Cancer- LECTURE 5
1/48
Earn XP
Description and Tags
Genetic basis and molecular themes
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
49 Terms
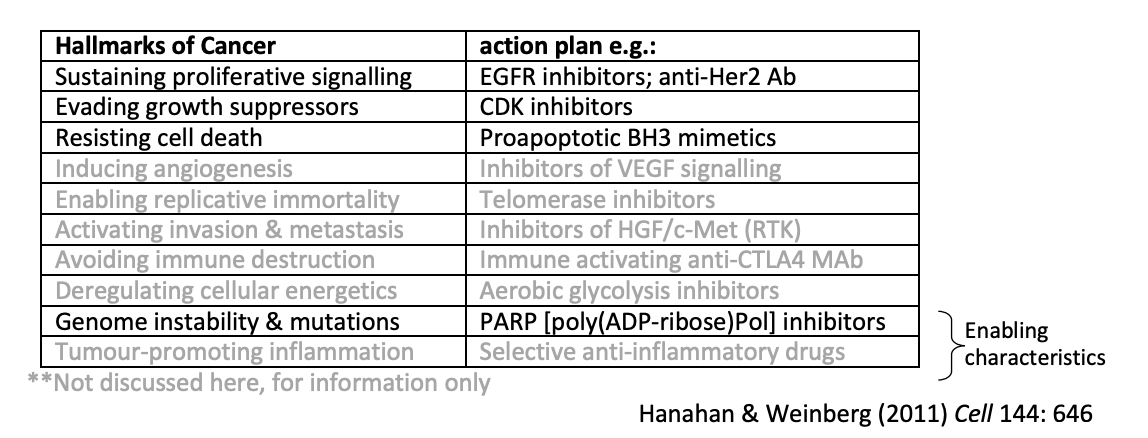
Research 1: finding the cuases of chicken sarcomas
Rous Sarcoma Virus (RSV)
retrovirus
contains +ssRNA genome
yet infection cycle required both DNA and RNA synthesis
identified as the transfomring agent in extracts of chicken sarcomas
Hypothesis as to why this happened?
provirus hypothesis:
DNA intermediate integrates into host cell genome
so messed up the chromosomes and caused cancer??
Research 1: finding the genetic cause?
Transformed cells with a virus mutant which was temperature sensitive
At normal temperate→ no cancer
At restrictive 35C temperature→ cancer
because virus manifested here
The gene that was for this ts mutation was found
v-SRC gene
Research 1: how was SRC mapped
based on a non-transforming mutant
carrying a deletion near the 3’ end of the viral RNA
These findings were exploited to generate a specific DNA probe for SRC
Research 1: Use of the SRC DNA probe
identified a gene present in normal cells
protooncogene→ c-SRC
closely related to viral oncogene v-SRC
THEREFORE: the gene was taken from host genome by cirus, then mutated and then when put back in, still has some host features but a bit wrong so messes up cell cycle→ cancer! So ‘protoncogene’ a gene that is invovled in cell-cycle probably and so if changed and sprad with virus= cancer!
Research 1: What this tells us about how cancer is caused?
oncogene is ‘activated’ by mutations
v-SRC encodes a de-regulated protein
RNA tumour viruses might capture cellular genes into their genome
They undergo genetic change
So when infecting normal cells
they drive them to malignant transformation!
i.e deregulated proliferation
Research 1: What is known about viral oncogenes
dominant
gain-of-function alleles
activated or over-expressed
Originally come from cellular protooncogenes
Research 1: what was SRC shown to encode
non-receptor tyrosine kinase
Research 1: what this helped to do…
Find many other viral oncogenes
and their cellular counterparts
Another cause of cancer?
Chromosomal aberrations
controversial:
is it a cause or an effect of cancer??
Research 2: Cytogenetics and Chromosomal aberrations
cytogenetic studies showed a given chromosomal rearragment was associated with a specific cancer
must be causative
Research2: first chromosomal aberration caused cancer found
Chronic myeloid leukaemia (CML)
found to carry a normal number of chromosomes but
one is too small
the Philadelphia Chromosome
THERFORE: must be the cause of this cancer?
