New Material Final Exam Lecture 4 & 5/5: DNA Analysis - Parts 1 and 2
1/50
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
51 Terms
There are a lot of details to high-throughput DNA sequencing that we
did not cover in class. You are only responsible for the level of detail
we covered in class.You should know the components of a plasmid used in transformation
experiments and why each is used.You should know how blue-white screening works.
You should know the components of a PCR and the steps in each cycle
what is the diff between cloning and molecular cloning
cloning → cloning entire individual
molecular cloning → using some sort of vector (eg plasmid) then putting it in a cell that can naturally make copies of it
recombinant plasmid is inserted into microbe
microbe reproduces, reproducing plasmid
what are endonucleases
cut phosphodiester bonds WITHIN nucleic acids (not on ends)
what are restriction enzymes / restriction endonucleases
subtype of endonuclease
named for enzyme’s natural role in restricting infection by phages
recognizes foreign DNA and cuts it into pieces
restricts what type of DNA can exist in the host
what are palindromic strands of dna
sequence reads the same in the 5′ → 3′ direction on both strands
what do type 2 REs (restriction enzymes) do
Recognize a specific short DNA sequence (usually 4–8 base pairs, often palindromic).
Cut both DNA strands within or very close to that recognition site.
Produce either:
Blunt ends (straight cut)
Sticky/cohesive ends (staggered cut with overhangs)
each RE has a unique recognition sequence
what are blunt and sticky ends respectively
