Lecture 3 & 4: Sickle Cell Anaemia
1/59
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
60 Terms
Sickle Cell Disease: Frequency of Disease
UK 10-15,000 affected in all ages
Worldwide 300,000 affected individuals born each year
In high incidence areas affects 2-3% of all births
Sickle Cell Disease: Life Expectancy
UK: 40-60 years
Improving year-on-year
Developing nations: 50-90% childhood mortality
Sickle Cell Disease: Economic Costs
Developed Countries (US) $27,779 per patient per year
Gene therapy £1 million
Developing – folic acid (£15)
Balanced Polymorphism
A (genetic) disease that causes a very serious illness, where the gene should have died out but hasn’t due to it providing heterozygotes with a genetic advantage
Sickle Cell Disease: Genetic Basis
A single base change (A to T) causes glutamic acid on the 6th position of the beta-globin chain to be replaced with valine.
Charged amino acid replaced with a neutral amino acid
mutation provides protection from malaria
Example of polymorphism
heterozygous - HbAS
homozygous recessive HbSS
homozygous dominant - Hb AB
Heterozygous State of Sickle Cell (HbAS) + Malaria
Advantageous: provides protection against malaria due to the interaction of sickle Hb and actin RBC cytoskeleton
Mutation has arisen on multiple occasions within malaria-affected areas.
highly advantageous in very high-frequency malarial areas (up to 40%).
Prevents parasite from altering the red cell cytoskeleton (and making it ‘sticky’ – an essential part of parasite life);
in _______ parasites cannot complete their life cycle as effectively.
Homozygous State of Sickle Cell (HbSS) + Malaria
Associated with severe disease
absence of modern health care - few affected individuals survive past childhood
No effective protection - malaria is a very severe disease in those with sickle cell disease as they are often already vulnerable to chronic ill health
Anaemia and poor nutritional status
Biochemistry of SCD: Disorder of Protein-Protein Interaction
Replacement of charged glutamic acid on the 6th position of beta-globin chain with neutral valine causes a structural change and charge change.
When haemoglobin is deoxygenated, the valine becomes exposed on the Hb surface.
Can interact with other Hb molecules
Significance of Change from GLU to VAL in B-globin chain
Charge of GLU prevents the chains from interacting,
normal haemoglobin: GLU molecules are charged and oppose interactions between the adjacent Hb molecules
Neutral VAL promotes interactions
sickle haemoglobin: VAL molecules have no charge and so permit hydrophobic interactions between the adjacent Hb molecules
Biochemistry of SCD: Polymerisation
Deoxygenated sickle haemoglobin may polymerise and form long fibrous linked chains
These chains distort the red cell, preventing normal flexing and producing a characteristic long-form cell with sharp ends (long sharp cell)
difficulties in flowing through capillaries and exchanging oxygen
Polymerisation of Hb in Healthy RBCs
The molecules of normal haemoglobin are separate within the erythrocyte and form a gel that can be shaped by the flexible cytoskeleton to form the typical flexible disk shape
Are RBCs Sickled all The Time?
No, Sickling reverses when red cells return to high oxygen, so initially the red cell shape is not permanently altered
reversibility of sickling is important to understanding the clinical presentation, damage to red cells, and treatment
Long chains only form following the loss of oxygen
Sickling only occurs in deoxygenated tissues
Most of the time not heavily deoxygenated
If sickled all the time, would not be able to through lungs = death
If all RBCs Form A Sickle Shape When Oxygen Is This A Problem?
Sickling process takes time to occur, so in health, most cells can return to the lung before major sickling occurs
Affected individuals do not have sickling at all times - dependent on crystalisation
Crystallisation
Not an immediate process - 2 parts
Slow nucleation occurs, where cells begin to stick together – unlikely for cells to sickle – happens too slowly – blood reoxygenated
Followed by rapid chain formation – gives rise to the sickled shape
The time taken for sickling of red cells is critical to the clinical illness of patients
How are Foetuses Protected Against Sickle Cell Disease
in the developing baby the beta haemoglobin chain is replaced by HbF to form foetal haemoglobin - no sickle haemoglobin present until after delivery.
