Single-nucleotide polymorphisms
1/14
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
15 Terms
SNP
A SNP (single nucleotide polymorphism) is defined as a single base change in a DNA sequence that occurs in a significant proportion (>1 %) of a large population.
Variations can be
Harmless (change in phenotype)
Harmful if it leads to a disease (diabetes, cancer, heart disease, Huntington's disease, and hemophilia)
Latent (variations found in coding and regulatory regions, are not harmful on their own, and the change in each gene only becomes apparent under certain conditions e.g. susceptibility to lung cancer)
Types of SNPs
Transitions:
– purine ↔ purine (A-G)
– pyrimidine ↔ pyrimidine (C-T; cytosine→uracil)
Transversions: – purine ↔ pyrimidine. Transitions are more common than transversions
Variation of the number of tandem repetitions and then insertions and deletions at the chromosomal level (more rare), even of a big portion of genome
What can SNPs tell us?
Causes of disease, dysfunctional protein
Correlation with disease prognosis, success of particular treatment
Useful genetic markers, to locate some gene of phenotypic interest; for instance, a gene correlated with a disease
Characterise individuals
Characterise populations (SNP distribution)
Applications in anthropology, tracing of migrations, human evolution
Microbial genotyping, because variations of microorganisms can be mapped Identification of geographical origin, phenotype
A criminal leaves a blood sample at a crime scene. How much can we tell about him or her? Not perfectly, but: Ethnic group, Eye and hair colour (hair colour easier to change), Family name?
Where do SNPs occur in the human genome? Distributed throughout the genome:
50% in non-coding regions
50% in the coding region:
nonsynonymous or missense mutations ( from amino acid substitution)
synonymous or silent ( from amino acid unchanged). Nothing detectable, Change in proportions of variable spliced proteins. Change in stability of mRNA (expression levels of proteins depend also on mRNA half-life), Effect on protein folding (translational pausing)
coding → stop codon. protein truncated: Example: common mutation causing phenylketonuria
stop codon → coding. protein extended: Example: haemoglobin Constant Spring. It has a α-chain variant, termination codon TAA is mutated to CAA (glutamine), produces extension of haemoglobin α-chain from 142 to 172 amino acids, causes mild anaemia.
Technologies used to study SNPs
One-dimensional technologies (electrophoresis) greatly limit the number of multiplexes that can be analysed for each lane and the number of lanes that can be used for each separation.
The massive, parallel two-dimensional approach greatly reduces the volume of reagents and the physical space required for information acquisition. (es. 96 wells or more than 1000 wells)
The zero-dimensional approach allows high throughput.
We can use two different methodologies for advanced SNPs typing:
Hybridation: High background and low specificity due to the difficulties in the standardization of the method: hybridization time and washing. Are used for known polymorphisms
Micro arrays
RT-PCR
Enzymes: High specificity thank to the possibility to performed standard protocols. Are used for known polymorphisms
Nucleotide extension
Cleavage
Ligation
Reaction product detection and display
For unknown polymorphisms we can use
Direct Sequencing
Microarray
Cleavage / Ligation
Electrophoretic mobility assays
Direct investigation for known snps
Hybridation
Allele-specific oligonucleotides (ASO)
ASO dot-blot with fluorescent probe
ASO reverse dot-blot with fluorescent amplicone
Photolithography
Fluorescence Resonance Energy Transfer (FRET).
Enzymatic reaction.
Restriction enzymes: PCR + digestion.
DNA polymerase with SNE (single nucleotide extension) or Allele refractory mutation system (ARMS).
DNA Ligase Oligonucleotide ligation Assay (OLA)
Allele-specific oligonucleotides (ASO),
general method. Target hybridization to a probe array. This approach uses allele-specific probe attached to a solid surface for hybridization of SNP-containing targets that carry a tag. The surface is washed to remove mismatched targets while perfectly matched target-probe pairs are detected by fluorescence for genotype determination.
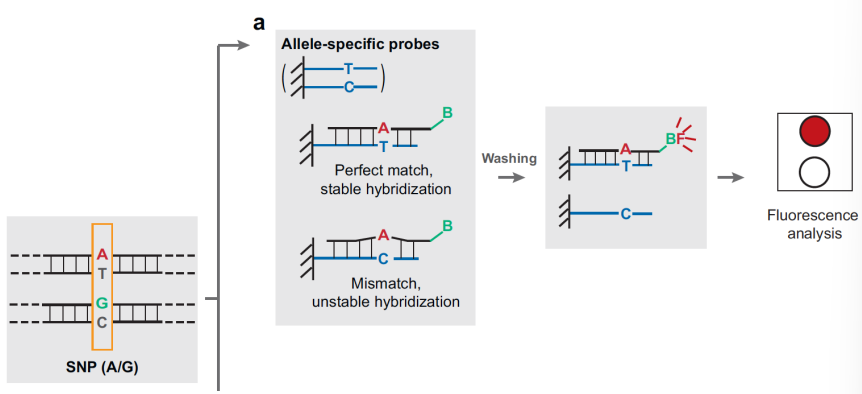
ASO dot-blot with fluorescent probe
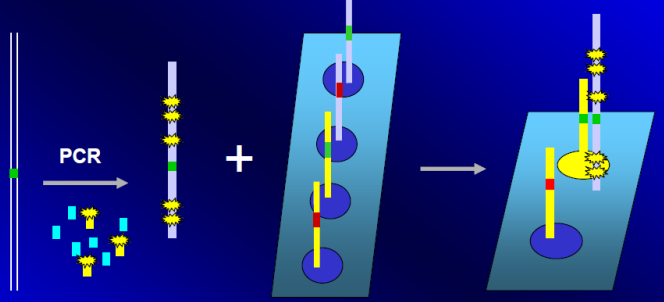
PCR products are blocked on the microarray, denaturated and then hybridized with fluorescent probes complementary to the region of interest. The multi-patient microarray permits to analyse a high number of patient in a single test for a relatively low amount of loci.