Research 2: What is Philadelphia chromosome
product of reciprocal translocation
between chromosomes 9 and 22
Note: translocations can easily be viewed with chromosome paining and spectral kayrotyping
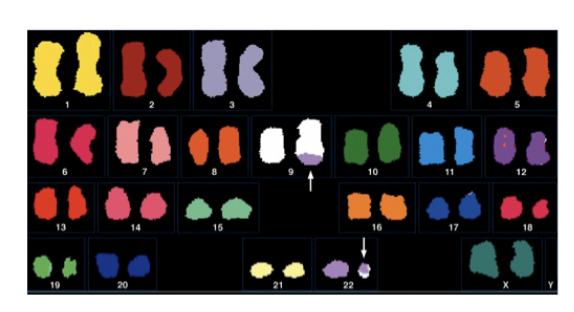
Research 2: Further anaysis showed
Translocation breakpoint was cloned
found to encode a fusion of
BCR-ABL
i.e two genes have kinda fused together when the chromosomes 9 and 22 got mixed up with eachother
Research 2: What are these genes?
c-ABL
human cellular homolog of
transforming sequence of Abelson murine leukaemia
encodes for a non-receptor tyrosine kinase
THERFORE: BCR-ABL encodes a deregulated tyrosine kinase
which will cause cancer!
Bringing together findings from virologists and cytogeneticists
viral oncogenes
found from RNA tumour viruses
also found at
sites of chromosomal rearrangements
or
actiavted by insertional mutagenesis in cancer cells
THEREFORE: viral oncogenes are also linked with chromosomal aberrations→ so must be some kind of cause of cancer!
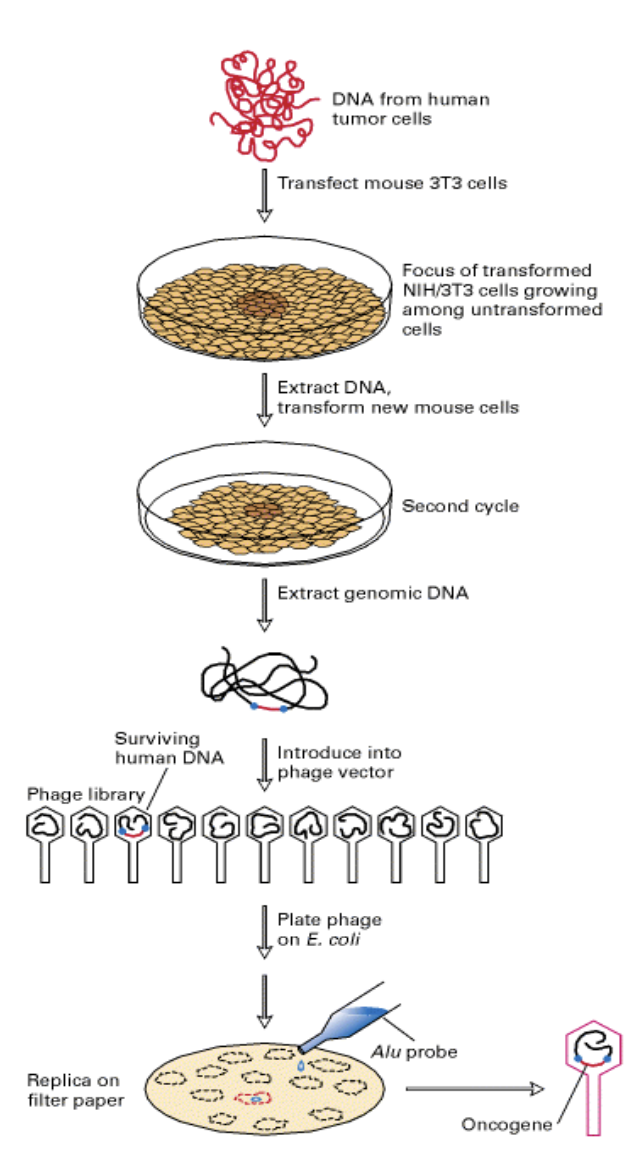
Research 3: Isolating human oncogene w/o knowing its sequence or strucuture
Humour tumour cell line transformed by transfection into
NIH 3T3 cells (immortalized mouse fibroblast cell line)
DNA extracted from resulting foci
from cells that were cancerous
Used in new rounds of transffection and extraction
Eventually getting rid of the rest of the human genome
leaving only the mouse genome and the human oncogene
DNA sequence could be cloned
