Corona Lab Final Study Guide
1/45
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
46 Terms
What is a virus?
A virus is a submicroscopic infectious agent that replicates only inside the living cells of an organism.
UniProt
Database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature.
ViralZone
Web resource for all viral genus and families, providing general molecular and epidemiological information, along with virion and genome figures. Has information about viral proteomes and their different protein components.
NCBI
Houses a series of databases relevant to biotechnology and biomedicine and is an important resource for bioinformatics tools and services. Major databases include GenBank for DNA sequences and PubMed, a bibliographic database for the biomedical literature.
How can a coronavirus infect a cell?
by attaching its spike proteins to ACE2 proteins that lie on the surface of the cells
How does a coronavirus replicate?
Uses the genetic molecule ribonucleic acid, or RNA, to provide instructions to infected host cells to build more copies of the virus.
How does a coronavirus make new proteins?
The first open reading frame of the coronavirus genome encodes a large polyprotein, which is processed into a number of viral proteins required for viral replication directly or indirectly.
How do the 7 human coronaviruses differ in infection severity?
HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1 cause mild upper-respiratory tract illness and contribute to 15%-30% of common cold cases. SARS-CoV, MERS-CoV, and SARS-CoV-2 are highly pathogenic and cause severe respiratory illness.
How many of the 7 human coronaviruses are betacoronaviruses, alphacoronaviruses, or gammacoronaviruses?
Betacoronaviruses: SARS-CoV, MERS-CoV, and SARS-CoV-2 (COVID-19) are all betacoronaviruses.
Alphacoronaviruses: Human Coronavirus 229E (HCoV-229E) and Human Coronavirus NL63 (HCoV-NL63) are alphacoronaviruses.
Gammacoronaviruses: Human Coronavirus OC43 (HCoV-OC43) and Human Coronavirus HKU1 (HCoV-HKU1) are gammacoronaviruses.
What does BLAST stand for?
Basic Local Alignment Search Tool
What is the purpose of BLAST?
Tool for comparing biological sequences like DNA, RNA, and proteins. It's used to identify similarities between sequences, aiding in tasks such as sequence alignment, annotation, and genomic analysis. By comparing query sequences against databases, BLAST helps researchers infer functional and evolutionary relationships, making it essential for various bioinformatics applications.
BLAST metrics:
a. E-value
represents the number of expected hits by chance alone when searching a database. A lower E-value indicates a more significant match, with values typically interpreted as the number of hits one would expect to see by chance when comparing a query sequence against a given database.
BLAST metrics:
b. Query cover
refers to the percentage of the query sequence that aligns with the subject sequence in the database. It indicates how much of the query sequence was included in the alignment, providing insight into the extent of sequence similarity between the query and subject sequences.
BLAST metrics:
c. Percent identity
represents the percentage of identical residues (nucleotides or amino acids) between the aligned regions of the query and subject sequences. It quantifies the level of similarity between the sequences, indicating how closely related they are in terms of their primary structure.
Scoring your alignment (BLOSUM62)
Scoring your alignment with BLOSUM62 involves using the BLOSUM62 substitution matrix, which assigns scores to amino acid substitutions based on their observed frequencies in related proteins. Higher scores indicate more conservative substitutions, while lower scores indicate less conservative substitutions. By summing the scores for each aligned pair of amino acids in your alignment, you can assess the overall quality and similarity of the alignment.
What type of format is BLOSUM62?
substitution matrix used in bioinformatics for sequence alignment, particularly in protein sequence alignments. It is a tabular format that assigns scores to amino acid substitutions based on their observed frequencies in related protein sequences.
What information does BLOSUM62 give us?
provides scores for amino acid substitutions during protein sequence alignment, reflecting the frequency of occurrence of these substitutions in related protein sequences. These scores guide the alignment process by indicating the likelihood of specific substitutions, aiding in accurately aligning sequences and inferring evolutionary relationships.
