unit 3 aos 2 chem
1/137
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
138 Terms
collision theory — for a successful reaction particles must…
collide each other
have enough energy to pass their activation energy barrier
collide with correct orientation for bond breaking
not all particle collisions cause reaction
particles might just bounce off each other if not enough energy or not correct orientation — only changes velocity
activation energy def
minimum amount energy needed for reaction to occur by breaking reactant bonds
symbol = Ea
in energy profile diagram this is diff between reactant energy line and peak
e.g. ways to measure reaction rates
volume of gas evolved
mass solid formed
decrease in mass bc of gas evolved
colour intensity of solution
precipitate formation
pH
temperature
— recorded as changes over time
the steeper the gradient in reaction rate graphs
the faster the rate of reaction
factors affecting reaction rate
surface area
concentration
pressure
temperature
catalysts
some cars park too close?
factors affecting reaction rate — concentration
as conc increases → number of particles in a given volume increases → frequency of collisions increase → number of successful collisions per sec increase → reaction rate increase
factors affecting reaction rate — pressure
can be increased by 1) adding more reactants to same volume or 2) decreasing volume
increased pressure → increase partial pressures/partial concs of all subs → increase frequency collisions → increase number of successful collisions per sec → increase reaction rate
factors affecting reaction rate — temperature
increased temp → increased kinetic energy particles hence increased speed of particles →increased frequency collisions → also increased proportion particles that have enough energy to pass activation energy barrier → increased proportion successful collisions per sec → increased reaction rate
why adding inert gas doesn’t increase rate of reaction
increases total pressure but not partial pressures of reactants → frequency of collisions increase but not frequency of successful collisions → no increase reaction rate
factors affecting reaction rate — surface area
for heterogeneous reactions (subs in reactions at diff phases)
increase surface area → increase particles exposure and contact area → increased frequency collisions → increased number successful collisions per sec→ increased reaction rate
why is increasing surface area sometimes dangerous
in combustion reactions → if powder instead of large solids just need spark → cause very fast reaction → explosion
catalysts def
subs that lower activation energy to increase reaction rate w/o being consumed
factors affecting reaction rates — catalysts
catalysts added → make reactant bonds weaker hence lowers activation energy needed to break them → increased proportion particles that pass activation energy barrier via alternative pathway → increased frequency successful collisions → increased reaction rate
catalysts added → provide alternative pathway with lower activation energy → increased proportion of particles with enough energy to pass activation energy barrier → increased frequency proportion successful collisions → increased reaction rate
closed system def
system allows transfer of energy but not matter with surroundings — reactants and products contained
open system def
system allows transfer of energy and matter with surroundings — reactants and products not contained
isolated system def
system doesn’t allow transfer of energy and matter with surroundings — e.g. well insulated calorimeter
why rate of evaporation in a closed system eventually reach 0
gas molecules colliding randomly everywhere in flask → some collide liquid surface and re-absorbed back into the liquid → as more evaporation occurs and more gas particles form → more chance of gas particles hitting liquid and re-absorbed → eventually reach when 2 processes have same rate → rate of evaporation measured is 0 (as what being produced, same amount being consumed)
in reversible reactions the yield of products…
is always less than the stoichiometric prediction
are all chem reactions equilibrium reactions
yes
but some of them the degree of reverse reaction is so small it can be ignored/negligible
equilibrium reaction def
reaction where forward and reverse reactions are significant
reversible reactions def
equilibrium reactions where products re-form into reactants to significant extent
eqs have double arrow
yield of products always less than stoichiometric prediction
irreversible reaction def
only occur in forward direction where products do not re-form into reactants
e.g. combustion of fuel
rate vs extent of reactions
rate: how fast it occurs (how long to establish position of equilibrium)
extent: the degree which reactants convert into products (the position of equilibrium/how far to the right)
equilibrium constant def
value indicates extent conversion of reactants to products at equilibrium
Kc
if high value = large amount reactants conv into products
if low value = less amount reactants conv into products
does catalyst affect equilibrium position (degree of conv of reactants into products)
no
it only affects rate of forward and reverse reactions (equally) and time taken to get to equilibrium
