank
Misprimes may be seen due to non-specific annealing of primers
Primers stick to some sequence nonspecifically and start making a product
Primer dimers may be seen due to annealing of primers to each other
optimizing PCR (general)
Critical PCR parameters
Primer design
Annealing temperature
Concentration of DNA template, nucleotide, divalent cations (Mg2+)
Type of polymerase (hot start enzymes require initial heat activation)
(optimizing PCR) primer design
CRITICAL for successful PCR
Make sure that your primers are at the correct location to amplify sequence of interest
Primers should be specific (i.e. don't want primers that target things that are repetitive)
Should avoid primers that anneal to each other
Primer melting temperature
(primer design) specificity/length of primer
Probability that any particular oligo sequence will occur randomly in a template decrease with length
If primers are too long:
Poor annealing
More likely to bind to each other
Secondary structure
Sweet spot: 18-30 nucleotides

(primer design) melting point of primer
Ideally between 60-65 degrees C
Melting temp of primers should be within 2 C of each other
Use an annealing temperature that is about 5 deg lower than the lowest Tm in the primer pair
Melting temp calculation/equation: Tm in C = 4(#C+#G) + 2(#A+#T)
melting point/temp (Tm)
temperature at which half of DNA duplex dissociate into single strands; temp where the primer and template start to come apart

practice problem (see pic)
1st strand: Tm C = 4(7+4) + 2(5+3) = 60
2nd strand: Tm C = 4(5+5) + 2(5+5) = 60
Answer: would make a good pair
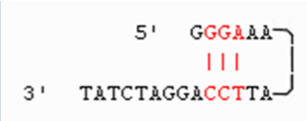
(primer design) complementary primer sequences
complementation within a single primer = hairpin
more than 3 base pairs = primer can binds to itself and not to template
complementation between primers
primers anneal to each other forming a primer dimer (DNA pol extends the primer and not actual template)

primer design process
Identify priming sites flanking a region/gene/variant of interest using NCBI gene database
Using a primer design software (e.g. Primer 3) and primer parameters, identify potential primers
Check primer specificity by in silico PCR
Check for SNPs under the primers
Primer may not bind as well
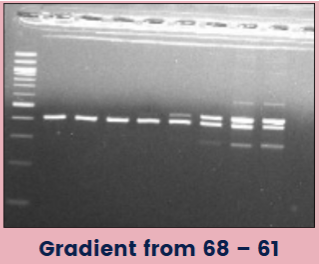
(optimizing PCR) annealing temp
Too low: products are more nonspecific; primers bind to things they aren't supposed to--bind randomly to sequences that aren't perfectly complementary
Too high: primers can't bind
Gradient protocols: programming the thermal cycler for a range of temperatures across the heat block, allowing for the ID of optimal annealing temperature
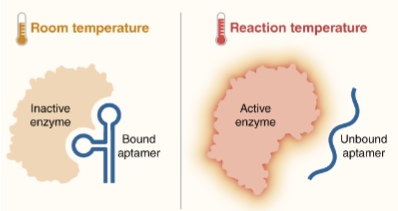
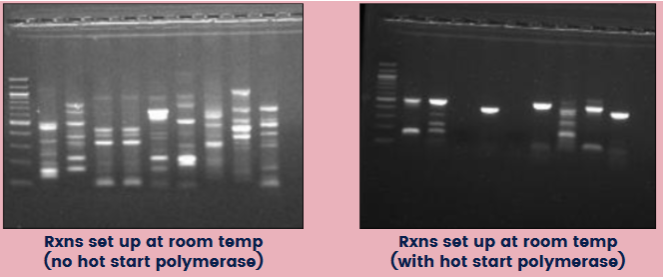
(optimizing PCR) hot start
Hot start polymerase: a sequestered enzyme that requires an initial heat activation
Benefits: starts the reaction at desired time and temp; avoid getting misprimes and unwanted products
If NOT using a hot start enzyme, reactions should be set up on ice
(optimizing PCR) additives
Can increase PCR yield and specificity