Nr database
[ ~600 million sequences)
• The largest database, but is redundant for proteins (many hits for the same organism)
• When refseq proteins return nothing for a species, check nr
• If you are looking for a sequence in a species that is not in refseq
Refseq database
[~150 mi. sequences)
• Main database for our use
• Not the largest, but typically 1 hit per organism
• Non-redundant, well-annotated set of reference sequences
PDB database
repository of three-dimensional structural data for large biological molecules, primarily proteins and nucleic acids. It provides researchers with access to experimentally determined atomic coordinates of biomolecular structures, facilitating studies on protein structure, function, and interactions.
How to run a BLAST search (may perform a BLAST search using a mystery sequence during the exam)
Access the BLAST Website: Go to the NCBI BLAST website (https://blast.ncbi.nlm.nih.gov/) or use a local installation if available.
Select the BLAST Program: Choose the appropriate BLAST program based on your query type (nucleotide or protein) and the database you want to search.
Enter Your Query Sequence: Paste or upload your sequence(s) into the query input box.
Choose Parameters: Adjust the search parameters as needed, including the database to search against, the type of search (e.g., blastn for nucleotide sequences), and any advanced options.
Run the Search: Click the "BLAST" button to submit your query and initiate the search.
Review Results: Once the search is complete, review the results, which typically include a list of hits ranked by similarity score, alignment statistics, and other relevant information.
Refine and Analyze Results: Refine your search parameters or further analyze the results, such as examining alignments, sequence annotations, and any additional information provided.