homogenous reaction def
reaction where all substances in same phase (physical state)
homogenous equilibrium def
all species in equilibrium mixture same physical state
heterogenous reaction def
reaction where substances involved in diff phases (physical states)
occurs at boundary between 2 physical states
can also happen between immiscible liquids (don’t mix and form homogenous mixture)
equilibrium def
when forward and reverse reactions occurring at same rate
when reaction at equilibrium…
reactants being formed and used at same rate
products being formed and used at same rate
— their concs don’t change
dynamic state not static
when considering getting to reaction equilibrium
consider:
starting point (initial concs of reactants and products) → what this means for net forward or net backward reaction → as more reactants or products consumed and more reactants or products produced eventually reaction rstes equal → equilibrium reached → no net change on conc of substances anymore
equilibrium and successful collisions between particles
at equilibrium frequency of successful collisions for forward reaction is equal to frequency successful collisions for backward reaction
representing equilibrium reactions with chem eqs
use reversible arrow
thermochem eqs with delta H value convey more info about eqs as equilibrium can be diff for endo or exo reactions
representing equilibrium reactions with rate vs time graphs
in this e.g. where we start with reactant high conc (can diff according to example):
as reactants used up → conc drops → hence rate forward reaction drops
as products produced → conc increases → hence rate backward reaction increases
this continues until reach a time where same rates of reactions → equilibrium reached
but note still in above e.g. there is a net forward reaction as still more forward than reverse happening even as forward reaction rate decreases
if situation flipped and more product conc at start then the graph labels would swap and reverse reaction would be on top instead (net reverse reaction)
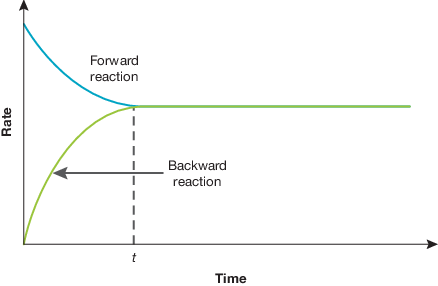
representing equilibrium reactions with conc vs time graphs
in this e.g. where we start with reactant high conc and net forward reaction (can diff according to example):
as reactants used up → conc drops until equilibrium point
as products produced → conc increases until equilibrium point
final conc at equilibrium depends on initial conc, equation stoich, value of equilibrium constant etc
the substances change according to their stoich molar ratios from equation (e.g. from graph = C increases twice amount than B does bc their molar ratios is 2:1)
note if catalyst added only change is that equilibrium (at t) reached at lower time → reached faster — final subs concs not changed
concentration fraction def
fraction from concentrations of substances in equilibrium reaction
that have a constant value (constant: unchanging over time at eq?)the concs of products divided by concs of reactants all to the power of their coefficients respectively
Q
equilibrium law def
relationship between concs of products and reactants, also considering their stoic ratios
equilibrium constant def
value of conc fraction at equilibrium
Kc or K
also gives extent which reactants conv into products
equilibrium constant formula
fraction
where numerator is product concs to the power of their corresponding stoic equation coefficients
over the denominator of reactant concs to the power of their corresponding stoic equation coefficients
using equilibrium constant to indicate reaction extent (whether net F or net R)
if K > 104 = net F reaction rate → has been more conv of reactants into products to get to equilibrium
if K is between K = 104 and K = 10−4 then extent of conv reactants into products moderate
if K < 10−4 = net R reaction rate → little conv of reactants into products to get to equilibrium
— to make this make sense rmbr that K is from a fraction where numerator is product conc → hence makes sense that if more forward reaction then more products conc hence larger K value
describing equilibrium qualitatively
use describing via ‘position of equilibrium’
if K is large then net F reaction rate to reach equilibrium → equilibrium lies to the right
if K is small then net R reaction rate to reach equilibrium → equilibrium lies to the left
what can affect equilibrium constant for a reaction
temp
how the equation for it is written (e.g. coefficients or direction)
what is the unit of K
depends on amount of M in fraction formula
if all M units cancel out K can be without units!
how K can change according to equation changes for same reaction
if equation coefficients double = square K value
if equations coefficients halve = square root K value
if equation is reversed = reciprocal K value
using stoich in equilibrium law calculations
ICE method must be used sometimes (e.g. if initial concs given and only one equilibrium conc known)
Initial amount - note usually if not given product amount it is zero as net F is occurring and at start it is not there yet duh
Change - can either be increase in amount from initial to equilibrium point (positive) or decrease in amount (negative) → note when product conc increases, the reactant conc correspondingly decreases according to their mol ratios!!