Dimethyl sulfoxide (DMSO)--use for when you have lots of C-Gs; prevents secondary structures in the DNA templates with more C=Gs
Formamide
Nonionic detergents
Polyethylene glycol
Bovine serum albumin
N,N,N-trimethyl-glycine (betaine)
multiplex PCR
Simultaneous amplification of multiple targets in a single reaction
Each target requires a separate primer pair
Requires extensive optimization for successful amplification of all targets
Differentiation of resulting amplicons:
Amplicon size
Fluorescently-labeled primers
Fluorescently-labeled probes
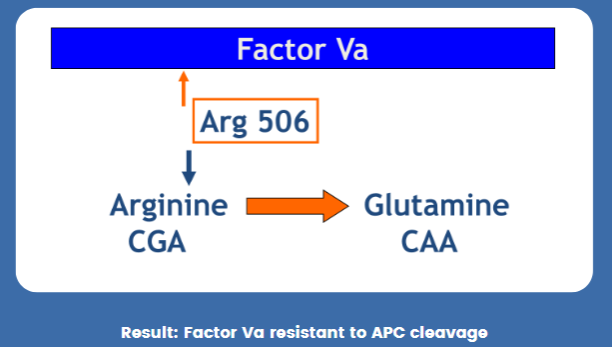
(PCR applications) Factor V Leiden
Point mutation in the Factor V gene
Arginine (CGA) → Glutamine (CAA) @ Arg 506
Inherited FVa resistance to APC cleavage
Prevalence in European ancestry ~3-8%
Autosomal dominant trait
Responsible for ~60% of familial thrombosis

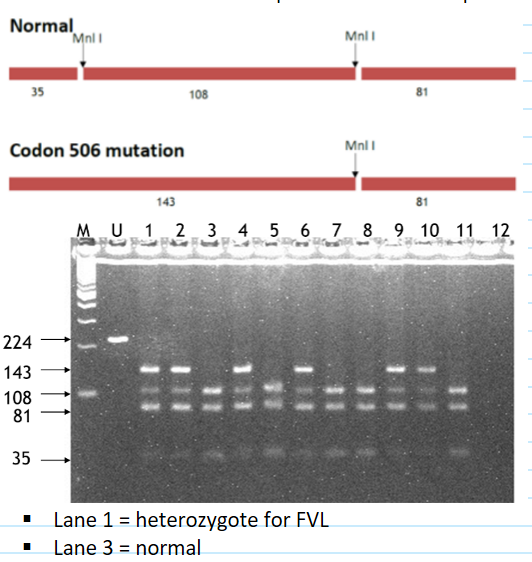
factor V leiden molecular diagnosis
method: Restriction fragment length polymorphism (RFLP): some mutations change a restriction enzyme site
PCR to amplify sequence of interest
Digest protein with enzyme of interest
Detect normal and mutant sequences with electrophoresis
(PCR applications) hereditary hemochromatosis (HH)
Autosomal recessive disorder where mutations in HFE gene = increased intestinal iron absorption and deposition of excessive amount of iron into the liver, pancreas, and other organs
HH is the most common genetic disorder in individuals of European ancestry
Heterozygote carriers: about 10%
Homozygous: 1 in 250 to 300
clinical features of hereditary hemochromatosis
hyperpigmentation, diabetes mellitus, liver cirrhosis
Other clinical features
Fatigue
Hepatomegaly (elevated serum aminotransferase)
Arthropathy
Hypogonadism (impotence in males)
Hypothyroidism
Cardiomyopathy
Symptoms typically occur late in the disease, when the total body iron content has reached as high as 20g (more than 5x normal)
Most patients are diagnosed earlier, when elevated serum iron levels are noted on a routine screening panel or screening is performed because of a relative diagnosed with HH
Approx 75% of pts are asymptomatic at presentation
hereditary hemochromatosis genetics
Affected gene: HFE
HFE protein interacts with cell surface proteins to detect amount of iron
HFE protein regulates hepcidin, which regulates iron
Two point mutations associated with HH:
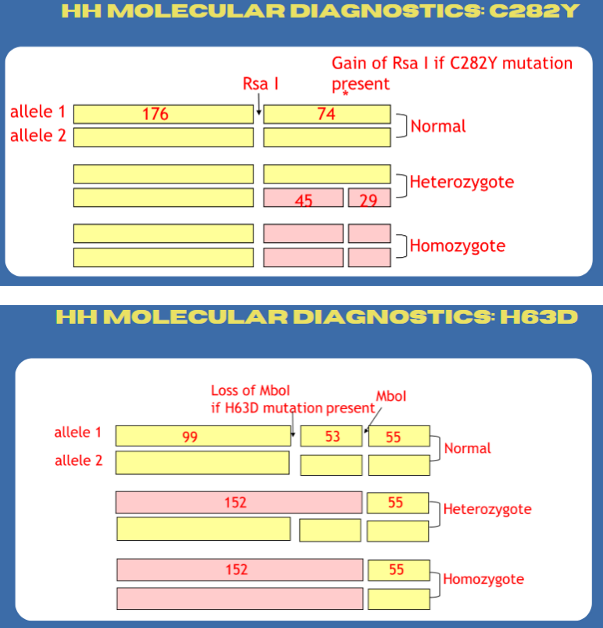
A cysteine to trypsin substitution at AA 282 (C282Y)
A histidine to aspartic acid substitution at AA 63 (H63D)
hereditary hemochromatosis molecular diagnostics
Two separate PCR-RFLP analyses using primers flanking C282Y or H63D
Molecular diagnostics cannot provide information about the degree of increased body iron stores or organ damage and thus cannot replace liver biopsy
(PCR applications) STR testing
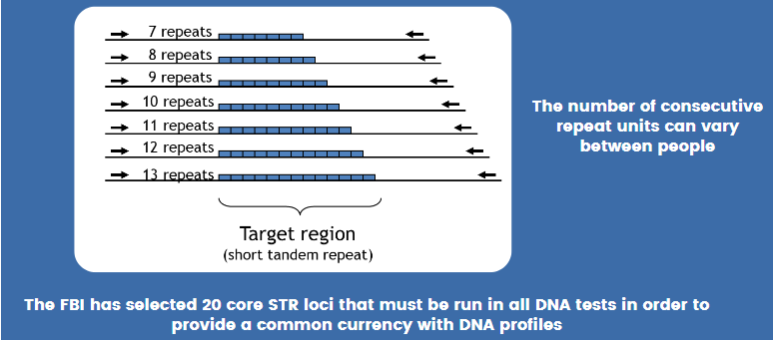
STR = short tandem repeat = microsatellite = simple sequence repeat (SSR)
Area or DNA where a short pattern of nucleotides is repeated multiple times
Individuals vary in how many repeats are present
types of STR repeat units (pic)

(STR testing) why can’t regular agarose gel be used for this kind of testing?