Human genes found with DNA hybridisation
using Alu sequence probe
recognises human Alu repeats
linked to human oncogenic sequence
against the backdrop of the mouse genome
OVERALL: find the human oncogenes
Research 3: What this proved
direct maligant transformation by a ‘human oncogene’
Isolated gene corresponded to a previously identified viral oncogene
RAS
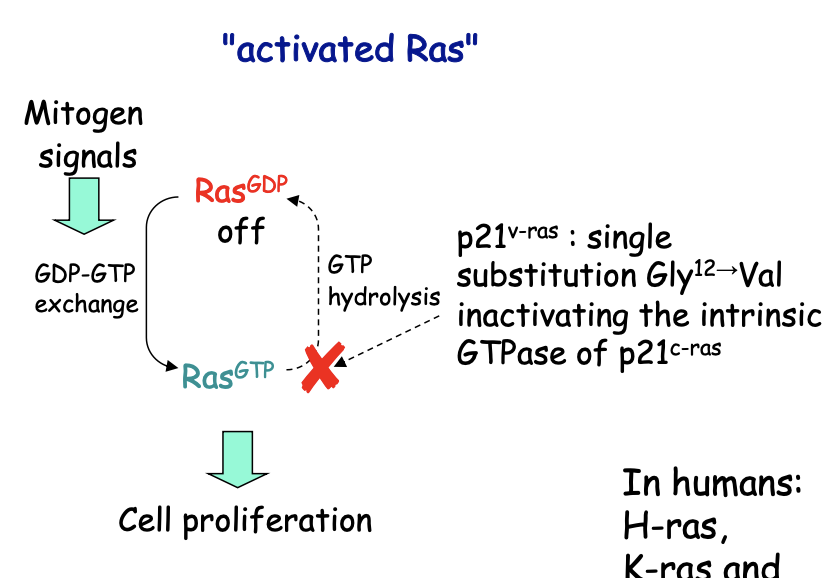
Research 3: What is Ras
Normal Ras
Mitogen signals
switches to its active GTP-bound form
via tyrosine kinase receptors
Signal mutations abolishing Ras GTPase
oncogenic
because signal transduction becomes constitutive
even if mitogens are present!
e.g common mutation→ Ras hyperactive (RasGly12→Val)
most common events in oncogenesis
humans:
H-ras, K ras, N-ras$
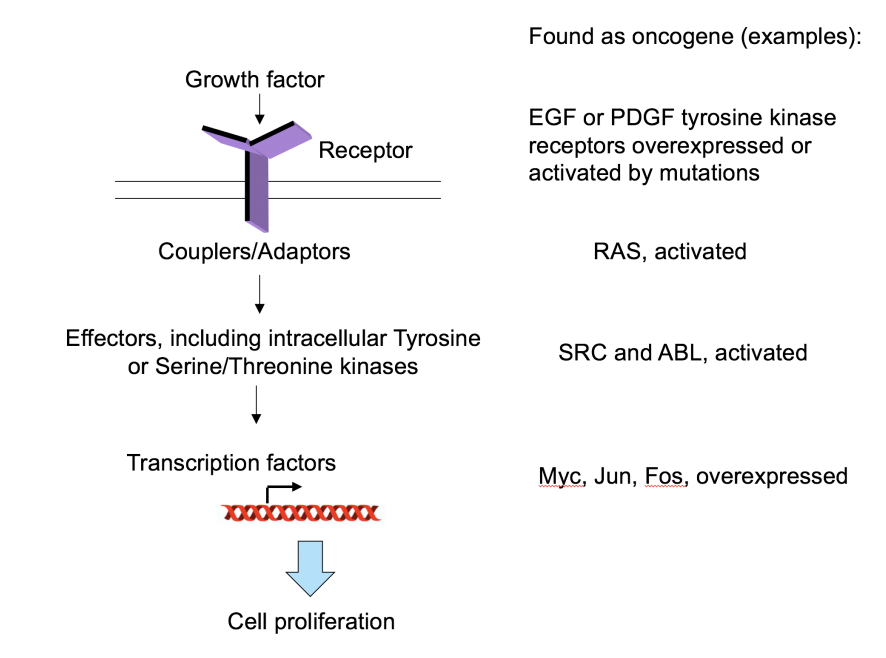
Research 3: How do oncogenes work to cause cancer
Involved in growth factor signalling pathways
Growth factor
Couplers/Adaptors
Effectors, inducing intracellular tyrosine or Serine/Threonin kinases
Transctiption factors
Cell proliferation
i.e found crucial insight into oncogenes affecting signalling pathways to cause cancer!
Research 4: finding info about genes which suppress tumours→ Experiment 1
Cell hybrids:
Malignant + normal→ hybrid
Observed: hybrid grows like normal
Continued experiment
hybrids are unstable
lose chromosomes
malignant phenotype returns
Conclusion:
maligant trait was recessive
BUT:
This is different to the idea that oncogenes are dominant!