Equilibrium - the final amount of substances at equilibrium based on change
— then conv this amount into concs and do further calcs as needed
reaction quotient def
concentration fraction at any other stage apart from equilibrium during reaction
Q
reaction quotient vs concentration factor
same thing? it’s just diff to K though
if Q = K
reaction is at equilibrium
if value of Q is diff to K
reaction has yet to reach equilibrium
if Q > K
net backward reaction is occurring (because K requires smaller value hence decrease in numerator hence decrease in product conc)
— aka backward reaction rate greater than forward reaction rate as reaction moves towards equilibrium
if Q < K
net forward reaction is occurring (because K requires bigger value hence increase in numerator hence increase in product conc)
— aka forward reaction rate is greater than backward reaction rate as reaction moves towards equilibrium
dynamic state of equilibrium def
reaction still taking place (hasn’t stopped) but substances concs kept constant as forward reaction rate equals reverse reaction rate
at equilibrium…
the forwards and backwards reactions are occurring at same rate not same extent
yield def
amount of product
le chatelier’s principle def
when change made to equilibrium system this system opposes this to restore an equilibrium state
le chatelier’s principle
any change that affects equilibrium position causes that equilibrium to shift in a way to partially oppose that change
ways a system at equilibrium may be disturbed
adding or removing subs involved (can be physical or chemical)
changing volume at constant temp
changing temp
le chatelier’s principle — adding substance to system
change: adding subs
effect: increased conc of reactant or product
hence system try to oppose change and re-establish equilibrium → reduce conc of either reactant or product (depend on what was added initially) → one direction favoured → equilibrium shifted to right or left
le chatelier’s principle — removing substance from system
change: removing subs
effect: reduced conc of reactant or product
hence system try to oppose change and re-establish equilibrium → increase conc either reactant or product (depend which was removed initially) → one direction favoured → equilibrium shifted to right or left
adding or removing subs at equilibrium — collision theory and equilibrium law
collision theory: subs added or removed → conc increase or decrease of subs → subs reaction rates affected accordingly → as reactions continue conc changes as usual and reaction rate changes until equilibrium reached
equilibrium law: subs added or removed → conc increase or decrease → affects Q formula accordingly → Q needs to increase or decrease to meet Kc → hence either net F or net R reaction rates until new equilibrium reached where Q = Kc
le chatelier’s principle — volume increase
change: volume increase
effect: all partial concs decrease hence ttl conc decrease
hence system try to oppose change by favouring direction that produces more particles → net F or net R reaction rates until equilibrium → equilibrium shifted to right or left
le chatelier’s principle — volume decrease
change: volume decrease
effect: all partial concs increase hence ttl conc increase
hence system try to oppose change by favouring direction produce less particles → net F or net R reaction rates until equilibrium → equilibrium shifted to right or left
increasing or decreasing volume at equilibrium — collision theory and equilibrium law
collision theory: volume increase or decrease → all partial concs increase or decrease → ttl conc increase or decrease → direction with reactants of more particles have increased collision → hence this direction has net rate → as reactions continue concs change accordingly and reaction rates change until equilibrium reached
equilibrium law: volume increase or decrease → all partial concs increase or decrease → affects either numerator or denominator more in Q formula → hence Q increase or decreased → Q must then increase or decrease to meet Kc value → reaction rates change accordingly until Q = Kc at equilibrium
when can volume not be used to change position of equilibrium
when both reaction sides same no. of moles → system cannot change conc of particles (and vol change didn’t disturb eq at all?)