agarose gel does not have enough resolution ; need to be able to tell the difference between things that are only 1-2 bp apart
STR testing (general)
Multiplex PCR used to copy several STR loci at once
Sensitivities to levels less than 1 ng of DNA
Ability to handle mixtures and degraded samples
Primers labeled with fluorescent dyes
Different fluorescent dyes used to distinguish STR alleles w overlapping size ranges
(STR testing) FBI CODIS DNA database
Used for linking serial crimes and unsolved cases w repeat offenders
Missing person/unidentified human remains
Launched October 1998
Links all 50 states
Requires 20 STR markers--13 original "core" + 7 new
other uses for STR testing
Forensic paternity
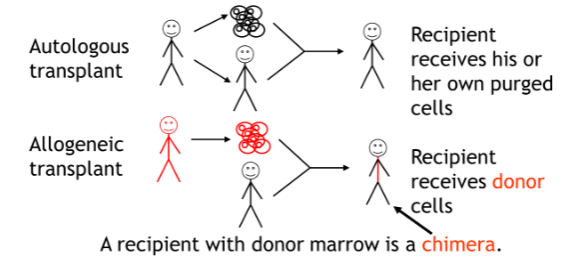
Chimerism
Allogenic bone marrow transplants are monitored using STR
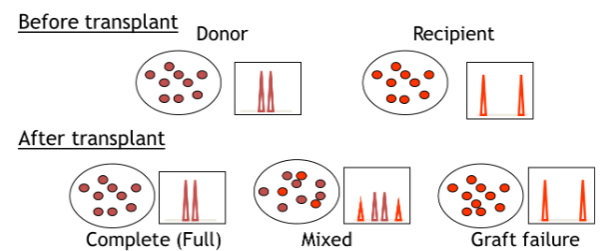
two parts of chimerism testing: pretransplant informative analysis and post-transplant engraftment analysis
modifications of PCR (list)
Multiplex PCR
Sequence specific PCR
Reverse transcriptase PCR
Nested PCT
Real-time (Quantitative) PCR)
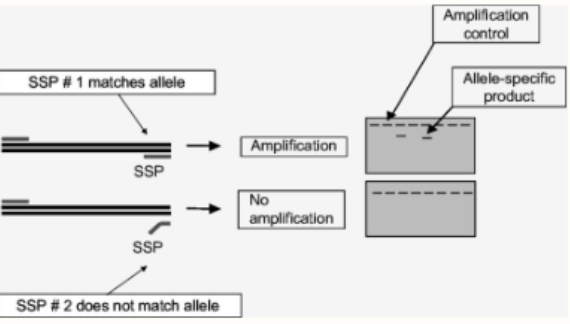
sequence specific PCR
In regular PCR, the primer can still bind to the template even if there are some mismatches
However, the primer MUST be complementary at the 3' end for annealing to occur
Can use to ID single base changes in DNA
Make primer that is complementary to mutant sequence at 3' end
Normal sequence = no amplification
Mutant sequence = yes amplification
Can detect mutant sequences; can go the other way too
reverse transcriptase PCR
use when starting material is RNA (viruses/diseases with errors in gene expression)
Use an enzyme called reverse transcriptase to turn RNA into DNA
Requires primers to generate DNA from RNA (oligo dT or random hexamers typically)
This results is a double stranded DNA copy of the RNA called cDNA
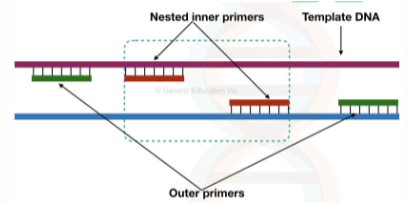
nested PCR
Increases specificity and sensitivity
Two pairs of primers are used in two separate runs
First pair of primers--amplify sequence of interest
Second pair--binds slightly inside of first pair
How would this increase sensitivity and specificity?
