Lecture 14: Pharmacokinetics - Metabolism and Excretion
1/53
Earn XP
Description and Tags
Block 5
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
54 Terms
Elimination
How a drug molecule is metabolised and excreted
Water-soluble drugs don’t require metabolism; they can be excreted in the urine.
The combination of metabolism and excretory pathways (assessed via clearance)
It is the removal of drug metabolites and water-soluble drugs from the body via urine >>>> bile >> faeces > lungs = skin
Clearance
Volume of plasma cleared by an elimination organ per unit of time
Influenced by multiple organs (e.g., liver, kidneys), which work together to eliminate the drug.
Used to assess elimination
Half Life
The time taken for the drug’s plasma concentration to decrease by 50%.
It is influenced by clearance.
Measured by injecting the drug, taking blood samples over time, and measuring the concentration.
Affected by the volume of distribution and clearance level
It can vary significantly, important when considering indications and drug use
Consider whether the half-life fits with the indication
Effect of Vd and CL On Half Life
t1/2 is directly proportional to the volume of distribution → the wider the drug distribution, the less contact it has with eliminating organs → cleared more slowly
t1/2 is inversely proportional to CL → the faster the drug is cleared the lowe the half life
First Order Drug Elimination Kinectics
Drugs are eliminated as a constant fraction of the drug per unit time
Elimination occurs at a rate proportional to the concentration.
Most drugs are eliminated via this route
When plotted on a semi-log graph, a straight line is seen.
This allows for easy extrapolation to determine the concentration at time 0, as the concentration decreases exponentially over time.
Zero Order Drug Elimination Kinetics
Drugs are eliminated as a constant quantity, regardless of the concentration.
When plotted on a semi-log graph, the concentration vs. time will not be a straight line.
This is because the rate of elimination is constant over time, and the concentration decreases linearly. e.g. ethanol
Often seen with eliminations that can become saturated at high [drug]
Used by some drugs
Kidney
Most important excretory organ, especially for water-soluble drugs.
It filters blood at the glomerulus: most lipid and water-soluble drugs (within a certain size) can be filtered into the nephron via the Bowman's capsule.
Water-soluble drugs are typically excreted in the urine.
Lipid-soluble drugs can be reabsorbed into the bloodstream through peritubular capillaries, surrounding the nephron
The kidney also helps remove metabolite and water soluble drugs
Other routes of excretion: bile (→ faeces), lungs (→ exhalation), skin (→ perspiration) — but the kidney is the most important.
Renal Elmination
Glomerular Filtration of Plasma
Passive Reabsorption
Active Tubular Secretion
Renal Elimination: Glomerular Filtration
Process where plasma is filtered into the nephron.
Only unbound drugs (i.e. not bound to plasma proteins) can be filtered.
The kidney receives oxygen-rich blood via the arcuate artery, supplying the glomerulus for filtration.
Renal Elimination: Passive Re-absorption
Lipid-soluble drugs can be reabsorbed from the nephron into the peritubular capillaries.
Molecules < 40 kDa can be freely filtered by the Bowman's capsule.
Reabsorption depends on urine pH and drug ionisation:
Weak acids in acidic urine = uncharged → more lipid-soluble → more reabsorption.
Similar for bases in basic urine.
Plasma protein-bound drugs (e.g., bound to albumin) are too large to be filtered; only free, unbound drugs are filtered.
Renal Elimination: Active Tubular Secretion
Organic Anion Transporter (OAT) proteins are expressed on the basolateral membrane (BM) of kidney tubule cells.
These transporters actively move drugs from the blood into the nephron for excretion.
Example: Penicillins are actively secreted into the tubules via OAT
Drug Removal
Drugs are removed via metabolism and excretion.
Most drugs are lipophilic → they tend to be reabsorbed into peritubular capillaries and recirculated in systemic circulation.
This recirculation continues until a sufficient amount is eventually excreted in the urine.
Drug metabolism involves enzyme-mediated conversion of lipid-soluble drugs into water-soluble forms for urinary excretion.
Factors Affecting Drug Metabolism
Drug metabolism can be a major liability for lead compounds:
First-pass metabolism in the gut lumen, enterocytes, and liver can reduce drug availability.
Speed of metabolism affects half-life – important for dosing in chronic treatments like analgesia.
Drug-drug interactions may alter metabolism rates or pathways.
Toxic metabolites may pose risks.