how to maximise yield of equilibrium reactions
by disturbing existing equilibrium so system opposes change by favouring one direction to produce more product amount while re-establishing equilibrium
le chatelier’s principle — increasing temp
change: giving heat
effect: increasing thermal energy / temp
hence system try to oppose change by decreasing thermal energy → favours endothermic reaction direction as converts thermal to chem energy → new equilibrium position and changed equilibrium constant
le chatelier’s principle — decreasing temp
change: removing heat
effect: decreasing thermal energy / temp
hence system try to oppose change by increasing thermal energy → favours exothermic reaction direction as converts chem to thermal energy → new equilibrium position and new equilibrium constant
ways system equilibrium can be disturbed — which cause sudden conc change
causes sudden conc change: adding/removing substances, volume changes
doesn’t cause sudden conc change: temp changes
ways system equilibrium can be disturbed — which cause diff equilibrium constant
cause diff equilibrium constant: temp
doesn’t cause diff equilibrium constant: adding/removing substances, volume changes
increasing or decreasing temp — collision theory
when temp increases → reaction rates affected → however endothermic reaction increases more as it absorbs thermal energy → hence endo net rate > exo net rate until equilibrium reached — image for this
unsure abt temp decrease? maybe temp decreases → reaction rates affected → however exothermic reaction increases more as produces thermal energy → exo net rate > endo net rate until equilibrium reached
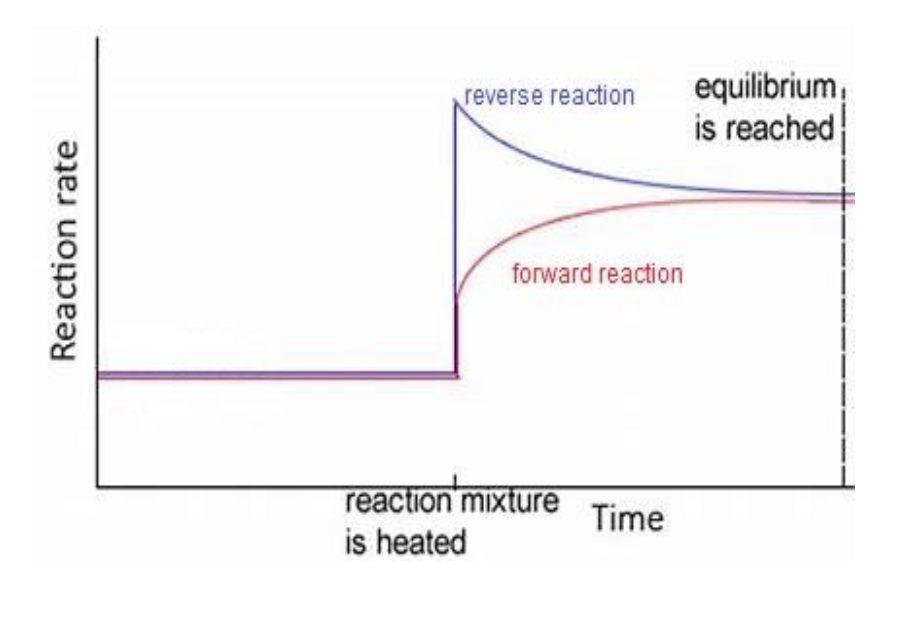
equilibrium shifting
to the right: forward reaction favoured
to the left: reverse reaction favoured
why can change in gases vol at constant temp also be interpreted using pressure not just conc
because pressure is proportional to conc → hence partial pressures may be referenced when vol changes
why does changing temp in system change equilibrium constant value
bc change in temp = change in energy available to system
changing conc by adding/removing or vol changes only causes redistribution of available energy and diff eq position but K value stays same
notes on conc vs time graphs
conc changes reflect stoic equation ratios
at equilibrium all concs become constant horizontal line at same time for all
sudden dip or spike in one conc means one subs added or removed
sudden dip or spike in all concs mean volume change
change in conc values without obvious spike or dip indicate temp change
one side equation all concs will increase or decrease and other side equation all concs vice versa
can Le Chatelier’s principle apply to open systems
yes
e.g. if open system and has a gas product in reaction the gas will keep escaping (subs removal) and equilibrium continually disrupted hence system oppose and more reactants consumed to replace it until all run out
how can extent and rate conflict for reaction from temp industrially?