Interpret and Save Results: Interpret the results in the context of your research question or hypothesis, and save relevant data for further analysis or reporting.
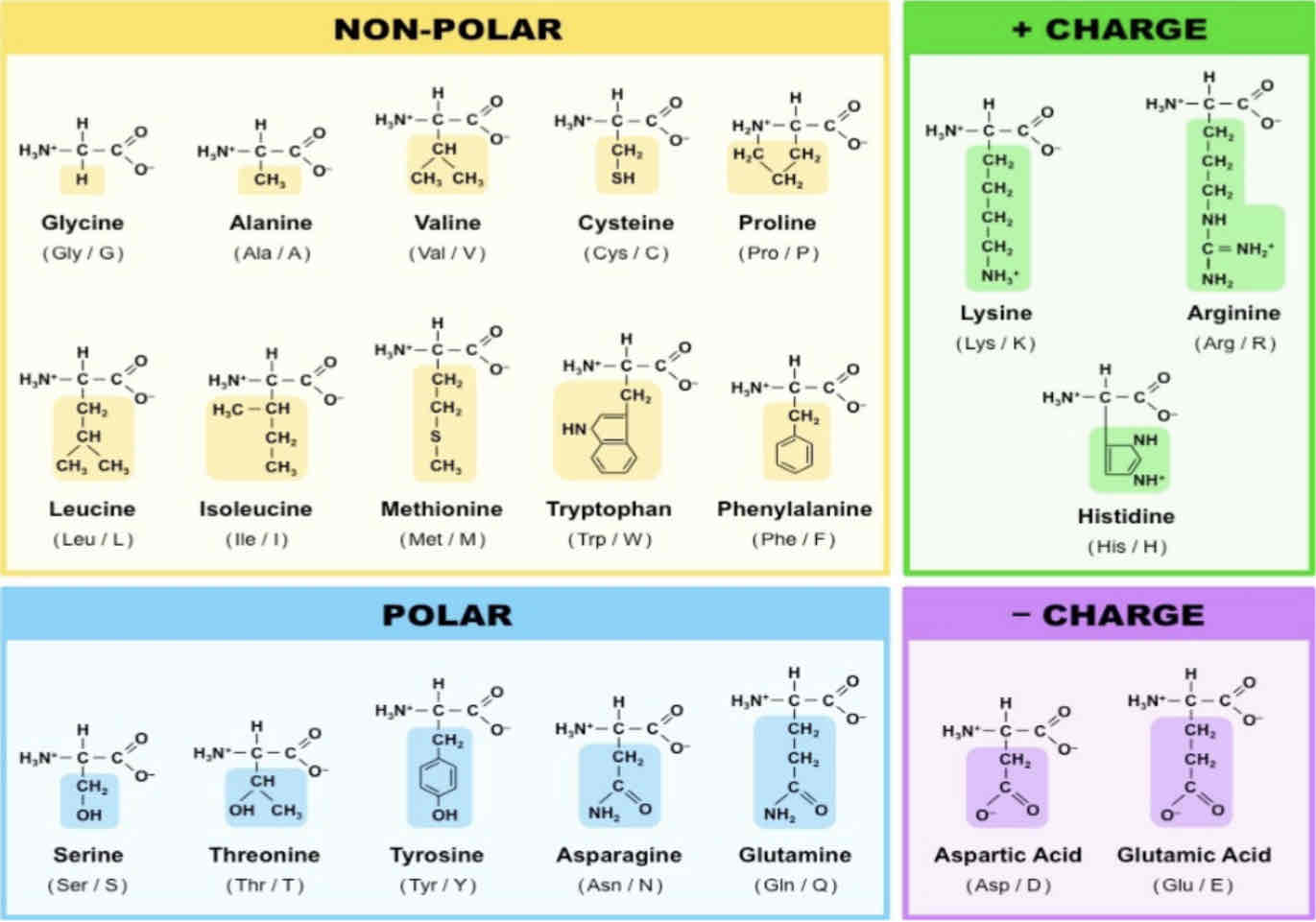
Amino Acid Properties, Names, and Abbreviations
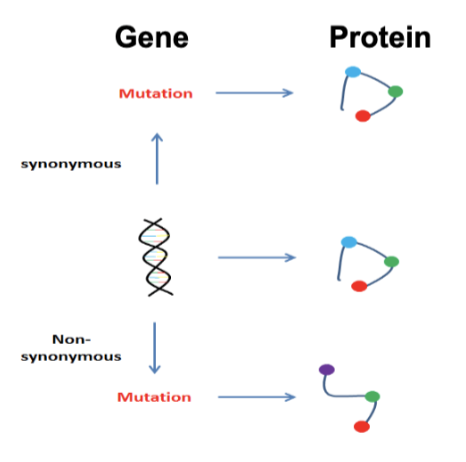
Evolution
Multiple Sequence Alignments:
a. MUSCLE
Popular tool for making multiple sequence alignments (MSAs). It's known for its speed and accuracy, using an iterative process to align sequences based on their similarities. MUSCLE is great for handling big datasets and producing alignments useful for evolutionary and functional genomics research.
Multiple Sequence Alignments:
b. Jalview
a user-friendly software for visualizing and editing multiple sequence alignments. It offers various tools for analyzing and annotating alignments, making it valuable for molecular biology and bioinformatics research. Its intuitive interface and extensive features make it a popular choice among scientists for exploring and interpreting alignment data.
What is the purpose of a Multiple Sequence Alignment?
Aligning multiple sequences enables us to compare the sequences to identify similarities in the sequences that may be important for structure and function
Phylogenetic Trees:
a. IQTree
tool for inferring phylogenetic trees from molecular sequence data using maximum likelihood methods. It's known for its accuracy, efficiency, and user-friendly interface, making it a popular choice among researchers for studying evolutionary relationships.
Phylogenetic Trees:
b. FigTree
software designed for visualizing and annotating phylogenetic trees. It provides various customization options and features for exploring tree structures and highlighting important evolutionary relationships, making it a valuable tool for researchers studying molecular evolution and phylogenetics.