Sensitivity = allows for more cycles to be run
Specificity = corrects for misprimes and other issues when the cycles are run; binding of two separate sets of primers to the same target template
real time PCR (general)
not only tells us if a sequence is present, but how much
Detects the accumulation of product during exponential phase which can be monitored in real time
No gel-based analysis at end of reaction
Computer based analysis based on fluorescence detection
standard PCR vs real-time PCR
Standard (qualitative)
Detects if disease is there or not
RT-PCT (qualitative/quantitative)
Viral load
Bacterial load
Amount of gene expression
(terminology) RT-PCR
RT-PCR: could refer to "real-time" PCR = DNA amplification is monitored during PCR process
RT-PCR: could also refer to "reverse transcriptase" PCR = method to detect RNA
(terminology) qPCR
"quantitative" PCR functionally the same thing as real-time PCR
(terminology) qRT-PCR
most likely refers to quantitative reverse transcriptase PCR = quantitating the amount of RNA present
(real-time PCR) chemistry + detection + analysis method
Chemistry: use fluorescent dyes, probes, or primers
Establish a linear correlation between PCR product and fluorescence intensity
Detection: fluorescence detection to monitor amplification monitored in real time
Analysis: software analysis and estimate of template concentration
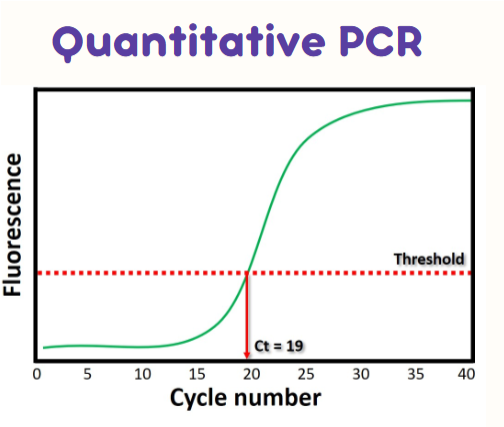
(real-time PCR) Ct value
cycle number at which the fluorescence intensity crosses the threshold line
if a sample has MORE starting material it will have a LOWER Ct value compared to something with less starting material
takes less cycles for it to cross the threshold
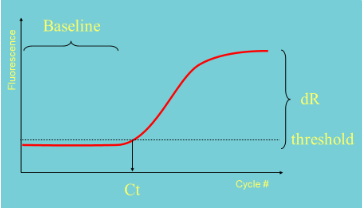
(real-time PCR) how to set a threshold
Set within the exponential phase of PCR
Needs to be set above background "noise"
(real-time PCR) standard curves
Serial dilution series of sample with known number of copies
Use this to establish the relationship between starting target copy number and Ct (cycle number)
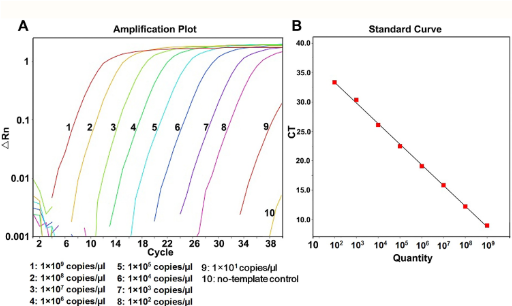
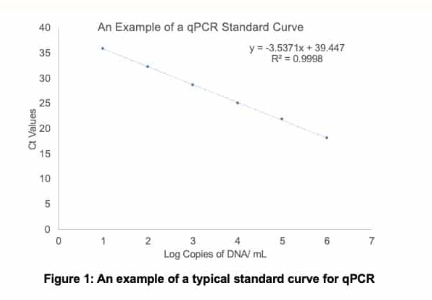
(real-time PCR) example qPCR standard curve problem: what is the starting number of copies of DNA in each sample?
Sample 1 Ct = 25
Sample 2 Ct = 18
Sample 3 Ct = 10
Sample 1: 12,148
Sample 2: 1,157,746
Sample 3: out of linearity; dilute and rerun, multiply by dilution factor
(real-time PCR) efficiency
How well a reaction doubles the target DNA sequence in each cycle
How to measure efficiency:
Perform a 10-fold dilution series
Generate a standard curve of log copy number vs. CT
E = -1+10(-1/slope)
Slope of 100% efficient reaction is -3.33
i.e., there are 3.33 cycles between CT values of each 10-fold dilution
(real-time PCR) what should the value for efficiency be?
in between 90 and 100%
Slope should be between -3.6 and -3.3
If below -3.6, then rxn has poor efficiency
Causes of poor efficiency:
Samples may contain PCR inhibitors
PCR primer and/or probe design not optimal
Inaccurate sample and reagent pipetting
Standard curve not properly analyzed
(real-time PCR) qPCR negative controls
NTC (no template control): should be negative—if positive, you may be detecting primer dimers
NAC (no amplification control): omit DNA polymerase—controls for background fluorescence
NRT (no reverse transcriptase): makes sure no genomic DNA is present when your starting material was RNA
(real-time PCR) qPCR positive controls
Exogenous positive control: external DNA or RNA carrying a target of interest
Endogenous positive control: target that is present in the experimental sample(s) of interest, but is different from the target under study (typically a "housekeeping" gene)
(real-time PCR) Your no template control is positive. What could be the cause? How would you resolve?
contamination/primer dimers
use different primers/different temp or run PCR again
(real-time PCR) Your endogenous control (positive) is negative. What could be the cause? How would you resolve?