Polypharmacy
The increased risk of drug-drug interaction
Commonly seen due to an ageing population, with patients being treated for chronic conditions, and so will take more mediications
Metabolic Reaction Sites
Liver – hepatocytes and SER
smooth ER (& cytosol & mitochondria)
kidney
lung
GI tract – enterocytes for first-pass metabolism
e.g. tyramine, salbutamol
brain
Plasma –enzymes present
e.g. succinylcholine by cholinesterase
skin – contributes to 10% of the drug metabolising capacity due to it being such a large organ
Phase 1 and 2 DME Reactions
Reactions can often occur sequentially
P1→P2: Drug → Metabolite → Conjugate (EXCRETED)
Doesn’t always occur as paracetamol can be independently be metabolised from either phase
There are different types of reactions
Phase 1 and 2 DME Reactions: Aspirin
Phase 1: Addition of OH (hydroxylation)
Phase 2: Addition of glucuronide (glucuronidation/ conjugation)
Larger and easier to excrete in the urine - larger and less water soluble
Phase 1 DME Reactions
Produces or uncovers chemically reactive functional groups (called functionalisation) that prepare drugs for Phase II metabolism.
Functional groups: –OH, –NH₂, –SH, –COOH
Alterations make drugs/metabolites slightly more polar → more water-soluble
Enables Phase II reactions by increasing drug reactivity
Important for pharmacological activation (e.g., prodrugs):
Reactions aim to create or reveal functional groups
Types of Phase I Reactions
Oxidation (e.g., cytochrome P450, alcohol dehydrogenase, MAO)
Reduction
Hydrolysis
Hydration
aim to create or reveal functional groups
Prodrugs
Inactive drugs until metabolised to active form
Increase lipid solubility → improves absorption and bioavailability
Examples:
Glyceryl trinitrate → nitric oxide (vasodilator)
Azathioprine → mercaptopurine
Cortisone → hydrocortisone
CYP Enzyme
A superfamily of haem proteins located in the SER
~57 are involved in drug metabolism - have complex nomenclature
CYP families 1-3 mediate 70-80% of all Phase 1 small molecule drugs
CYP3A4 and CYP2D6 metabolise >50% of all drugs
Individual variation of metabolism via enzymes is significant
CYP450 DME Reaction
Enzymes contain heme iron in either ferric (Fe³⁺) or ferrous (Fe²⁺) form.
Requirements for drug metabolism: Substrate (the drug); Cytochrome P450 enzyme; Molecular oxygen (O₂); NADPH; NADPH–CYP450 reductase
Mechanism:
Drug binds to the CYP450 enzyme.
NADPH–CYP450 reductase transfers an electron to the enzyme.
Molecular oxygen is added, converting iron to its ferrous form.
The enzyme undergoes a series of reactions, including protonation generating water and a ferric-oxide–drug complex.
The enzyme is recycled after the reaction.
Produces a hydroxylated drug product (DOH).
Process is important in in vitro drug metabolism testing.
Phase 2 DME Reactions
Mostly occurs in the liver and involves conjugation reactions, attaching a large polar molecule to the drug or its Phase 1 metabolite.
Makes the molecule larger and easier to excrete.
Drugs/metabolites with –OH, –SH, or –NH₂ groups are more susceptible to conjugation.
Types of Phase 2 Conjugation Reactions
Glucuronidation (most common)
Sulphation
Amino acid conjugation
Glutathione conjugation
Fatty acid conjugation, etc.
Involves transferases enzymes
Products of Phase 2 DME Reactions
They are generally:
Water-soluble → enhances urinary excretion.
Have increased molecular weight (MW).
Pharmacologically inactive:
↓ Receptor affinity (↓ ability to bind to drug target).
↑ Excretion rate.
Enterohepatic Circulation
Involves the re-circulation of drugs between the liver, bile, intestines, and bloodstream.
After oral administration andabsorption, drug enters the liver via the bloodstream.
In the liver, the drug undergoes glucuronidation → forming a drug-glucuronide conjugate.
This conjugate is excreted in bile into the GI tract.
Some is lost in faeces, but:
Gut bacteria express β-glucuronidase, which cleaves the conjugate, producing the active drug again
The active drug is regenerated, reabsorbed across the GI tract → returns to the liver/blood.
This cycle can prolong drug action, delay clearance, and lead to ~20% of the drug being recirculated.