enzymes that prod blunt ends: cut straight through both strands at the same position, leaving no overhangs
enzymes that prod sticky ends: cut offset between the two strands, creating short single-stranded overhangs
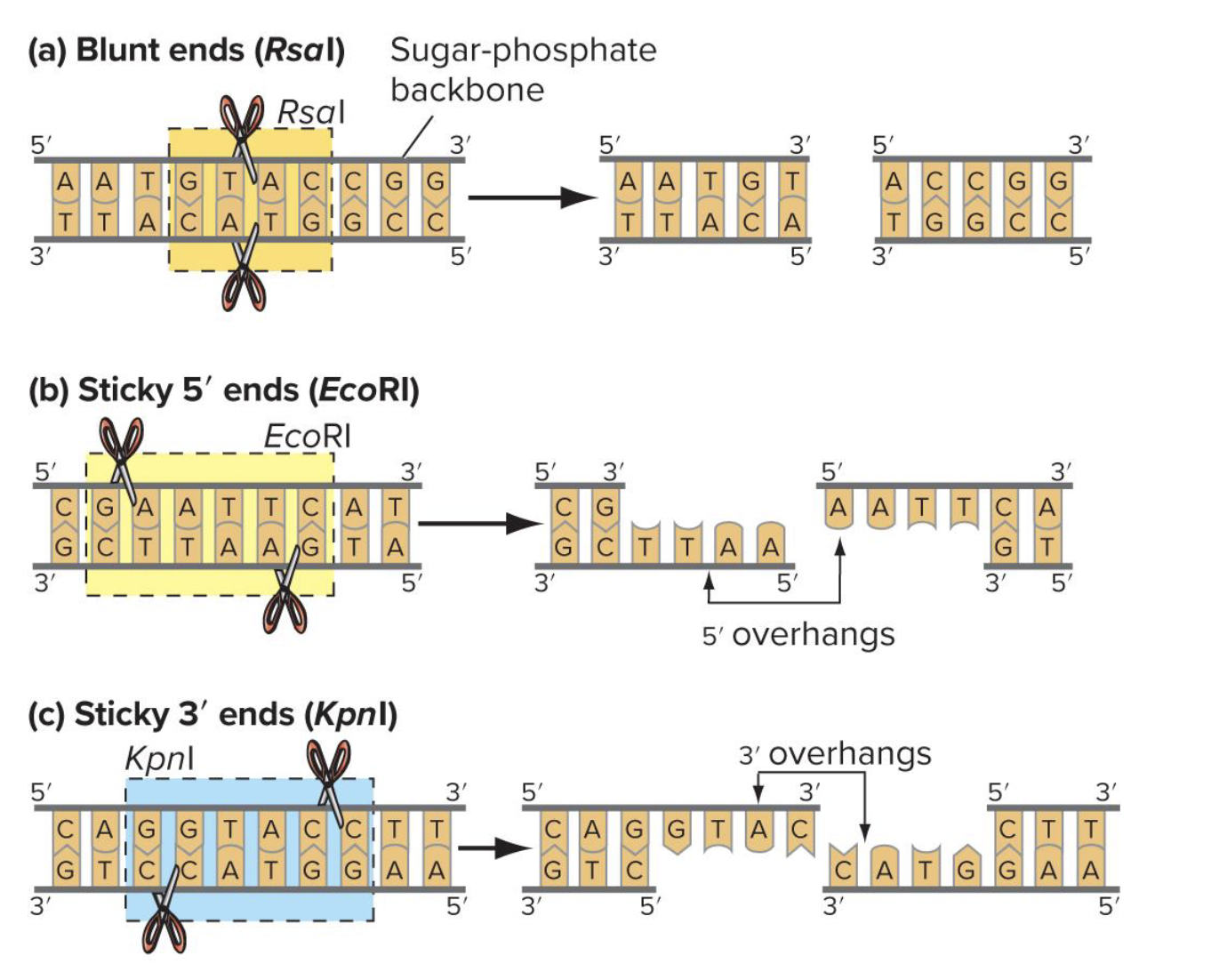
whicj direction is the recognition site for restriction enzymes always read in
5’-3’
what does gel electrophoresis do? how does it work
separates dna fragments based on size
have aggross gel w wells filled w dna-binding blue dye in it
the matrix is mostly made of aggros
has + and - electrodes
during electrophoresis, blue dye moves down the gel
the dna moves down too (diff distances depending on size)
DNA migrates from - charged end to + (bc dna is - charged)
the larger the dna molecule, the slower it moves through the gel matrix
DNA-binding dye lights up DNA bands under UV light
do larger or smaller molecules move further towards the + side of the
smaller
what does each band represent in gel electrophoresis
many dna fragments of the same size
what is molecular cloning
the process of using living cells to make many exact replicas of a DNA fragment
how are dna fragments purified and inserted into vectors (eg plasmids)
purify a DNA fragment (eg by gel electrophoresis)
insert the dna fragment into the vector
digest a pre-existing vector (eg plasmid) AND the dna fragment with the same RE (opens them up, leaves them w same exposed complimentary regions of dna)
creates complimentary sticky ends that can H+ bond w each other
DNA ligase joins the dna fragments tg (covalently links them)
cell divides and reprod plasmid with gene of interest interested in it
explain the steps to molecular cloning