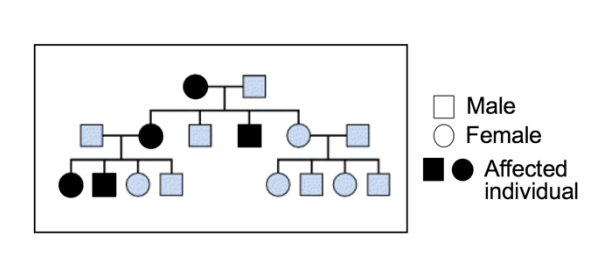
Research 4: Experiment 2- Familial predisposition
Looked at Hereditary retinoblastoma
mutation is recess
BUT
predisposition to the mutation is autosomal dominant trait
based on likelihood of inactivation of the remaining gene copy
Research 4: Experiment 2- How was RB locus identified
mapping deletions in chromosome 13 of patients
Clues to function of RB protein came from studies of DNA tumour viruses
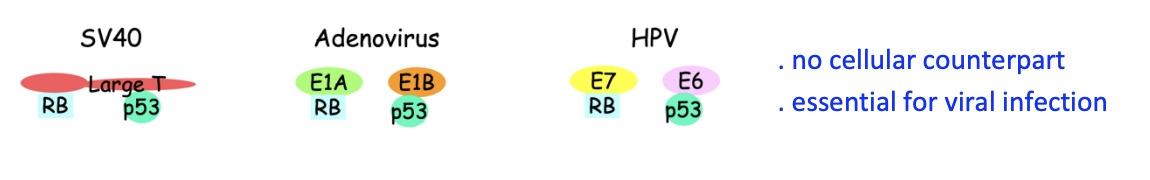
Reserach 4: Experiment 3- Findinding what causes RB inactivation
DNA tumour viruses encode oncoproteins
these inactivate RB and p53
Are essential for viral infection
Research 4: What is RB
Repressor of E2F
which is a transcription factor that controls S-pase gene expression
SO usuually used to stop moving into S-phase
member of ‘pocket proteins’ family
with p107, p130
Key cell-cyle target is
S-phase transcription factor E2F
Research 4: How RB works
RB binds and inhibits E2F
Upon mitogen signals
RB is a substrate of G1/S CDK
so undergoes sequential phosphylation
Hyperphosphylation inactivates RB triggering E2F-dependent transcription
Among S-phase E2F transcriptional targets are
E2F and Cyclins E and A
Their up regulation sets up a positive feedback loop
for RB inactivation unleashing the S-phase transcriptional programme
OVERALL: mitogens→ G1/S-CDK phosphylation inactivate RB→ go into S-phase
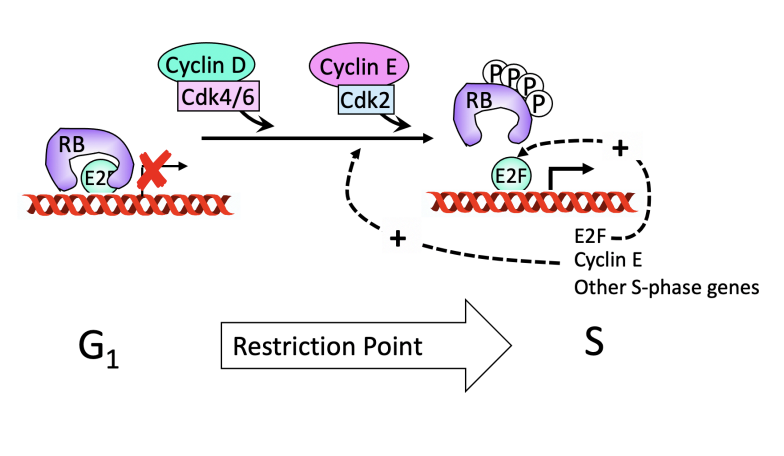
The pathway of RB inactivation for cell proliferation shows
Shows requirement of mitogens to pass restriction point!
Research 4: How RB links to cell cycle progression
with build-up of G1/S-CDK activity
in response to a constellation of mitogen signals
required to pass the restriction point
RB inactivation is key to cancer progression!