bad sample/missed step or reagent/bad reagent
recollect, run again OR add reagent/replace reagent
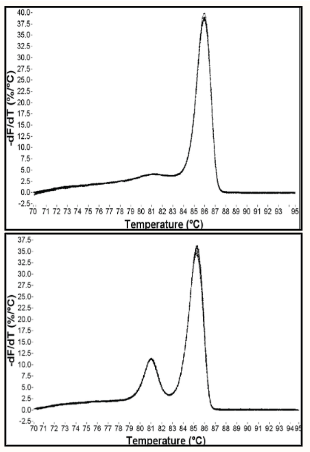
(real-time PCR) melt curves
Something that you can add on after amplification is complete
Typically used with SYBR green chemistries
Answers if you only amplify one product or not
Slowly heat the end product--as DNA denatures, the fluorescent signal goes away
Larger products = more heat to denature
(real-time PCR) technical replicates
Running multiple reactions on the sample (usually 3)
qPCR highly sensitive to pipetting errors
Ideally replicates should be tightly clustered
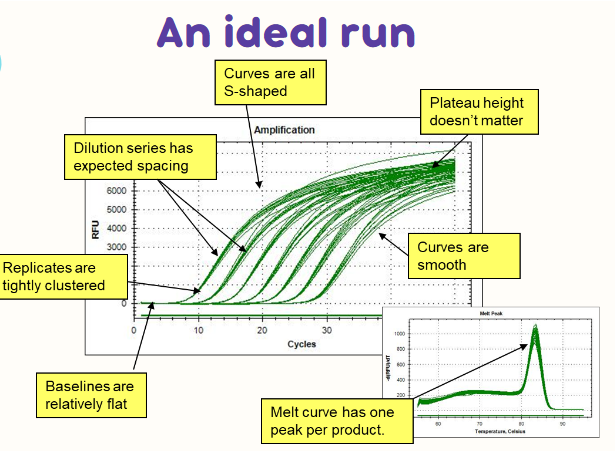
(real-time PCR) ideal run graph (pic)
curves are all S shaped
dilution series has expected spacing
replicates are tightly clustered
baselines are relatively flat
plateau height doesn’t matter
curves are smooth
melt curve has one peak per product
(real-time PCR; troubleshooting) replicates
If replicates aren't tightly clustered, suspect:
Pipetting error
Poorly optimized PCR reactions
Sample evaporation
Unknowns outside of range of detection
Instrument calibration
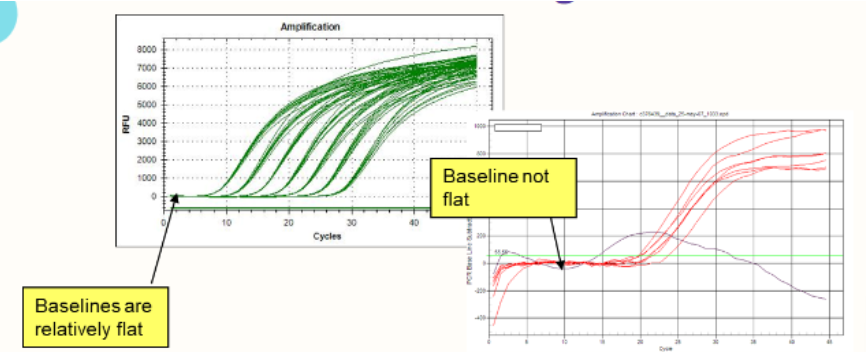
(real-time PCR; troubleshooting) baselines
If baselines aren't flat, suspect:
Sample evaporation
Bubbles
Reagents not thoroughly mixed
Baseline "window" not properly set
(real-time PCR; troubleshooting) dilution series
If the dilution series comes out "compressed" or "stretched," suspect:
Pipetting
Too much DNA (for your assay)
PCR inhibitors
Too little DNA (for your assay)
Poor PCR efficiency
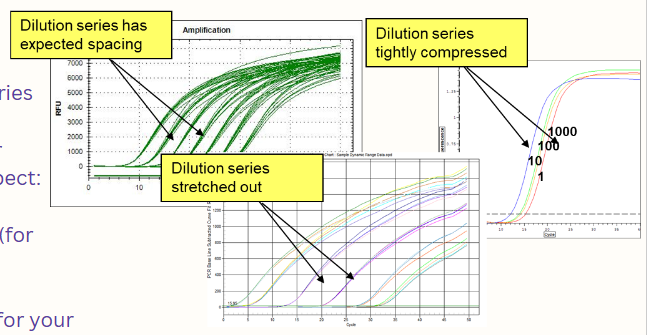
(real-time PCR; troubleshooting) curve shape
If curves are not S-shaped, suspect:
Curves are not actual PCR products!