purify dna fragment (eg by gel electrophoresis) and insert it into a vector (eg plasmid)
transfer vector w its insert into “compitent” cells (cells that can incorporate foreign substances into it and replicate foreign dna)
copies will be made through dna replication
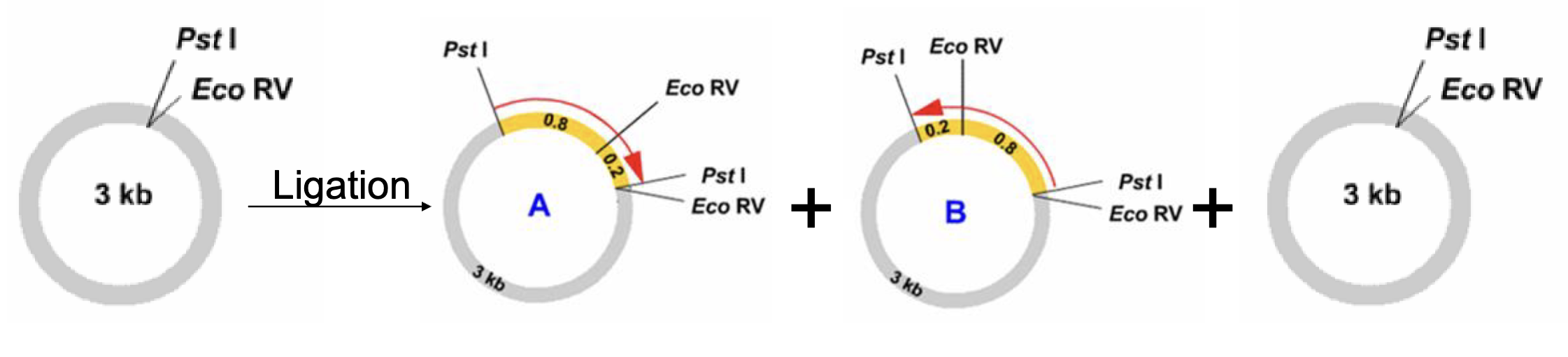
will inserting a segment of dna into a plasmid yield identical new plasmids
no → you will end up w a combination of diff plasmids
some proportion of plasmid will have insert in one orientation or the other
some will have no insert (not 100% effective)
what are the 3 main features of plasmids used as cloning vectors
origin of replication
selectable marker gene (eg antibiotic resistance gene)
some way to differentiate btwn cells that uptook plasmid vs ones that didn’t
if they took in the antibiotic then you can treat the cells created w the antibiotic and the surviving ones would have uptaken the dna
polylinker (DNA sequence containing multiple restriction enzyme recognition sites)
so could open up the plasmid
what is bacterial heat shock transformation
process of inserting the plasmid vector into the bacterial cells
making bacterial cell walls leaks so they let in plasmids
make test tubes a bacterial cells of interest really cold then really hot suddenly → damages cell walls
what is done to the bacterial cells of interest after heat shock transformation
shake them to whatever temp the bacteria grows best in in their test tube with NO ANTIBIOTIC IN THE INITIAL GROWTH MEDIUM
gives cells an hourish to develop / incorporate resistance genes from plasmid
add them to a growth medium +antibiotic
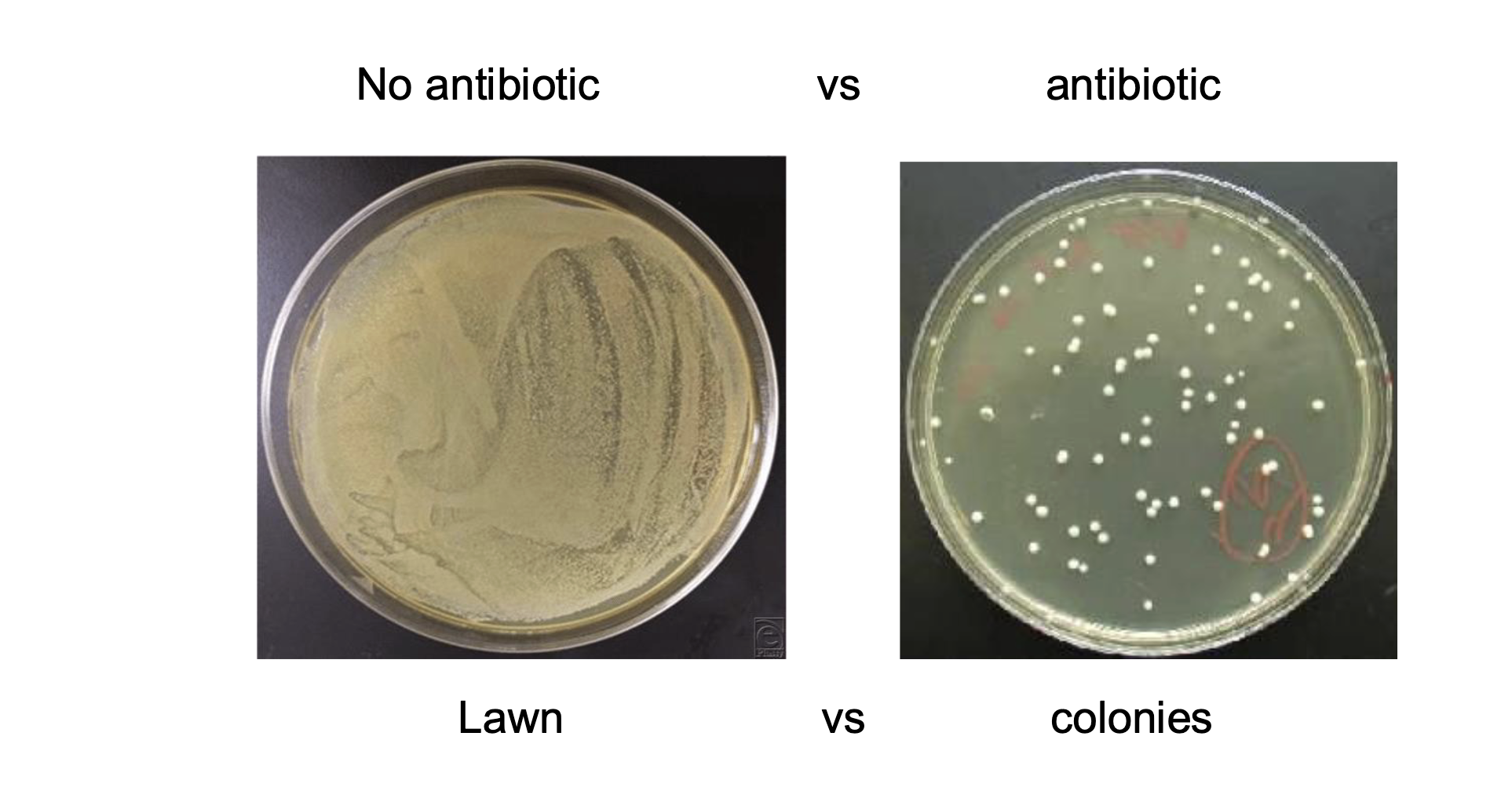
so only cells that have properly incorporated the plasmid survive