Research 4: what is p53
Crucial tumour suppressor
Protein made from TP53 gene
transcription factor
Plays a role in regulating cell growth, preventing uncrontolled cell division and triggering cell death
‘guardian of the genome’
first discovered as a target of DNA transforming viruses
inherited predisposition to cancer
one defective copy of TP53
Somatic mutation in > 50% cancers
Research 4: How does p53 normally work
DNA damage of mitogenic stress
Causes inhibition of E3 ubiquitin ligase Mdm2
Triggers accumulation of p53 and transcriptional activation
Induction of p14ARF by excess/out-of context mitogen signals
due to oncogenic activation
This impedes inappropriate proliferation
promotes cell death
unless p53 has been inactivated by mutation (in oncogenesis)
OVERALL: prevents cell cycle progression upon DNA damage
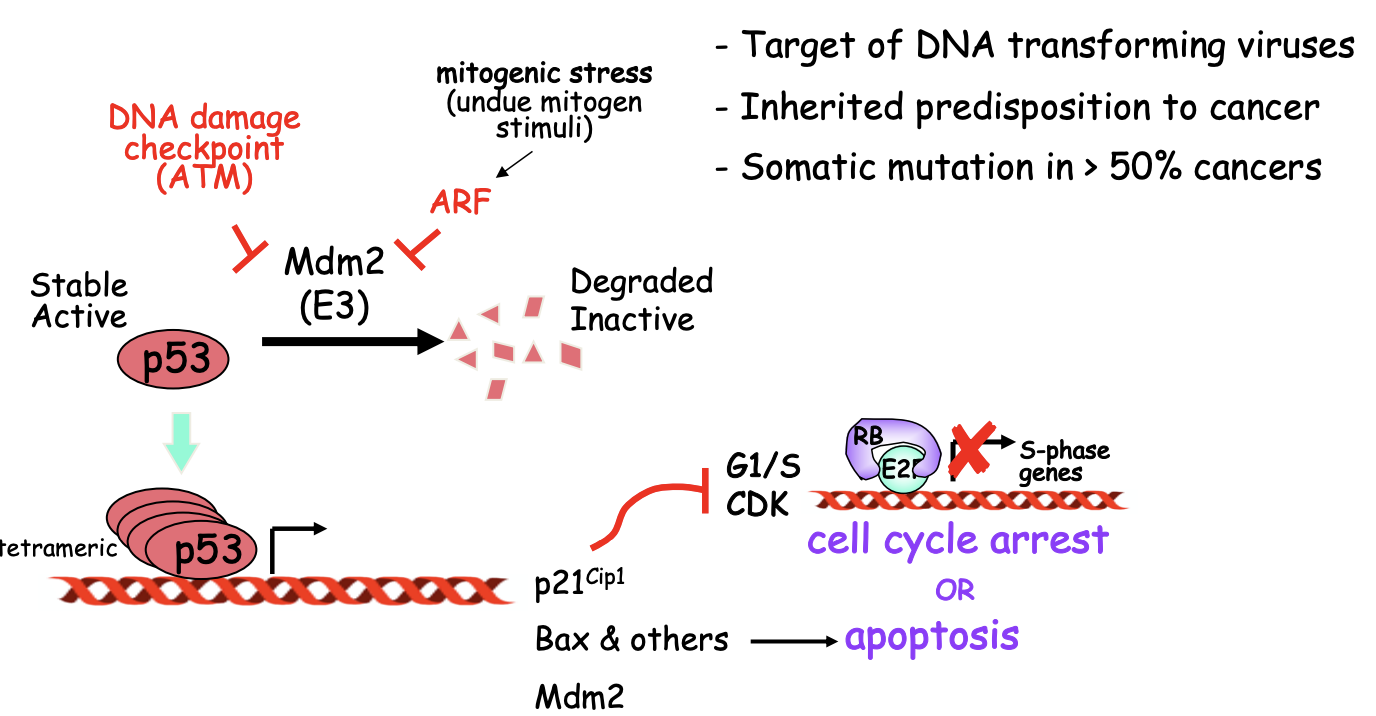
Research 4: What is p14 ARF?
protein encoded by CDKN2A locus
Tumour suppressor
Binds to MDM2 which stabilises p53!
means p53 can do its job and cause cell death
Research 4: How does p53 trigger cell cycle arrest or apoptosis?
Cell cycle arrest
vis CKI p21Cip1
Apoptosis
Via BAX
MDM2 is also a transcriptional target of p53
and forms a negative feedback loop
Whether chose between arrest and apoptosis depends on extent and persistence of the DNA damage!