Phylogenetic Trees:
c. Tree Anatomy and the meaning of the terms in the tree
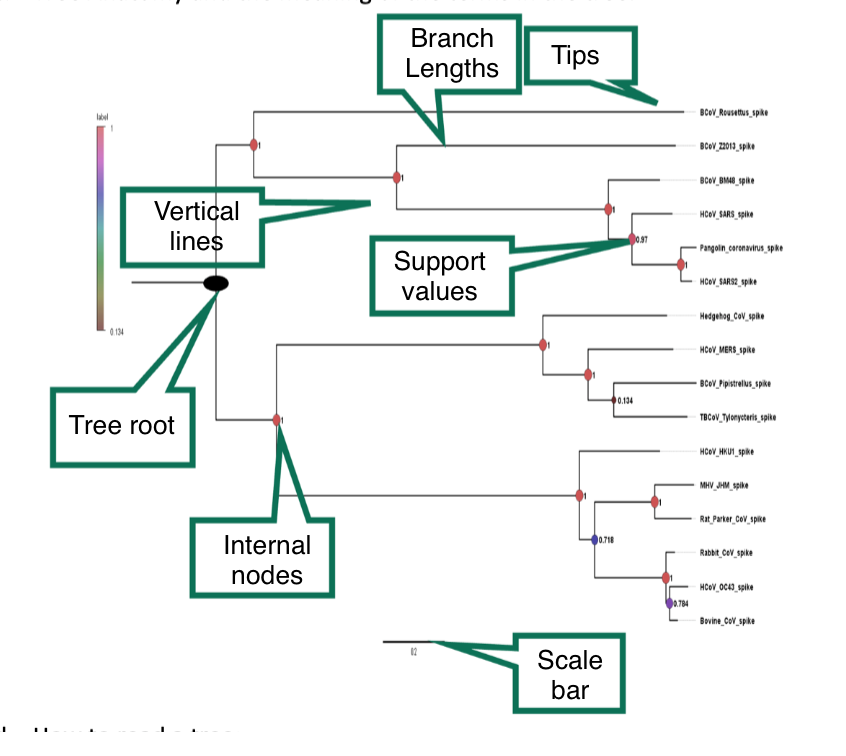
How to read a tree:
i. Who is more closely related?
In a phylogenetic tree, taxa (species or groups of organisms) that share a more recent common ancestor are considered more closely related. This relationship is depicted by branches that are closer together on the tree. Therefore, taxa located closer on the branches or connected by shorter branches are more closely related to each other than taxa located farther apart or connected by longer branches.
How to read a tree:
ii. What is a clade?
a group of organisms in a phylogenetic tree that includes an ancestral species and all of its descendants. They are depicted as branches or subtrees in the tree diagram. They represent evolutionary relationships based on shared ancestry, with each clade forming a distinct evolutionary lineage.
How to read a tree:
iii. What is a last common ancestor?
the most recent individual or species from which all the taxa (species or groups of organisms) depicted in the tree have descended. It represents the point in evolutionary history where the lineages leading to the depicted taxa diverged from a common ancestor. It is typically located at the base of the tree, and its branches represent the subsequent evolutionary divergence into different lineages or clades.
How are the human coronaviruses related? Which are more similar to each other, and which are less similar?
Human coronaviruses share a common evolutionary history but vary in their relatedness. SARS-CoV and SARS-CoV-2 are closely related betacoronaviruses, while MERS-CoV is a separate lineage within the same genus. The other four human coronaviruses belong to different genera and are less closely related to SARS-CoV, SARS-CoV-2, and MERS-CoV.
Protein Structure:
a. Structure → Function
The three-dimensional structure of a protein dictates its function by defining key functional domains and active sites that interact with other molecules. This structural arrangement determines the protein's role in biological processes such as enzyme catalysis, molecular recognition, and signal transduction.
Protein Structure:
b. Structure is more conserved than sequence
In protein structure, conservation refers to the similarity in the three-dimensional arrangement of amino acids among related proteins or across different species. It's commonly observed that protein structure is more conserved than sequence, highlighting the importance of structural integrity in maintaining protein function across evolutionary variations.