creates colonies instead of a lawn
what are inserts
the piece of foreign DNA you WANT to put into the plasmid vector
NOT the antibiotic resistance gene → that is added in conjunction to that
what are some challenges with creating molecular clones
not all cells will take up a plasmid
not all plasmids will have an insert (self-ligation)
some cells will ligate before plasmid can be incorperated
how do you distinguish between cells without inserts from those w inserts
the lacZ gene (codes for B-galactosidase; breaks down lactose)
on some plasmids the multiple cloning site is located within the LacZ gene
lacZ gene is split by fireign DNA insert
no LacZ product (no B-galactosidase)
B-galactosidase can also cleave things other than lactose → can cleave X-Gal (analogue of lactose) and make it from white → blue
put vectors onto plate with X-Gal → ones with LacZ gene in tact will show as blue and ones w no LacZ (and WITH insert) will be white
how do you distinguish between cells…
without plasmids
without inserts
with plasmid with insert
without plasmids → antibodies
without inserts → blue
with plasmid with insert → white
what are some applications of molecular cloning
gene expression
prod a lot of protein product of interest
study gene fx
introduce and study specific muts
link promoter regions to reporter genes to study gene regulation
etc
what is a polymerase chain reaction (PCR)
allows us to amplify regions of DNA we’re interested in
in vitro (tube) DNA synthesis reaction
creates many copies of a segment of dna
generally used to amplify segments of dna ~200-1000 bps long
needs very few starting dna molecules