Research 4: How can you have genetic predisposition to cancer?
A loss-of-function mutation in genes for:
negative regulators of cell cycle
checkpoint components
apoptotic pathways
With one of these mutations inherited, only need one inactivating ‘hit’ to cause cancer (in the other allele?)
THEREFORE: increased susceptibility
These genes are tumour suppressor genes
Research 4: mutations involved in different cancers

Research 4: Getting cancer from gain-of function mutations
A state of genomic instability causes
loss of critical control genes and emergence of gain-of-function mutations
Gets worse over generations
translates into gene amplification
boosting positive regulators in cell cycle
e.g cyclins D and E
Or cause even more chromosomal rearragments
can cause more oncogenesis
OVERALL: gain-of-function mutations→ cell proliferation and in a positive feedback loop
Research 4: How can this genomic instability occur?
Aneuploidy
incorrect number of whole chromosomes or portions due to segregation errors
instability at the nucleotide sequence level
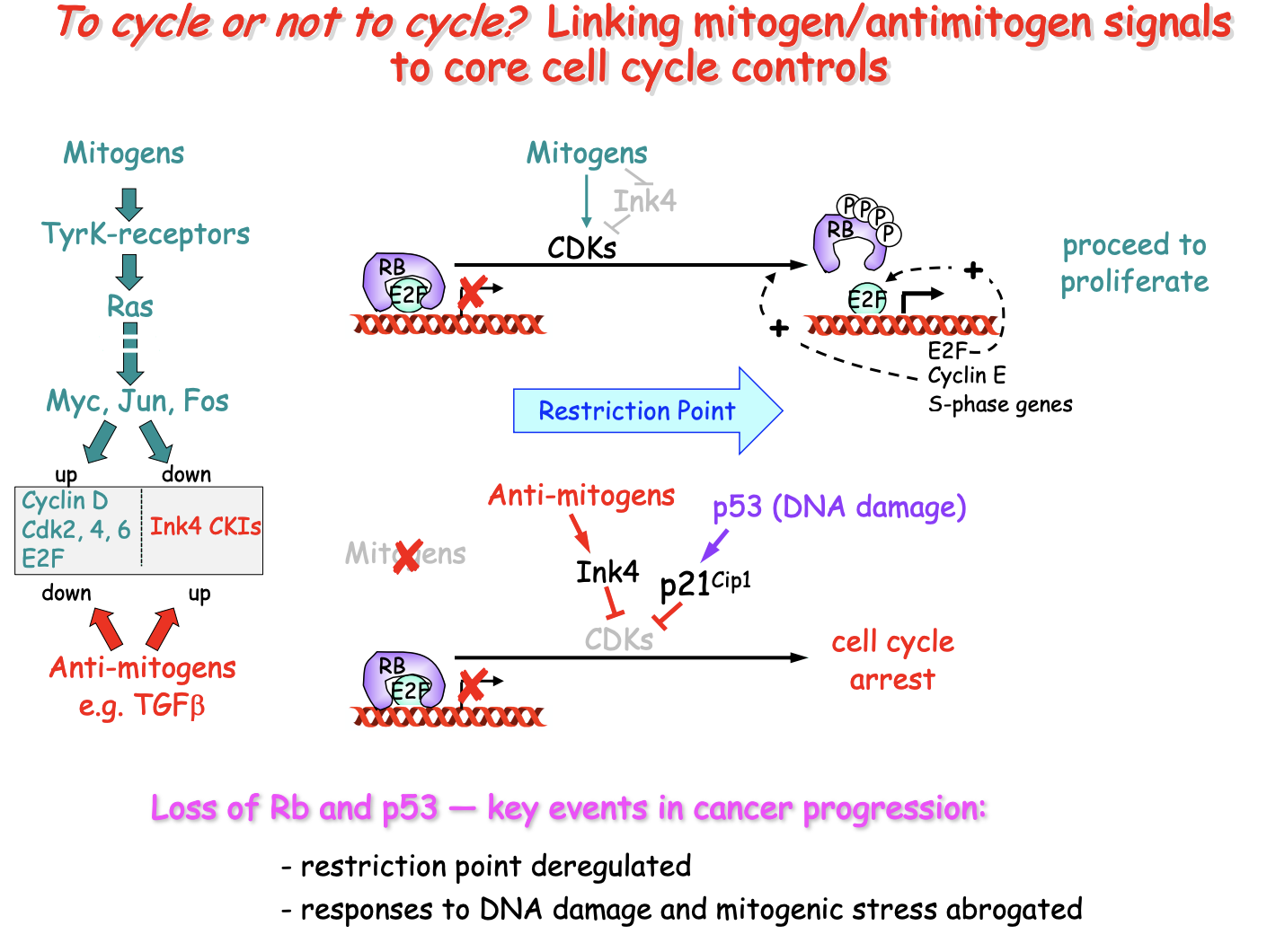
Linking mitogen/antimitogen signals to core cell cycle controls
Note:
Ink4→group of CKIs
TGFb→ transforming growth factor beta
used in cell growth, differentiation and apoptosis
Jun, Myc, Fos→ protooncogenes
crucial role in regulating gene transciption and cell growth
abnormal expression= cancer
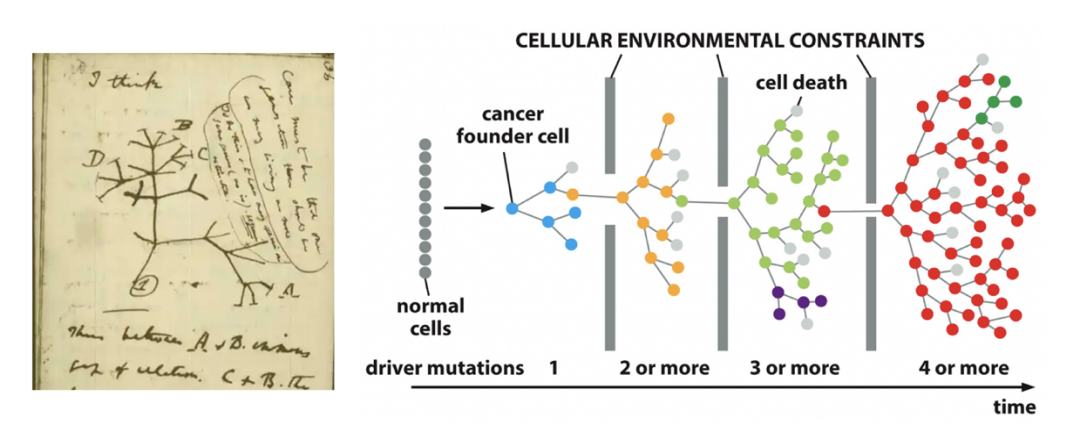