Protein Structure:
c. Difference between Primary, Secondary, Tertiary, and Quaternary structures
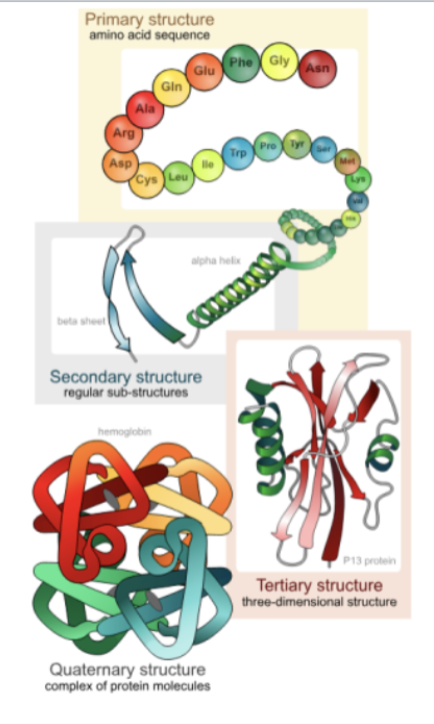
Protein Structure:
d. What are the different types of secondary structures?
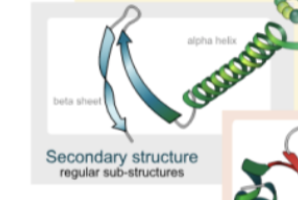
Protein Structure:
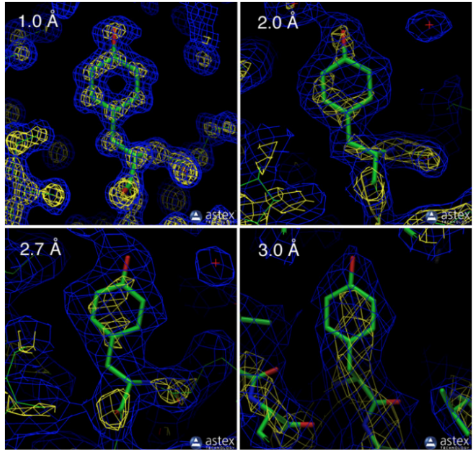
e. Protein Structure resolution, which is the structure with the highest resolution?
1.0 Å
PyMOL Colorations - Know what they represent
Open PyMOL
type fetch 6WZQ to load protein in PyMOL session window
select amino acid number 291 in chain A
Explain the difference between evolutionary rate vs conservation fraction.
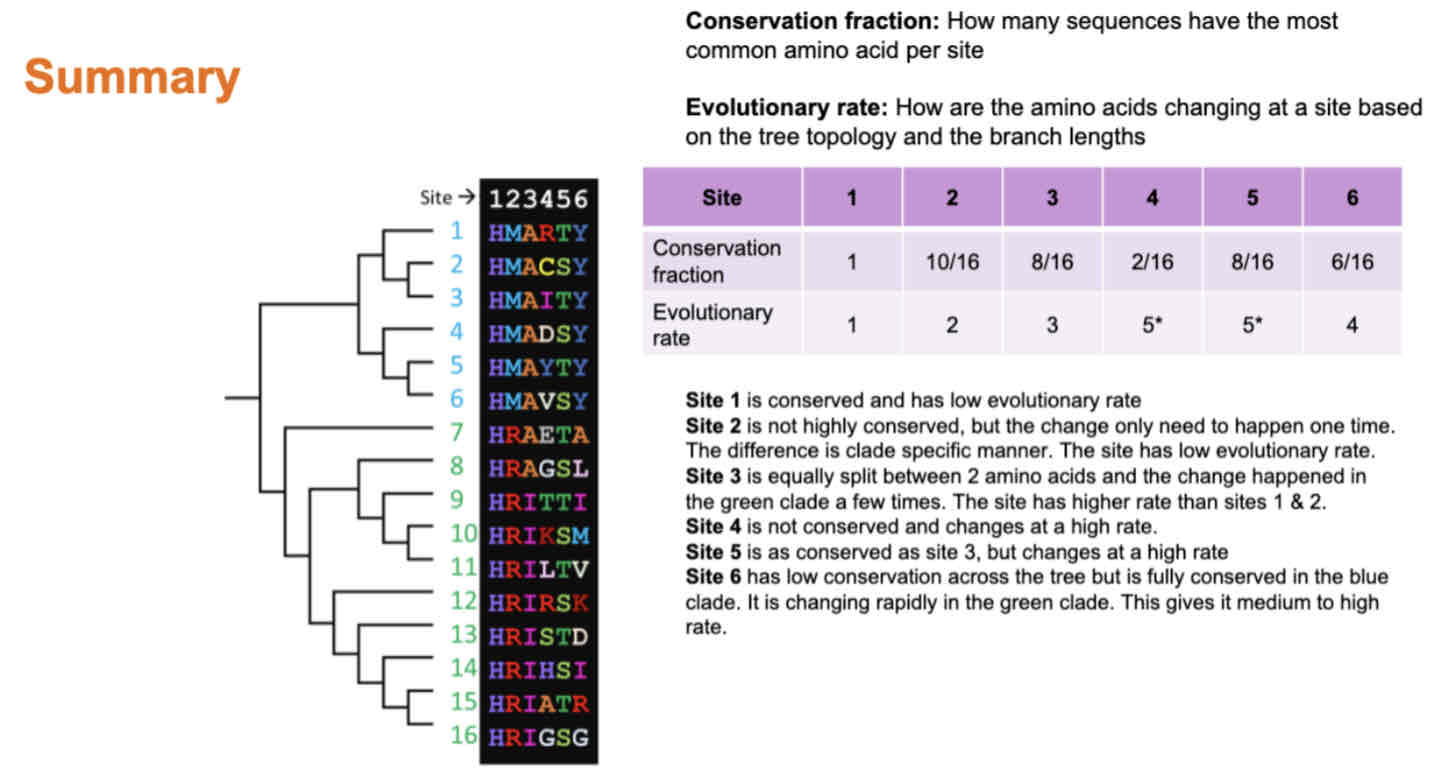
Why are some sites evolving fast and some slow? How is the evolutionary rate at different sites important for the structure and function of the protein? Describe how this is important for the fitness of the virus.
Different sites in proteins evolve at varying rates due to factors like selection pressures and structural constraints. Fast-evolving sites may adapt to environmental changes, while slow-evolving ones are often functionally or structurally critical. This variation in evolutionary rates impacts protein