Hbf not lost until 3-6 months after birth – B chains not initially produced - no sickle Hb – gives babies protection
has a role in treatment and disease severity
Variation in Sickle Cell Phenotype
Not all patients with sickle cell disease are equally affected - some have much milder disease
All have the same sickle cell disease mutation but have co-inherited factors that change disease severity
Hereditary Persistence of HbF In Sickle Cell Disease (HPFH)
Hereditary persistence of foetal haemoglobin (HPFH) in affected individuals the foetal haemoglobin remains in adult cells at a high concentration (up to 20%)
Has two effects:
fewer sickle haemoglobin molecules, but
HbF does not form polymers and interferes with the formation of long chains of haemoglobin - sickling is less severe
Interfere with crystallisation proteins
Cell Biology of Sickle Cell Disease
A disease of
Chronic red cell damage
Acute crisis
Sickle Cell Disease - Chronic Damage
Damaged to Hb in response to repeated cycles of polymerisation and depolymerisation
Eventually, the polymerisation does not reverse, and haemoglobin may become denatured
Cell may become ‘irreversibly sickled’ – can’t enter lungs
The repeated cycles of shape-change then reversal damage the cells causing permanent partial shape change and damaging and degrading membrane pumps so that cells become dehydrated
Unable to transport oxygen or sit in gel
Sickle Cell Disease- Anaemia
Red cell survival in blood is greatly reduced
often from 120 days to as little as 8 days.
Production is increased to compensate but cannot replace cells rapidly enough, so patients are chronically anaemic.
Typical haemoglobin levels are 70-80g/l compared with normal around 130-140g/l.;
low level is compensated for by other systems
Spleen
Responsible for removing damaged cells and circulating pathogens in the blood
Sickle cells enter and are destroyed – shortening their lifespan
Blood Appearance in SCD (General)
Red blood cell damage:
Repeated cycles of sickling and recovery damages red blood cells.
Damaged cells are prematurely destroyed.
This leads to anaemia.
Most cells are not typically sickle-shaped, but few have a normal shape
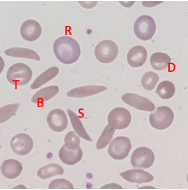
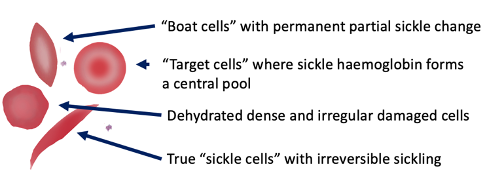
Sickle Cell - Blood Appearance: True Sickle Cells
Permanent, irreversible sickling
Pointy ends
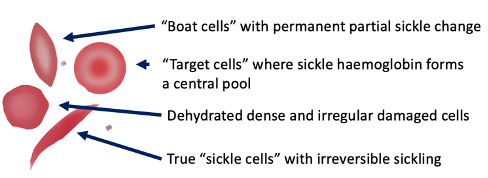
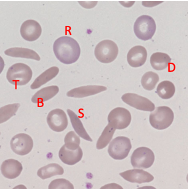
Sickle Cell - Blood Appearance: Boat Cells
Permanent partial sickle change
elongated but not spikey
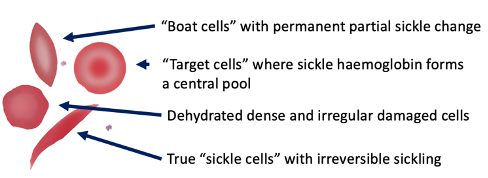
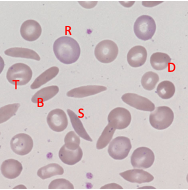
Sickle Cell - Blood Appearance: Dense Irregular Cells
Dehydrated cells
Hb is squashed due to repeated cycles of polymerisation/depolymerisation
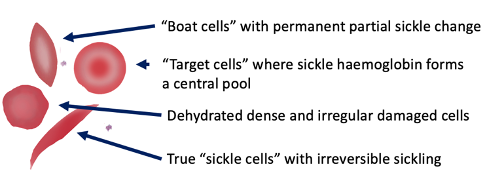
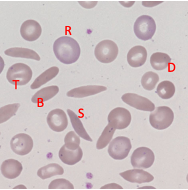
Sickle Cell - Blood Appearance: Target Cells
Sickle Hb forms a central pool
Disruption to the Equilibrium Between RBC and Sickle Shape
Gives rise to acute crisis
Don’t readily form sickle shapes in capillaries but equilibrium can be shifted and can occur more readily and rapidly - blocks blood flow
Hypoxia, fever, and dehydration favour sickle-shape
Reason for Hospitalisation
Why does Dehydration Favour Sickle Shape and Assit Acute Crisis
Affects peripheral circulation - slows down blood flow
This give a longer time for the sickling process to occur
In some physiological conditions, the sickle cells may form in small vessels
Acute Crisis
Sickle cells block the small vessels causing a failure of blood supply and tissue damage
Cells can pass round bends and stick to other cells and form meshes
Can cause the activation of white cells in response to blockages – release cytokines
Process affects tissue beyond the site of blockage as blood supply is lost – tissue death
Clinical Effects of Acute Crisis
Damage to many organs
Bone pain and damage – oxygen supplied to bones via small vessels – blockages occur here
Chest crisis – pain in ribs – less likely to take deep breaths – hypoxic conditions - sickle
Stroke – loss of blood to the cerebrum
Sepsis – due to loss of blood supply to tissue – infection more likely as tissue dies