Two ways to view the expansion of cancer
Clonal view
Hierarchical view
Clonal view of cancer
Viewed as a multistep evolutionary process driven by mutations
Mutations are the driving force
of iterative cycles of selection and clonal expansion
Due to proliferative advantage!
constantly changing and evolving with mutations to get better at proliferation
Hierarchical View
cancer organised like normal tissues
some cells are ‘cancer stem cells’
give rise to the diverse set of cells making up a tumour
but: what is the nature of the cell of origin in this views??
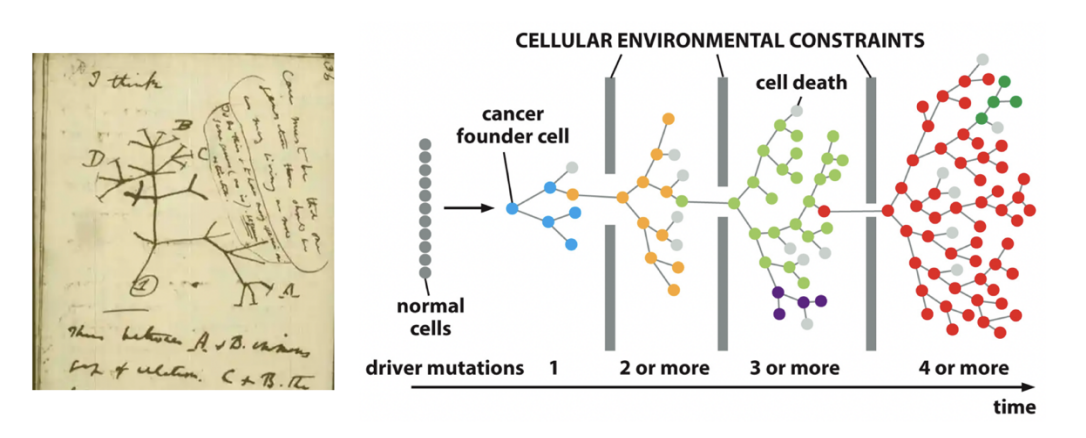
In either model: malignant transformation is enhanced by…
Increased rates of DNA damage
Loss or rearrangement
Disruption of DNA repair
loss of checkpoint mechanisms
ALL LEAD TO: increase above the spontaneous rate of mutations in normal cells
Cause of cancer→ Drive by mutations in cell cycle control:
upreg core elements for cell cycle progression
i.e activate oncogenes
inactivate core elements for cell cycle brakes
i.e inactivate TSGs
altering signalling pathways
at receptros, transducer or effector levels
Others
Overall causes of these mutations
chromosomal instability (CIN)
e.g lose, gain, rearrange whole or portion of chromosomes
e.g anueloploidy
both an outcome and further instigator of CIN
Genetic instability (GIN)
increased rate of point mutations
overall: These affect protooncogenes and TSGs→ to cause cancer!