Outcomes of Metabolism: Pharmacological Activation
E.g. prodrugs
levodopa → dopamine
azathioprine → 6-mercaptopurine
codeine → morphine (active)
Outcomes of Metabolism: Pharmacological Inactivation
e.g Paracetamol → paracetamol glucuronide
Outcomes of Metabolism: Change in Pharmacological Response
E.g. diazepam → oxazepam;
The ‘metabolite’ has a different therapeutic effect to its precursor following metabolism
Outcomes of Metabolism: No Change in Pharmacological Activity
e.g. lidocaine → monoethylglycylxylidide (MEXG) – both produce the same effect
Outcomes of Metabolism: Change in Drug Distribution
Change from a lipid-soluble drug than is widely distributed, with a high volume of distribution, to a water-soluble metabolite, more confined to the bloodstream → alters volume of distribution
Internal Factors Affecting Drug Metabolism
Species differences: Challenge in extrapolating in vivo testing to humans.
Genetics: Individual variation in enzyme expression/activity.
Age: change in drug metabolising enzyme expression (infant = low; increase with age, then declines)
Sex: Minor differences,
Disease (e.g. hepatic dysfunction):
Liver = key organ for metabolism.
Dysfunction lowers clearance → drug may accumulate → risk of toxicity.
Pharmaceutical companies must assess how these factors impact metabolism.
May require dose adjustments in vulnerable populations (infants, elderly, liver disease patients).
External Factors Affecting Drug Metabolism
Lifestyle: cigarette smoking induces the metabolism of, e.g.
theophylline, caffeine, tacrine, imipramine, haloperidol, pentazocine, propranolol, flecainide, Estradiol
Environment – chemicals present can act as inducers or inhibitors and can increase in the expression of enzymes or inhibit their actions
diet – impact drug metabolism
(BBQed meat, Brussels sprouts ↑, grapefruit juice ↓)
Effect of Inducers and Inhibitors on Drug Half-Life
Inducers decrease half-life and increase drug metabolism;
Inhibitors increase the half-life and decrease drug metabolism
CYP3A Enzyme and Drug Interactions
Expressed in enterocytes (intestinal cells).
Felodipine (anti-hypertensive) undergoes first-pass metabolism in the GI tract and is metabolised by the enzyme, with only 15% reaching systemic circulation after a set dose.
Grapefruit Juice: Inhibits the enzyme, leading to increased drug absorption (high levels in the circulation) and higher bioavailability.
Toxicity Risk: The interaction of grapefruit juice with CYP3A4 can lead to toxicity testing in drugs.
Example: Terfenadine (prodrug) metabolises to fexofenadine via CYP3A4.
Terfenadine at high concentrations can be cardiotoxic.
Grapefruit juice + Terfenadine: Increases risk of cardiac toxicity due to interaction with hERG Kv1.1 (voltage-gated potassium channel).
Metabolic Assessments
Metabolic Sttability assess the speed of metabolism.
Rapid metabolism → short t½ → frequent dosing (problematic for chronic pain treatment).
Slow metabolism → long t½ → less frequent dosing.
Metabolite Identification:
Identify reactive/toxic metabolites and enzymes involved in metabolism.
Affects drug-drug interactions
Drug-Drug Interactions (DDI):
Which drug-metabolising enzymes are involved?
Drugs eliminated by a single DME are more vulnerable to DDIs when taken with enzyme inhibitors/inducers.
Co-administration of drugs
Does the new drug induce or inhibit DMEs?
Toxicity:
Assess the reactivity of metabolites.
Minimise risks by understanding pharmacological activation and inactivation to avoid reactive metabolites
Traditional Approach to Assess ADME
Introduced late in the assessment process: ADME factors are assessed in in vitro and in vivo tests after initial drug design.
Linear process: Often reveals the need to re-design and re-synthesise molecules and then assess activity, making the process lengthy and slow.
Low throughput: Not ideal for rapid testing.
Requires re-synthesis of compounds.
Time-consuming and expensive.
Marginally predictive: Poorly predictive of drug candidates and their efficacy.
Current Approach in ADME Testing
High throughput – test many compounds at once to see what can be eliminated early on
Relatively rapid – save money
Only quality molecules re-synthesised
Selective > reduced attrition rate
Less expensive
Metabolic Stability Assessment: In Sillico
Assess sites of instability in compound structures
‘Soft spots’ – areas on drug molecules where they may be more susceptible to instability
Metabolic Stability Assessment: Metabolite Structure Elucidation
Uses spectroscopy to assess the structure of metabolites during optimisation or late discovery
Metabolic Stability Assessment: Reaction Phenotyping
Assess which DMEs are involved?