WBC protection lost
Clinical Effects of Sickle Cell: Chronic Anaemia
Red cell damage and premature destruction
Adaption through increased heart output to increase O2 transport - symptoms less apparent when young
RBC don’t survive as long
When older it is problematic - chronic problems to heart and lungs e.g. acute chest syndrome, pulmonary hypertension and renal diseases
this along with effects of repeated crisis limits life expectancy
Clinical Effects of Sickle Cell: Bone Pain
Blockage of smell vessels supplying the bones causes sudden onset of very severe pain in children and adults.
This can sometimes be managed at home but may need admission and strong painkillers.
Chronic pain can be debilitating and areas of dead bone can become a focus for infection e.g. necrosis and ulceration
Dactylitis
Severe swelling and pain
Can impair bone growth and dead areas of bone can be prone to infection
Long term treatment - antibiotics
Clinical Effects of Sickle Cell: Skeletal Deformity
Occurs in response to blockage of small vessels that supply bones in children can damage the ‘growth plate’ preventing their normal growth and resulting in long-term problems
Can result in shorter fingers
Clinical Effects of Sickle Cell: Stroke
Rare but severe
the blockage of small vessels in brain leads to death of the brain tissue they supply
People at risk must be treated much more intensively to reduce the chance of further ____ and permanent disability
Risk due to abnormal circulation
Clinical Effects of Sickle Cell: Spleen and Infection
Removes damaged cells but is full of tiny capillaries – resulting in damage
Repeated sickling destroys the tissue leading to fibrosis and calcification.
Organ becomes non-functional, increasing the risk of sudden and severe infection with particular types of bacteria (encapsulated bacteria)
Organ becomes smaller and shrinks as a result of damage
Clinical Effects of Sickle Cell: Chest Crisis
Severe sickling can cause the blockage of small vessels in the lungs – results in fluid leaking into tissues - results in inflammation and damage
Poor gas exchange - increases chance of sickling
Lung tissue and heart enlarge to compensate
Reduces oxygen further and the pain can prevent individuals from taking full breaths
Leads to a cycle of further hypoxia and sickling.
a medical emergency and is one of the major causes of death in sickle cell disease.
In the long term, can result in chronic lung disease.
General Aim of Treatment (For Crisis)
To slow the sickling process by reversing any causes and improving oxygenation and circulation through fluids and removing pain
Tissue damage can then be limited allowing the body to heal itself
Treatment for Mild/Simple Cases of Crisis:
Pain relief: for comfort and to allow better movement breathing and self-care
Individuals in pain don’t drink/ inflate lungs well – further problems
E.g. paracetamol, ibroberaphen, morphine
Hydration: adequate fluids will preserve blood flow and red cell hydration
given a drip in hospitals or drink 3L of water at home
Treat causes: reverse causes to prevent ongoing sickling e.g. high altitude or infection – give antibiotics, treat pain
Assess: who is seriously ill and may need hospital care
Return to normal life: as soon as possible
Treatment for Severe Cases of Crisis
Use simple measures to treat the bone pain and crisis and
Transfusion
Exchange transfusion
Experimental therapies of clinical trial
Transfusion
Used to treat severe cases of crisis if Hb levels are very low
Raising Hb increases oxygenation – reducing likelihood of sickling
Can’t have too many transfusions – can develop antibodies – difficult to cross-match
Exchange Transfusions
Treatment for those with severe chest crisis
Used if life-threatening
It aims to replace most of the circulating sickle cell blood with normal blood – if the % of sickle blood is lowered to less than 30% sickle damage is prevented
Replacement of sickle blood with normal blood → take one unit and replace with a new unit
Long Term Treatments: Preventative Measure
Used to reduce the frequency of crises and maintain health:
Education (primary approach) – safety!