What causes GIN and CIN
mis-regulation of cell-cycle
through loss of brakes or checkpoints
→ tumourgenesis
E.g (2) Mutations in TSGs
Due to mutations in Tumour suppressor genes (TSGs)
Normally:
encode cell cycle brakes
checkpoint elements
apoptosis stuff
DNA repair pathways
Cancer when
recessive loss-of-function mutation occur in both alleles
→This can be caused by either GIN or CIN
TSGs genetic predisposition to cancer
May inherit an already defective TSG allele
only new one further alteration to cause cancer in the other allele
it is the predisposition that is dominant over the actual gene that will cause the cancer
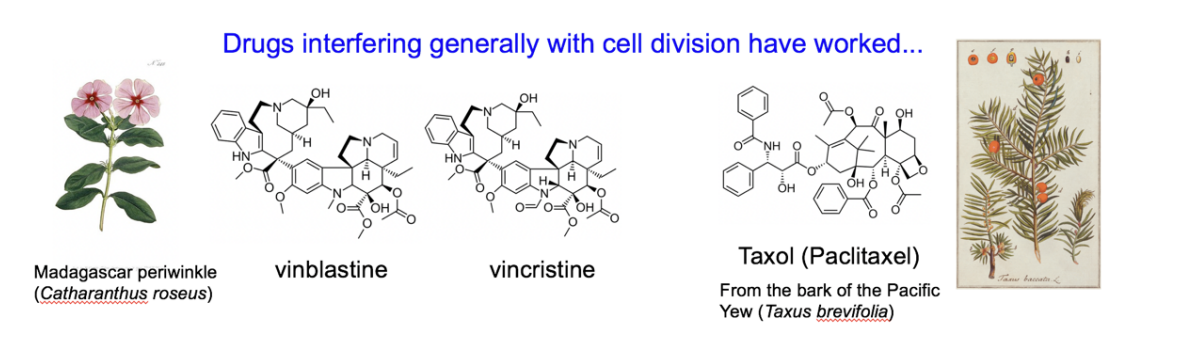
Why studying the molecular basis of cancer is important
Lead to novel therapies for
general or
unique signatures!
Cancer drugs for general cell division
Cancer drugs which are more specific due to molecular understanding
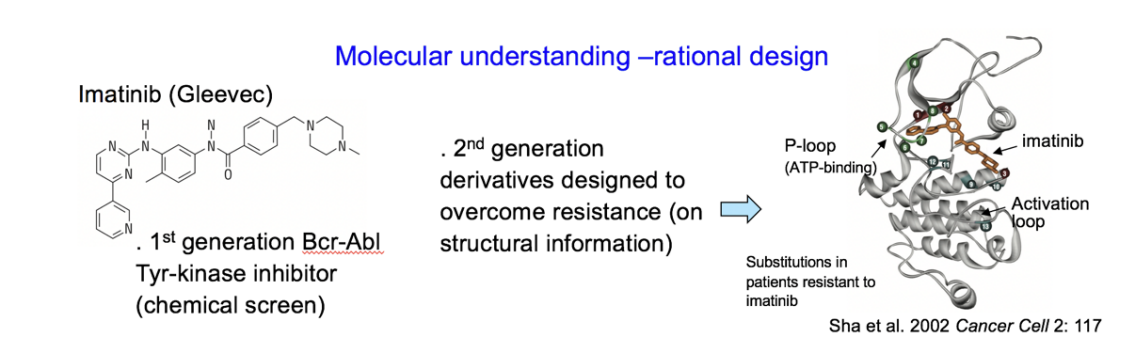
How make it easier to make new target strategies
Look at individual hallmarks of cancer
holistic view
see what they affect
form a drug/action plan which can target these specific things