e.g. in exothermic reaction
might want high temp so high rate of reaction occurs and more efficient product formation
however high temp also disturbs equilibrium → hence favouring reverse endothermic reaction and product conc actually decreases too → lowers equilibrium constant value
electrolysis def
non-spont chem reaction occurs by passing electrical current thru subs in solution or molten state
electrolytic cell def
electric cell where non-spont redox reaction occurs by applying external voltage across electrodes
electrolytic cell diagram
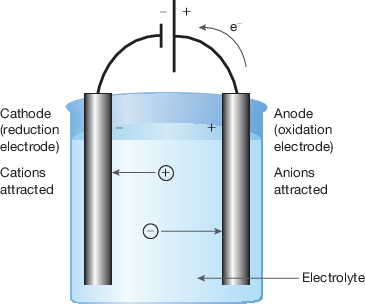
3 features of electrolytic cell
freely moving ions
— can donate/accept electrons and allow them flow thru external circuit → circuit completed and solution (?) electrically conductive
— cations attracted to cathode and anions to anode
2 electrodes where redox reactions at
— positive ions reduce at cathode (-ve electrode)
— negative ions oxidise at anode (+ve electrode)
external electrons source
— electrons flow in one direction to cathode (negative electrode) so reduction can occur — from cations
— electrons given to power source from anode (positive electrode) so oxidation can occur — from anions
electrolysis of ionic compounds: molten NaCl (l)
heating solid NaCl cause ions to separate and freely move as pure liquid subs → molten liquid = called a melt → electric current passed thru → electrons flow to cathode (inert electrode) → becomes negative → sodium ions attracted to cathode and reduce here → positive anode forms → chloride ions attracted to anode and oxidise here
why does applying current with two electrodes in pure water cause no electrolysis?
not enough freely moving ions in pure water to carry electric current so no current flow→ no electrolysis
electrolysis of water
low conc of electrolyte like KNO3 added → freely moving ions available for current flow → cathode is negative with available electrons → water reduced here to form hydrogen gas and hydroxide ions (turns area more alkaline) → anode is positive and no available electrons → so water oxidise here to form oxygen gas and protons (turns are more acidic) → note if allowed to mix freely the protons and hydroxide can form water again (neutralisation reaction)
— reaction: 6 H2O (l) → 2 H2 (g) + O2 (g) + 4H+ (aq) + 4 OH- (aq)
but protons and hydroxide ions form water so final equation:
2 H2O (l) → 2 H2 (g) + O2 (g)
using electrochem series for spontaneous vs non-spontaneous redox reactions
if spontaneous = clockwise circle
if non-spontaneous (forced by energy input) = anticlockwise circle
galvanic cells vs electrolytic cells
g cells = spont redox reaction prod electrical energy, e cells = electrical energy needed to drive non-spont reaction
g cells separation into half cells needed as spont reactions and electron travel needed but e cells no need separation (into diff containers) as non spont
g cell chem energy converted to electrical, e cell electrical energy converted to chem
g cell anode is negative but e cell anode is positive
g cell cathode is positive but e cell cathode is negative
g cell Eo is positive, e cell Eo is negative (g cell produce voltage, e cell consume it?)
min amount voltage needed for electrolysis to happen
more than what is produced for those subs if they were to react spontaneously
Eo cathode - Eo anode
direction of electron flow in electrolytic cell
— anode to cathode
note that it is always this way regardless of cell type
why are states in molten electrolytic cell equations diff to electrochem series states sometimes?
bc need to consider temp here for molten conditions (usually very high temp so some subs may be liquids, gases etc) — higher than the databook series temp of 25 degrees?