Rapid screen in the discovery phase and definitive testing in the preclinical phase
Look at drug metabolites and enzymes are important in drug metabolism
Metabolic Stability Assessment: Microsomal Stability Assays
Phase 1 stability (t½ ); plan in vivo studies; optimization
Microsomes – fragments of ER and either attached ribosomes – can use those isolated from hepatocytes
Metabolic Stability Assessment: S9 Or Hepatocyte Stability Assay
assesses microsomal and extra-microsomal metabolic reactions
selected compounds in late discovery
S9 fraction = cytosol + microsomes
Metabolic Stability Assessment: Assays
Different assays can be used to assess different components
In phase 1 many of the oxidization reactions occur in the SER – use of microsomal assay
Phase 2 assays largely occur in the cytosol – use of S9/ hepatocyte assay
Metabolic Stability Assessment: Phase 2 Stability Assays
Examines the compound susceptibility to conjugation
Phase 2 t1/2
Generation In Vitro Models: Rat Liver
Hepatocyte generation: Use rat liver to generate 109 hepatocytes.
Liver is infused with an isotonic buffer containing Ca2+ chelator to loosen boundaries between cells.
Collagenase treatment to break down collagen to produce hepatocyte suspension (can be plated or preserved);
Cells lose structure and activity after a few hours - time limited model
Fractionation: homoginised tissue is spum down at a low speed to separate nuclei and debris, supernatant is then taken and spun down to produce S9 fraction and the spun down to yield microsomes and cytosol.
Use of Rat Liver Hepatocytes
2D monolayers or sandwich cultures.
3D liver spheroid cultures: Mimic native structure and function.
Limitations: liver-on-a-chip:
Cells lose structure after a few hours, limiting long-term use.
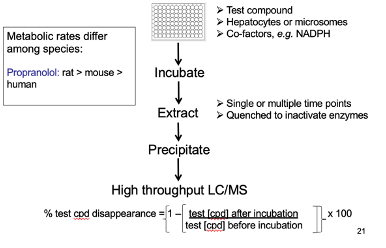
Assessment of Metabolic Rate
Metabolic rate differs by species.
Example: Propranolol metabolism — rat > mouse > human.
Method:
Incubate test compound with hepatocytes and required co-factors.
Extract and measure the amount of parent drug remaining.
Decrease in parent drug = metabolism to metabolites.
Can be assessed through
Liquid Chromatography (LC)
Mass Spectrometry (MS)
Drug Drug Interactions: Inducers
A test drug molecule is added to a hepatocyte culture and is removed after a few days.
After removal, CYP450 enzyme substrates are added.
If these substrates are metabolised more rapidly, with increasing [drug], it suggests that the test drug induced drug-metabolising enzymes.
Induction confirmed when:
→ Higher concentrations of the test drug = More substrate metabolised
→ More metabolites are generated due to enzyme upregulation
Drug Drug Interactions: Inhibitors
A test drug molecule is added to a hepatocyte culture and is removed after a few days.
After removal, CYP450 enzyme substrates are added.
Measure amount of metabolite produced
ResultL increasing concentrations of the drug results in a decreased amount of metabolites generated
In Vivo Methods Used in Late Discovery/ Early Preclinical Stages
species comparison to correlate with in vitro studies
single/multiple/IV dose in mouse, rat, dog or monkey
measure AUC, Cmax (max conc), Tmax (time to get peak), t½,Vd, F
allometric scaling to predict human dose and PK – extrapolation of findings
In Vivo Methods Used in Preclinical Stages
testing formulation (assess AUC, Cmax), safety & toxicology
Use radiolabelled compound
How is cyclophosphamide used in in vivo studies, and what does its clearance indicate?
→ Used as an immunosuppressant (e.g. rheumatoid arthritis)
→ Used as a chemotherapeutic (e.g. for some leukemias)
In in vivo models, clearance correlates well with body weight
R value (correlation coefficient) is strong, indicating predictive value for human translatio
Key Features of Drug Metabolism and Its Assessment
Two phases of metabolism:
→ Phase 1 = Functionalisation (e.g. CYP450 enzymes; prep for Phase 2)
→ Phase 2 = Conjugation (adds groups to increase water solubility)CYP450 enzymes are vital in Phase 1 reactions for many prescribed drugs
In vitro and in vivo methods are used to assess metabolism
Drug metabolism is influenced by many factors:
→ Non-modifiable (e.g. genetics, age)
→ External (e.g. diet, disease, other drugs)Drug-drug interactions (DDIs) must be considered during drug development