very sensitive
can be visualized using agarose gel electrophoresis
what are amplicons
amplified dna regions in PCRs
what are some applications of PCRs
make many copies of dna sequence of interest
research
genetic engineering
cloning experiments
used in research and diagnostics
prenatal genetic testing
testing food for pathogens
HIV test
genetic fingerprinting
etc
which reagents are needed in PCRs
polymerase
do NOT want primase (will create primers on random pieces of dna)
instead want to provide specific primers in the mixture
DNTPs needed
need template
need buffer
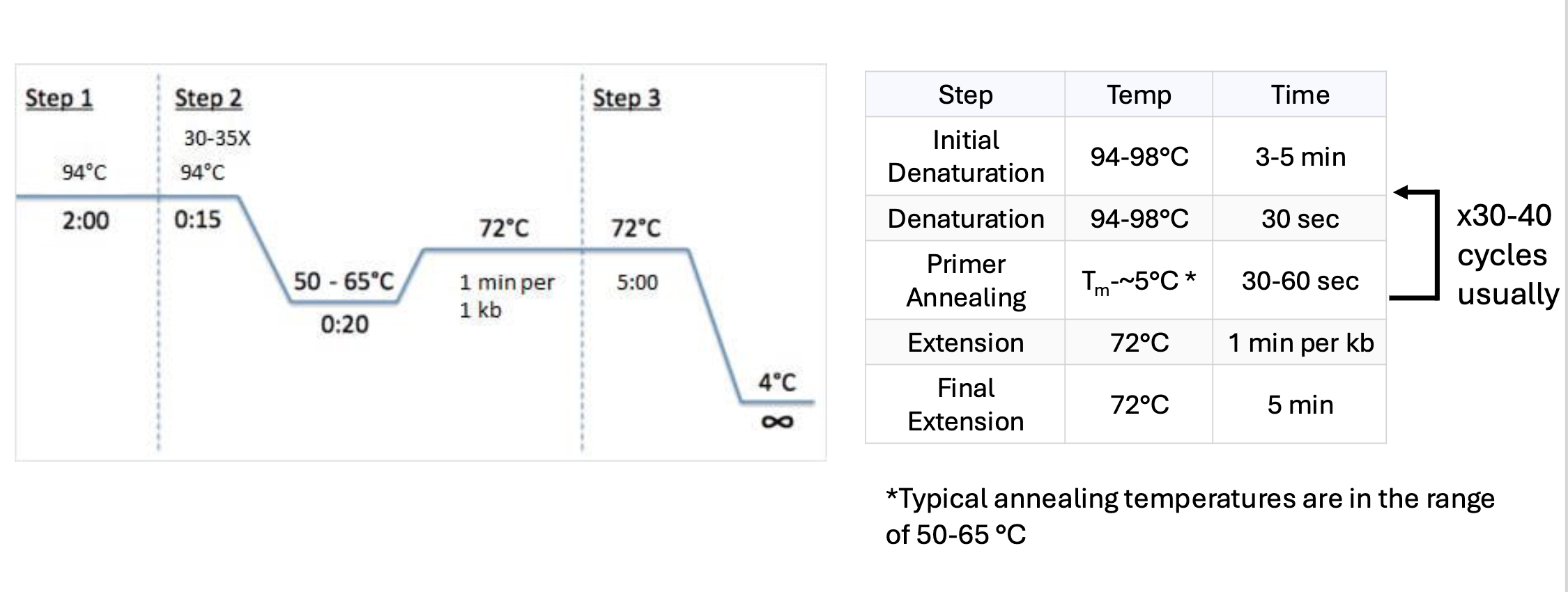
what are the 3 steps in each cycle of polymerase chain reactions (PCRs)
denaturation
seperate double strands using temperature
annealing
make conditions so primers can add
extension
synthesizing new dna
explain the denaturing step in PCR reactions
around 95-98 degrees C
double stranded dna breaks apart
mixture contains prokaryotic dna pol from thermophiles so they don’t denature
explain the annealing step in PCR reactions
around 50-65 degrees C (depends on length and sequence of primer)
primers now can/do bind to unwound single stranded dna
explain the extension step in PCR reactions
around 68-72 deg
dna pol binds and extends off primer
what does PCR result in? why is that the case
exponential amplification
dna doubles each time
longer you keep PCR going the more times there will be more replications
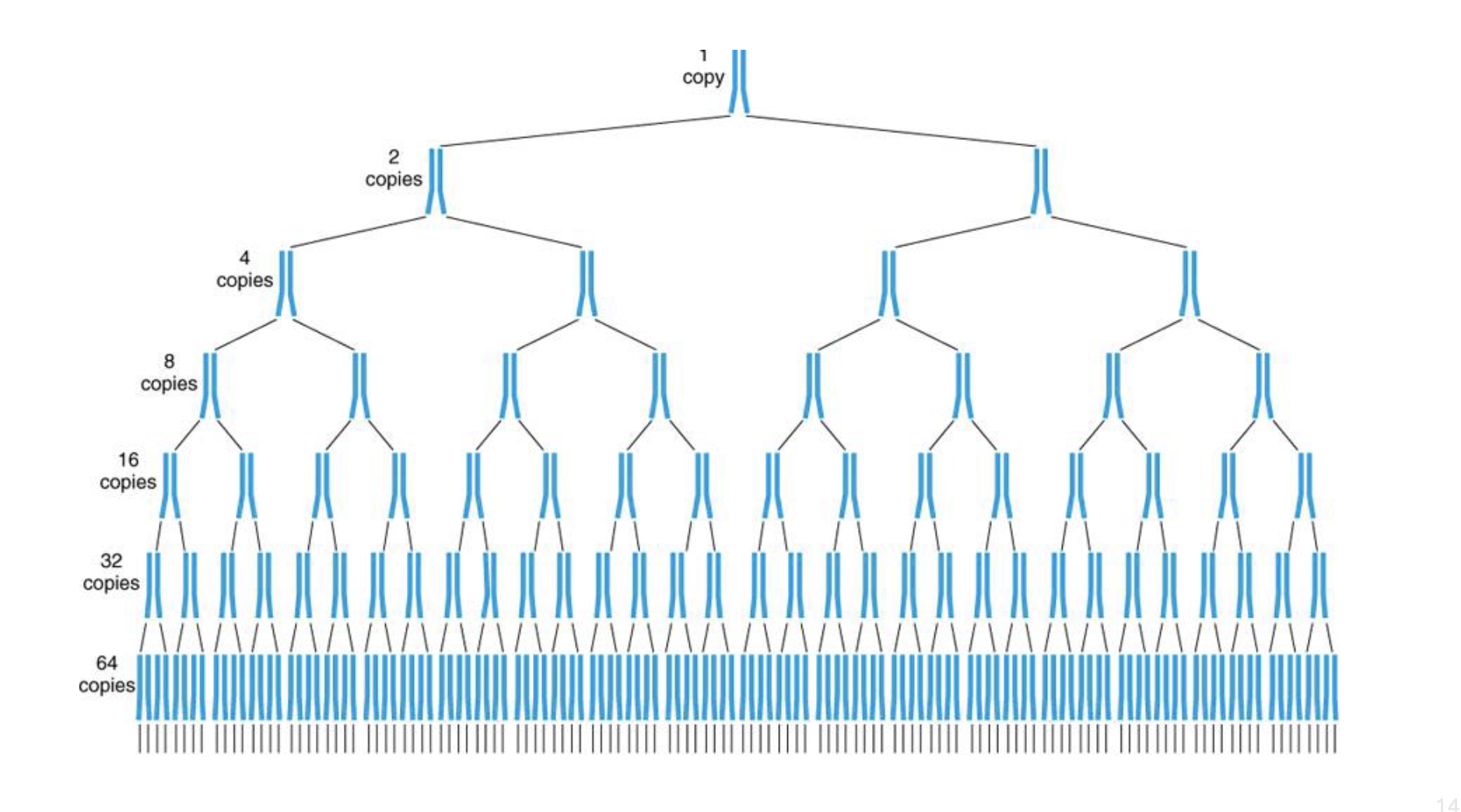
what is the general PCR thermal cycle
keep temp around 95 to 98 deg C at first for around 2 mins so dsDNA breaks apart
reduce temp (to around 50-65 C) for around 20 secs so primers can bind