standard cell potential of galvanic cell vs electrolytic cell
g cell = positive (makes sense as voltage produced)
e cell = negative (makes sense as voltage consumed)
why is using electrochem series important for electrolysis
sometimes e cell has more than one possible reaction around an electrode as diff substances present → need electrochem series to predict redox reactions that occur among other competing possibilities based on their oxidising and reducing strength → to predict products of electrolysis
predicting the products of electrolysis
list species present (include electrode metals, potential oxidants and reductants, water if there, impurities)
write half-eqs of these species in descending order of Eo
circle species present in e cell that can participate
select oxidant (undergoing reduction) with highest Eo → bc needs less energy for reduction than an oxidant with lower Eo which ends up giving an overall higher voltage
select reductant (undergoing oxidation) with lowest Eo → bc needs less energy for oxidation than a reductant with higher Eo which ends up giving an overall higher voltage
write the reduction and oxidation half-eqs and combine to form overall full eq
find min voltage required by Eo cathode - Eo anode
— the two half eqs should make the smallest possible anticlockwise circle on electrochem series (as needs the least energy amount)
using pH to infer redox reactions occurring
sometimes questions present an indicator and the colour it turns around electrodes to indicate what is produced there from a half eq
hydronium prod → pH decrease — more acidic
hydrogen ions → pH decrease — more acidic
hydroxide ions → pH increase — more alkaline
factors affecting electrolysis of solutions
electrolyte conc
electrolyte nature
electrodes nature
factors affecting electrolysis of solutions — conc of electrolyte
when conc changes and not 1 M → this changes their electrochem series order → bc in databook all species recorded in order at 1 M
when orders are changed this means the stronger oxidants and reductants may now be diff to what shown in databook
factors affecting electrolysis of solutions — nature of electrolyte
when diff electrolytes present (e.g. dilute sodium nitrate solution vs dilute copper nitrate solution) → changes which products form in cell as subs to consider for strongest oxidant and reductant differs
factors affecting electrolysis of solutions — nature of electrodes
electrodes can be inert (graphite or plat usually) or metal that participates in reaction as strong reductant to undergo oxidation (doesn’t occur at cathode as solid metals don’t gain electrons) → changes products of e cell
how half eqs can change as e cell progresses
note sometimes half-equations are just initial reactions → however as reaction progresses the strongest oxidants and reductants may change → for example as solid copper oxidises → after some time copper ions replace previous subs undergoing reduction and it reduces instead at cathode → here note that copper mass is shifting from anode to cathode!
commercial electrolytic cells example — the downs cell
electrolysis of molten sodium chloride
must be molten rather than in aqueous solution form as here water would reduce instead of sodium ions
products of electrolysis (sodium and chlorine gas) continually removed so they do not react and reform sodium chloride — electrodes also separated by iron mesh to keep them apart for this
added chems reduce melting point of sodium chloride
density diffs to collect liquid sodium
current can flow thru molten NaCl bc it exists as separate sodium and chloride ions
fresh NaCl can be added when required
chlorine gas at anode collects in a compartment as valuable by-product
molten sodium form at cathode → less dense than electrolyte → floats to surface → overflows into separate container
operates at high temps to maintain salt in molten state
commercial electrolytic cells example — the hall-héroult cell
electrolysis of alumina dissolved in molten cryolite to prod aluminium
occurs in steel vessel
occurs at very high temps (around 980 degrees celsius — to melt alumina)
anode: carbon blocks above the cell partially immersed in electrolyte → oxide liquid ions form oxygen gas → reacts with carbon anode to form CO2 → hence consumption of anode material means it needs replacement every now and then
cathode: carbon lining of cell → al liquid ions drawn here and reduce to form al liquid
cannot occur with soluble al salt in water as water stronger oxidant that al ions hence preferentially reduced
cryolite: solvent and electrolyte, much lower melting point than alumina hence letting aluminium be deposited
alumina: fed into electrolyte at regular intervals where it dissolves into ions (al ions and oxygen ions — both liquid), current applied moves ions
al liquid and cryolite have density diffs → allow separation as al settle at cell bottom and drained for collection
v specific amount alumina needed
operates at low voltage but needs high current