Sample evaporation
Fluorescence drift in unamplified samples
Something seriously wrong with assay
If curves are not smooth, suspect:
Poor pipetting (bubbles)
Sample evaporation
Poor assay (low fluorescence reagents)
System malfunction
(real-time PCR; troubleshooting) melt peaks
If melt curves have more than one peak:
More than one product
Possible normal primer-dimer
Using too low an annealing temperature
Primers need to be redesigned
advantages of qPCR
Quantitative
Amplification and detection all in one
No post-PCR processing
Reproducible
Potentially not influenced by non-specific amplification
Many chemistries are sequence specific or use melt curve analysis
Rapid
Very sensitive
disadvantages of qPCR
A more limited capacity for multiplexing than standard PCR
Development of protocols needs high level of technical skill and/or support (requires R&D capacity and capital)
High capital equipment costs
(qPCR applications) HCV
Blood borne virus
Initial infection is often asymptomatic, but can result in chronic infection leading to cirrhosis of the liver and liver cancer
No vaccine ; curative treatment = direct acting antivirals
diagnosed through reverse transcriptase and qPCR
HCV reverse transcriptase-PCR viral quantitation detection
Convert the RNA to DNA using reverse transcriptase
An HCV-specific primer is used to prime the cDNA synthesis reaction
After cDNA synthesis, a PCR reaction is performed using HCV-specific primers
Taq DNA polymerase
Two primers: the primer used for cDNA synthesis and a second HCV-specific primer
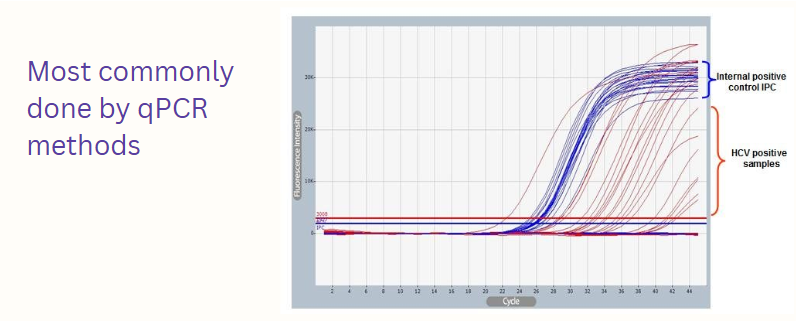
how does real-time PCR detect the formation of products?
fluorescence (SYBR green, TaqMan, FRET, Molecular Beacon, isoG:C, Scorpion)
fluorescence
Absorption of light cause excitation of electrons
When electrons return to the ground state, light is emitted--fluorescence
Light emitted is always lower energy (longer wavelength) than light absorbed
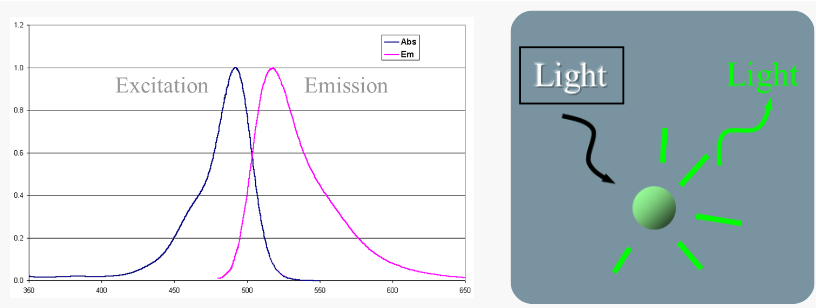
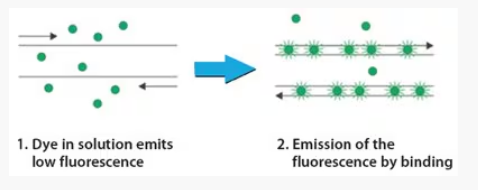
(qPCR chemistries) SYBR green
SYBR green is a fluorescent dye that binds to dsDNA
Low fluorescence when not bound to dsDNA
Higher levels of fluorescence with DNA binding
Absorption maxima 497 nm (blue)
Emission maxima 520 nm (green)
what stage of qPCR is fluorescence detected with SYBR green?
during extension! (last stage)
little to none during denaturation
little bit during annealing
when is maximal fluroescence detected with SYBR green? when is the measurement taken?
at the end of extension step
taken at the end of a cycle/extension
pros/cons to SYBR green
pros
Easy ; cheap
No need to design/buy probes
cons
Nonspecific
More background fluorescence compared to other methods
types of probe-based detection chemistries (3)
TaqMan
FRET probes
Molecular Beacons
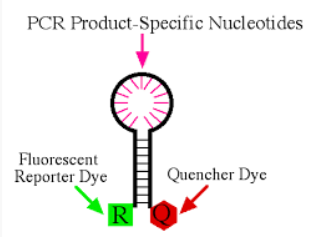
probe
a single stranded synthesized DNA molecular, often about 18-25 bases in length
Complementary to target—anneals between primer binding sites
Labeled with fluorescing and quenching nucleotides
probes vs primers
probes are NOT designed to be the starting place for Taq pol—no 3’ end to attach bases to
usually only one probe is used compared to 2 primers
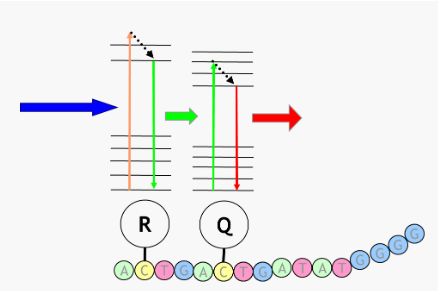
(probe based detection) fluorescence resonance energy transfer
aka FRET ; basis behind most probe technologies
Reporter (R) absorbs light (blue) and fluoresces (green)
Emitted green light is absorbed by the Quencher (Q)
Quencher then emits light of still longer wavelength (red)
This energy transfer only occurs when the Reporter is next to the Quencher
(FRET) quenching nucleotides
Absorb the energy emitted by fluorescing nucleotides
Light quenchers: absorb fluorescent energy from the "reporter fluor" and then emits fluorescent light at a longer wavelength
Dark quenchers: absorb fluorescent energy from the "reporter fluor" and then do not emit light
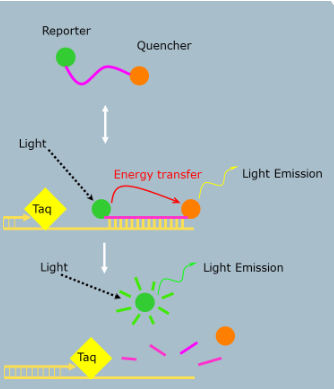
(probe based detection) TaqMan probes
Unbound probe free in solution (no reporter fluorescence)
During annealing, probe and primer bind target (no reporter fluorescence)
During extension, Taq hydrolyzes (eats up) probe and reporter can now emit fluorescence
when is the fluorescent signal detected in TaqMan probe based methods?