inc temp again to around 68-72 deg C so polymerase can bind and extend off primer
repeat around 30-40 times
dec temp to around 4 C when done
what was the first DNA sequencing method? explain what it is used for and what it is a combination of
Sagner sequencing
older method
used for smaller sequences (not whole genomes → although USED to be used for that but took ages)
combines chain termination with florescent labeling
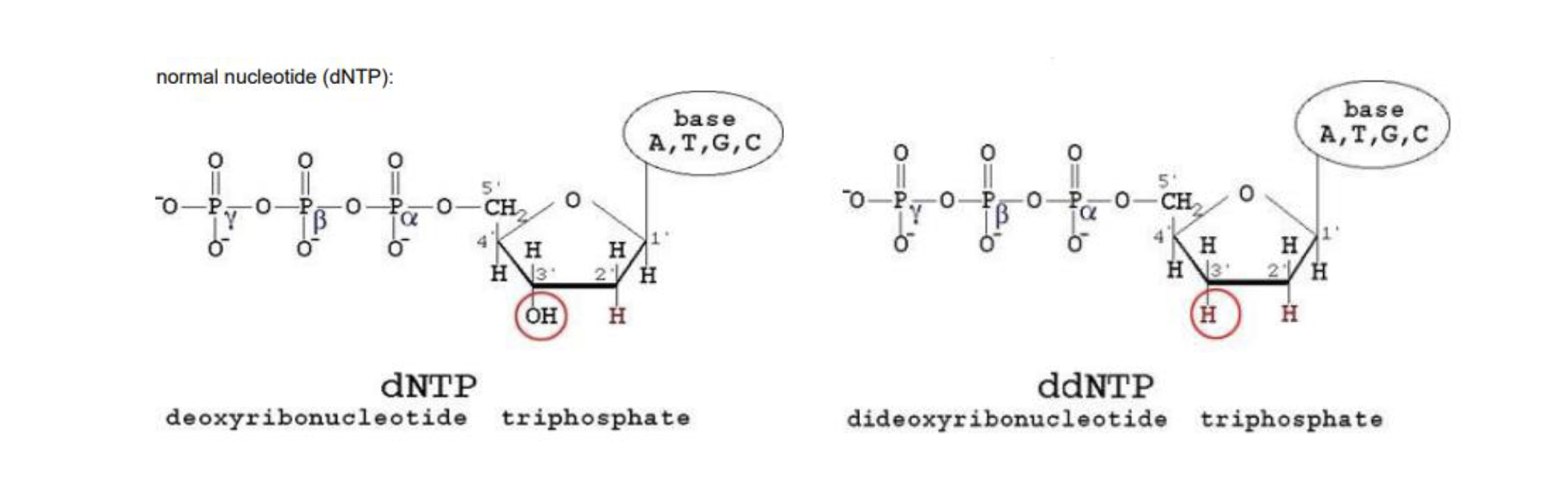
what is chain termination
DNA pol connects the 3’ OH to the next nucleotide at the 5’ phosphate
strand cannot elongate without the 3’ OH
cant add other nucs if that OH is turned into an H instead
what are dNTPs vs ddNTPs
dNTPs are standard → they have a OH group on the 3’ end
ddNTPs (deoxi nucleotide triphosphate) → has OH switched with an H can’t add another nucleotide after these
what is fluorescent labeling
each ddNTP labeled w a diff colour
dNTPs are NOT labeled
the ID of the last base can be determined based on how the nucleotide fluoresces (which colour it is)
are dNTPs or ddNTPs more abundant in Sanger sequencing
dNTPs → higher conc so more likely to encorperate
how do you get so many diff dna sequences of diff lengths in Sanger sequencing to test from
PCR with fluorescent, chain terminating ddNTPs
what happens when a ddNTP is incorperated into a nucleotide sequence during dna repilcation? what does this help with
replication stops → nothing else can bind
you end up w a mixture of strand lengths each ending where the ddNTP has been incorporated
can tell which is where in the seq based off colour
if only dNTP, would all be same length and be harder to tell which nuc is where