structure and function, with changes in critical regions potentially disrupting protein function. For viruses, maintaining functional proteins is crucial for survival and spread, emphasizing the importance of evolutionarily conserved sites for fitness.
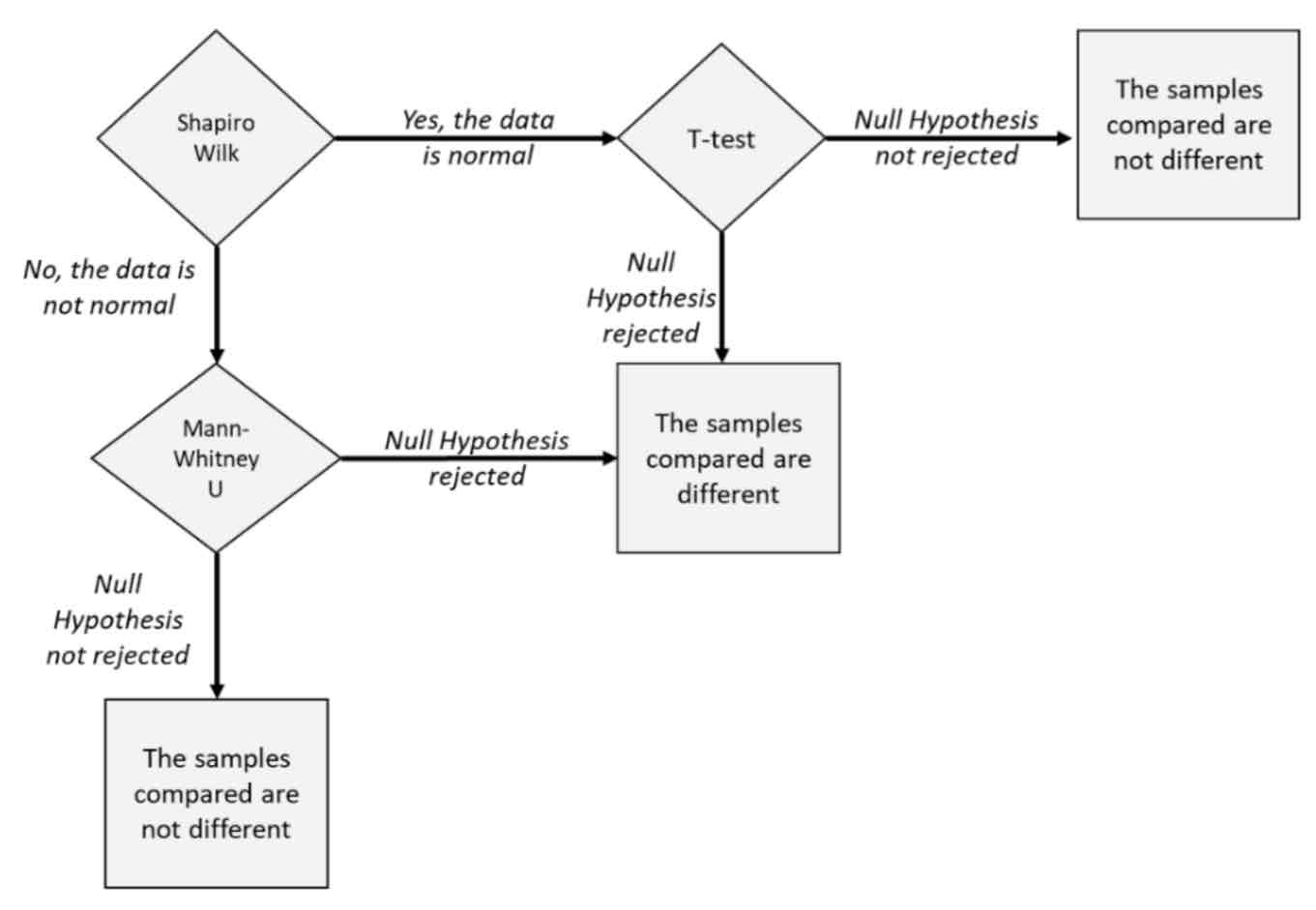
Know the main difference between all the statistical tests (Shapiro Wilk, Mann-Whitney, T-test) and when to use them.
How to interpret a boxplot:
i. What do the box and whiskers represent?
ii. The middle line?
iii. The dot?
iv. The asterisk/n.s. on the brackets?
The box in a boxplot shows the interquartile range, with whiskers extending to the minimum and maximum values within 1.5 times the IQR. The middle line represents the median, and dots may indicate outliers. Asterisks or "n.s." denote statistical significance between groups being compared.
What are the qualities of a protein pocket that make it a good drug target?
Good drug targets in proteins are characterized by specificity to the target protein, accessibility to small molecules, evolutionary conservation, favorable physicochemical properties for binding, functional importance in protein function, association with disease pathways, and structural characterization. These qualities enhance the likelihood of developing safe and effective therapeutics.
Why are we concerned about
a. Accessibility
b. Conservation
c. Druggability
when identifying drug targets for coronaviruses?
When identifying drug targets for coronaviruses, we're concerned about:
a. Accessibility: Ensuring that the target protein's binding site is accessible to drugs for effective binding and inhibition.
b. Conservation: Confirming that the target site is conserved across different strains of the virus to ensure broad efficacy.
c. Druggability: Assessing if the target site possesses favorable properties for drug binding, aiding in the development of effective therapeutics against coronaviruses.
Why are highly conserved drug pockets potentially broadly-neutralizing antiviral targets?
Highly conserved drug pockets are potentially broadly-neutralizing antiviral targets because they are present across different viral strains, reducing the likelihood of resistance and increasing drug effectiveness against various variants. Targeting these regions disrupts essential viral functions, making them promising for broad-spectrum antiviral therapies.