during extension!
Taq pol will hydrolyze the probe and the reporter/quencher are now unbound and far away so the quencher doesn’t absorb up all the signal
(probe based detection) FRET probes
uses two adjacent hybridization probes:
One attached to donor fluorophore (reporter)
One attached to accepter fluorophore (quencher)
Designed to bind near each other
No signal from the acceptor fluorophore without binding near the donor fluorophore on the DNA template
When they bind near each other, the donor fluorophore excites the acceptor fluorophore which then emits fluorescence that can be measured
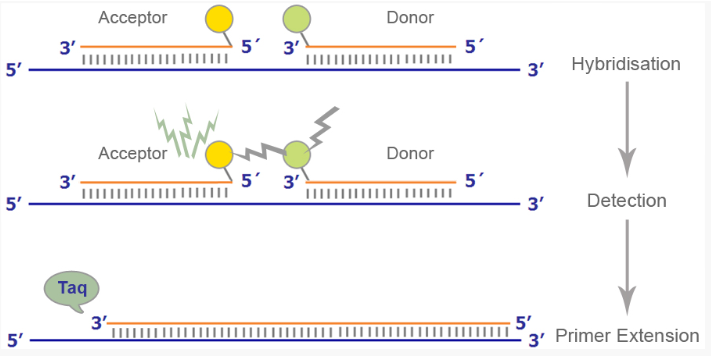
at what point in the PCR cycle should fluorescence be measured for FRET probes?
at the annealing step
probes can be resused and are not destroyed
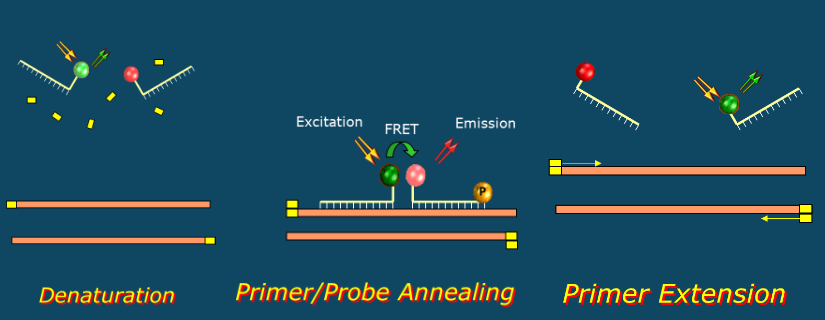
(probe based detection) molecular beacons
Hairpin shaped DNA probes
Labeled with reporter and quencher at 5' and 3' end
Added to PCR reaction mix
Hybridize to target during PCR annealing step
Hybridization physically separates fluor from quencher allowing reporter fluorescence
Maximal fluorescence during ANNEALING
what two chemistries is the signal gathered at the annealing step?