t/f: incorporation of the ddNTP is random
true
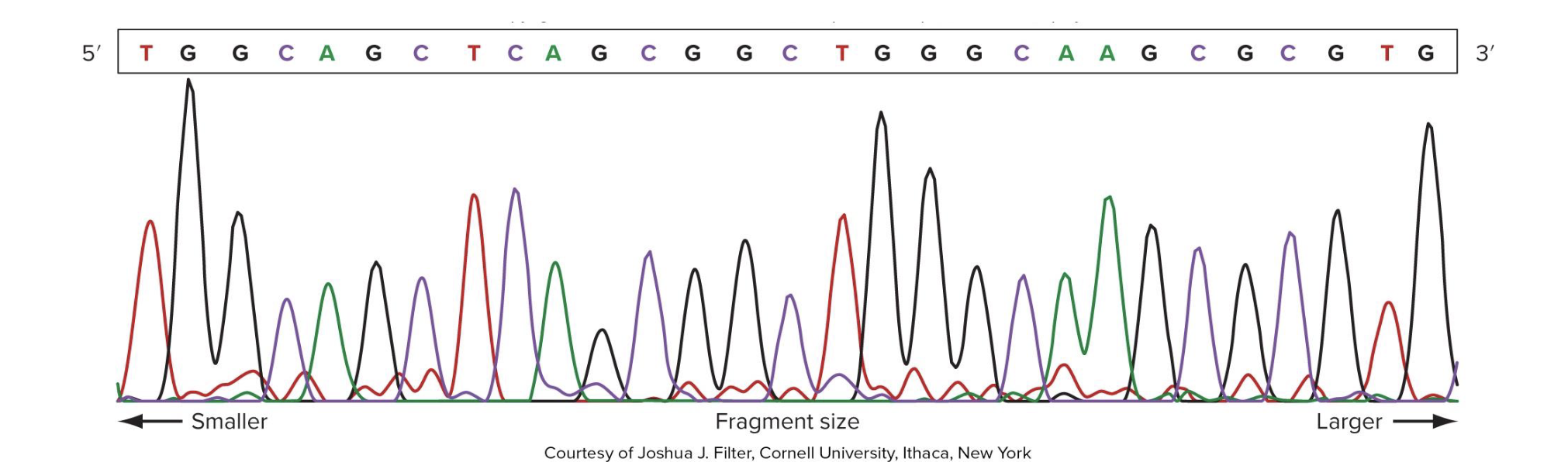
what are 2 potential outputs of Sanger sequencing
sequence chromatogram
text file
computer’s interpretation
errors common near the start and end (bc peaks are closer tg → less chance ddNTPs bind there)
note: the pic is a sequence chromatogram
what is capillary electrophoresis similar to? what does it do?
similar to gel electrophoresis BUT not agarose and in a thin tube
reaction products separated by size
each fragment size has a fluorescent label
how does capillary electrophoresis work?
PCR w fluorescent, chain-terminating ddNTPs
size seperation like in capillary gel electrophoresis (but recognized
laser excitation and detection by sequencing machine (tells which colour each ends w → which nucs are where in the overall sequence)
which method would you use if you wanted to sequence more than one DNA segment at a time - like a whole genome or all the mRNA in a cell
high-throughput sequencing
CAN use sanger but takes forever (have to break up whole genome into segments and separately sequence each)
what are the 2 main technologies used for high-throughput sequencing
Sequencing by synthesis (Illumina)
PCR-based dna sequencing on a flow cell
diff machines exist that have diff read lengths, cycles, number of samples that can be included, and amount of data you get out (sequencing depth)
(Oxford) Nanopore technology
based on characteristic current disruption each nuc produces when a dna molecule flows through a nanopore
diff machines exist → including the Minlon which is the size of a usb key
nanopore looks mostly like Sanger