Vaccination – protection for spleen
Antibiotics -prevents pneumonia and meningitis
Folic acid – promotes RBC production
Long Term Treatment: Modifying Red Cell Physiology
Raise HbF
the drug hydroxycarbamide is recognised to increase intracellular HbF and can reduce the frequency severity of crises
Reduces instances of sickling
Long Term Treatment: Chronic Transfusion
Used in severe disease
Transfusion done monthly to replace all sickle blood with normal blood, or bone marrow transplant for very selected case
Problem with Bone Marrow Transplantation
Dangerous
Long term problems
Mortality risk
Experimental Therapies
Gene therapy - replace HbSS/ increase HbF
Anti-sickling molecules - prevent HbSS protein-protein interactions → prevent crisis
Anti-adhesion molecules - stop crisis by making Hb less sticky and block vessels
Modify vascular tone - relax capillaries and improve blood flow
nitrous oxide improves vascular tone
Treatment of SCD in Developing Countries
Most cases are in developing countries
Low-cost interventions are critical!
Screening – to identify cases early
Education – to discuss what can be done
Folic acid – cheap and supports blood cell production
Vaccination, antibiotics – reduce crises
Malaria treatment – as needed – biggest treatment of homozygous
Hydroxycarbamide (£250 per year)
Factors Affecting Choice of Diagnostic Test
Speed – sometimes you need a fast result e.g. surgery
Accuracy – most often accuracy is more important than speed
Expertise – simplicity makes tests easier to provide
Throughput – high capacity may need test that can be automated
Cost – the cost of the health service must always be considered
Types of Diagnostic Test
Cell biological approach: Look for sickle cells
Physiological approach: Detect sickle haemoglobin polymerisation
Biochemical analysis: Detect the abnormal charge properties of haemoglobin
Gene analysis: Detect the typical mutation
Blood Smear
Looks for sickled cells themselves – distinct characteristics can be seen on a blood film
Cheap and (fairly rapid)
ROLE: may spot an unexpected diagnoses
Problem of Using Blood Smear as A Diagnostic Test
Expertise required – may be overlooked
Relatively low sensitivity in healthy patients such cells can be rare – can’t see characteristics cells
Not good for complex diagnoses where there are multiple other problems affecting cell appearances
Doesn’t consider Hb or genes
Haemaglobin Solubilty Test
Detects SHb – forms polymers
Use detergent to lyse red cells then add a reducing agent
normal haemoglobin forms a clear suspension of monomers - relatively transparent.
Sickle haemoglobin forms polymers - solution is cloudy and cannot be seen through
ROLE: Great in emergencies e.g. Pre-operative
Advantages of Using Haemoglobin Solubility Test
Cheap,
Quick,
very sensitive,
Little training is required
Problem of Using Haemgolbin Solubity Test as Diagnostic Tool
Too broad
Test doens’t distinguish between homozygous and heterozygous → not good for complex diagnoses
Test Based on The Altered Charge of Sickle Cell
Different charge of sickle haemoglobin means it will move at a different rate than normal haemoglobin when placed in an electric field.
Shown using flatbed electrophoresis
relatively slow with lower capacity
Hb lysed and placed at the end of the paper
Normal Hb – glutamic acid – charged
More charge – moves faster
Sickled Hb – valine (neutral) moves slowest
ROLE: Used for non-urgen diagnosis
Use of Chromoatographic Machines
Analysis done using a high-pressure column chromatography detecting different haemoglobin forms as they emerge under high pressure from a charged column.
Differentiates betwen Homozygous and heterozygous
one band healthyHeterozygous – 2 bands (middle)
One band further right – disease – moves slowly in chromatographic column
Machines are fast
Advantages of Using Chromatographic Machienes
Accurate and intermediate cost
Insufficiently rapid for very urgent samples, and requires expertise to interpret
Great for complex cases and can be scaled up for large sample numbers – differentiates between homozygous and heterozygous
Genetic Testing
Used to detect the abnormal sickle gene - if detected, diagnosis is proven
Accurate and useful in very complex cases where results of other tests are unclear
Generally retained for particular circumstances where diagnostic material may be limited, and accuracy is essential – particularly pre-natal testing
Problems of Using Genetic Testing As A Diagnositic Tool
Cost
Time
Expertise