FRET probes
Molecular Beacons
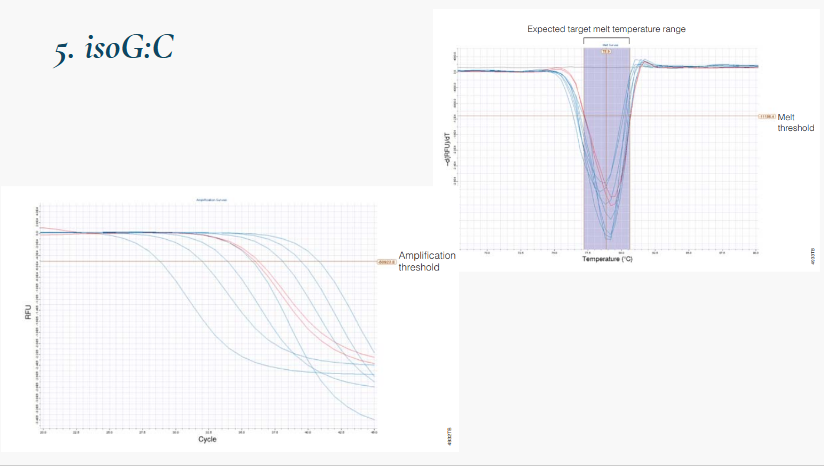
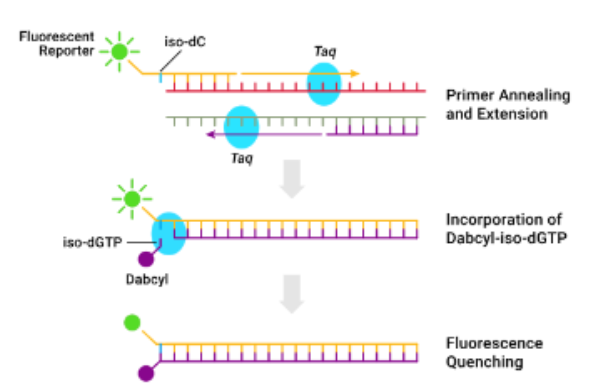
(fluorescently labeled chemistries) isoG:C
Forward primer labeled at 5' end with fluorescent iso-C
Reverse primer is unlabeled
Dabcyl-iso-dGTP (quencher) in dNTP mix
During elongation, dabcyl-iso-dGTP incorporated opposite iso-C
Proximity of dabycl to reporter quenches fluorescence
Signal decreases as product accumulates
(fluorescently labeled chemistries) scorpion primer/probes
Similar to molecular beacons but a primer and a probe are contained in one molecule
After extension of the primer, the target-specific sequences fold over to hybridize with the newly synthesized target sequences, separating the reporter from the quencher
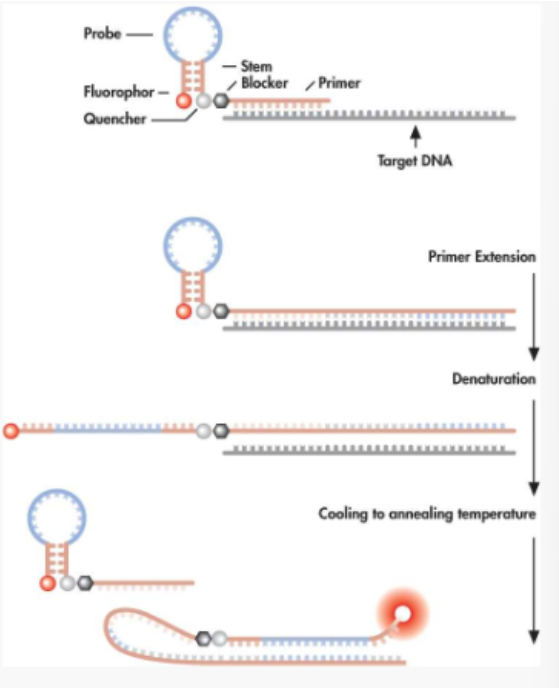
list of real-time PCR applications
Pathogen detection and genotyping assays
Microbial/viral load assays
SNP detection, allele discrimination, genotyping, haplotyping
Clinical diagnostics (cancer, therapy response)
DNA target quantification (nuclear, mitochondrial, residual DNA in protein preps [QC])
Gene expression (and microarray validation)
DNA methylation, apoptosis analysis
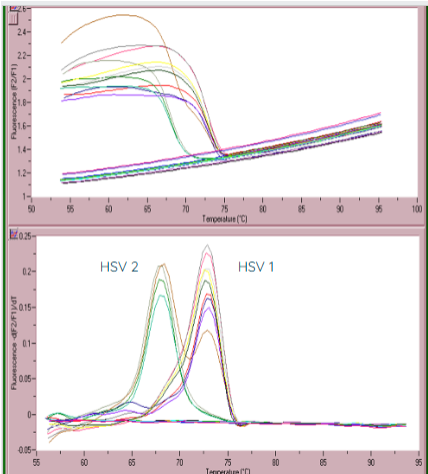
(qPCR applications) HSV detection
can make FRET probes to HSV-1 and HSV-2
if the probes attach to the amplicon DNA without a mismatch, then HSV-1 is suspected
HSV-2 is suspected when there is a mismach in the FRET probes
can be confirmed with a melt curve (measuring the change in fluorescence)
due to the probe having a mismatch with HSV-2, the probe will dissociate from the product at lower temperatures
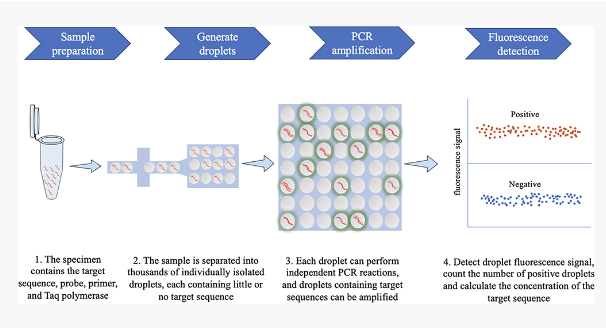
digital droplet PCR
Digital PCR that is based on water-oil emulsion droplet technology
Each droplet is read in a flow cytometer to determine the positive droplets in sample
Data is then analyzed using Poisson statistics to determine the target DNA template concentration
Provides and absolute quantification of target DNA copies without need for a standard curve
Particularly useful for detecting a small number of tumor cells among lots of wild-type cells/rare mutations