what is a library and the library preparation process in sequencing by synthesis
library: the collection of DNA (or cDNA) fragments from your sample that have been processed so they’re ready to be sequenced
contain adapters that help w the process and primers
a sequencing run can include many samples, each usually consisting of many DNA fragments w different sequences
one index per sample NOT one index per type of fragment
can have a mixture of 10s of hundreds of diff sample
what does sequencing by synthesis use
reversible dye terminator nucleotides
have reversible blocking group → can be manipulated chemically to regenerate an -OH allowing synthesis to continue one step at a time
a diff fluorescent dye for each nuc
like Sanger sequencing but reversible
t/f: each sequence in synthesis sequencing will all be the same length at any given time
true
what does the output of sequencing by synthesis look like
each dot is many identical copies of a single dna fragment from the library
diff channels for each nuc w a bunco of dots on it
tells you which place in the sequence it is
each dot lights up according to the nucleotide just incorporated
all samples are sequenced at the same time
explain nanopore sequencing
flow cell contains an array of tiny holes (nanopores) embedded in a mem
each nanopore has an electrode connected to a sensor
when a molecule passes through a nanopore, the electrical current is characteristically disrupted → as DNA moves through it will influence nanopore differently and computer connected to electrode will tell you which is nucleotide is which (diff electrodes connected to diff nanopores let off diff signals which tells which nuc it is)