ASO reverse dot-blot with fluorescent amplicone.
Complementary probes are blocked on the microarray and hybridized with the amplicons from different loci of a single patient, marked with fluorescence. With the single patient microarray is possible to analyse in a single est many variant regions of the same sample.
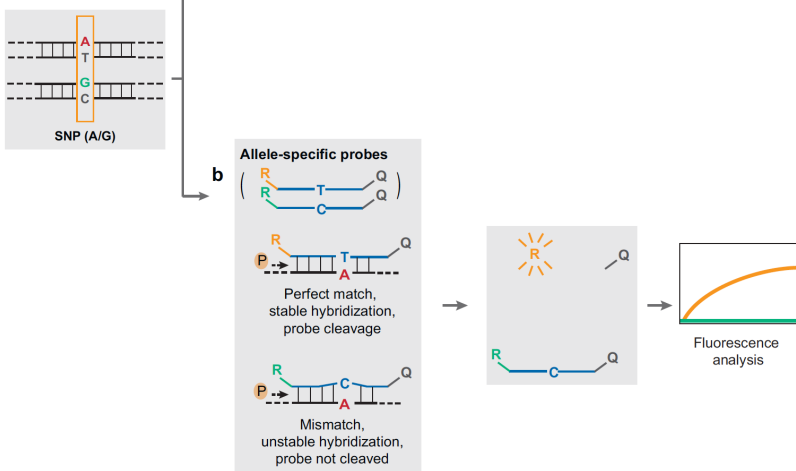
Fluorescence Resonance Energy Transfer (FRET).
The probes need to be specifically designed and is a more specific process. We can use molecular beacons with the same principle
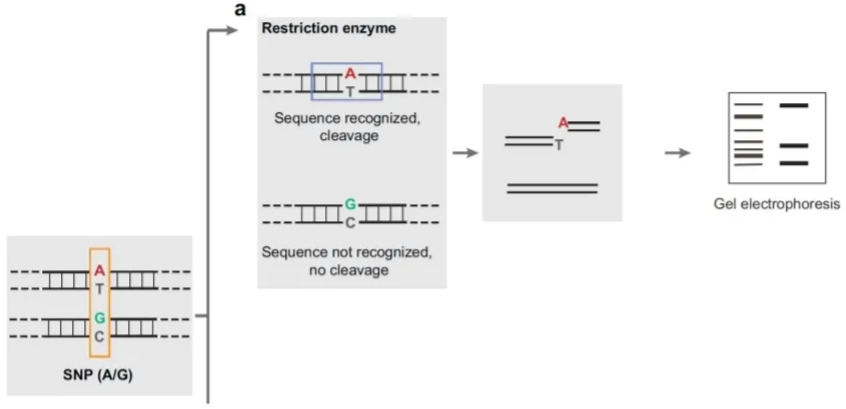
Restriction enzymes: PCR + digestion
This approach uses a restriction enzyme that cleaves only one of the alleles. Products from the restriction digest are un on a gel and the SNP genotype is inferred based on the number of fragments of different size. If we have both alleles with the same gene, both fragments will be cut. If both alleles are the same and are mutated they won’t be cut (one band), if we have 3 bands they are not mutated or the gene is non homologous for the alleles, so the enzyme can/can’t cut, both things can happen.
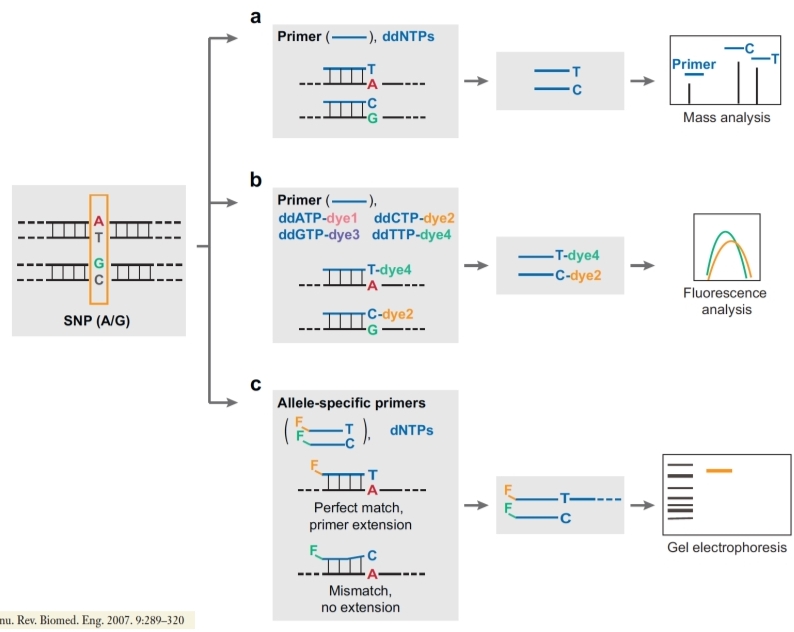
DNA polymerase with SNE (single nucleotide extension) or Allele refractory mutation system (ARMS).
The last base of the primer is complementary to the base of interest in the SNPs; the second primer is complementary to the mutated allele of snp. We perform a PCR and then we have to do it again using the specific primers used here: if they have a perfect match the DNA polymerase will start the elongation, if they don’t match perfectly no amplification is done, so we can visualize the results with gel electrophoresis (the reaction happened if we see the bands) or we can analyse our amplification by mass spectrometry (unusual because it’s very expensive). It’s better to do a fluorescence analysis or we have to link a dye to the probe, two different dyes if we want to see both probes.
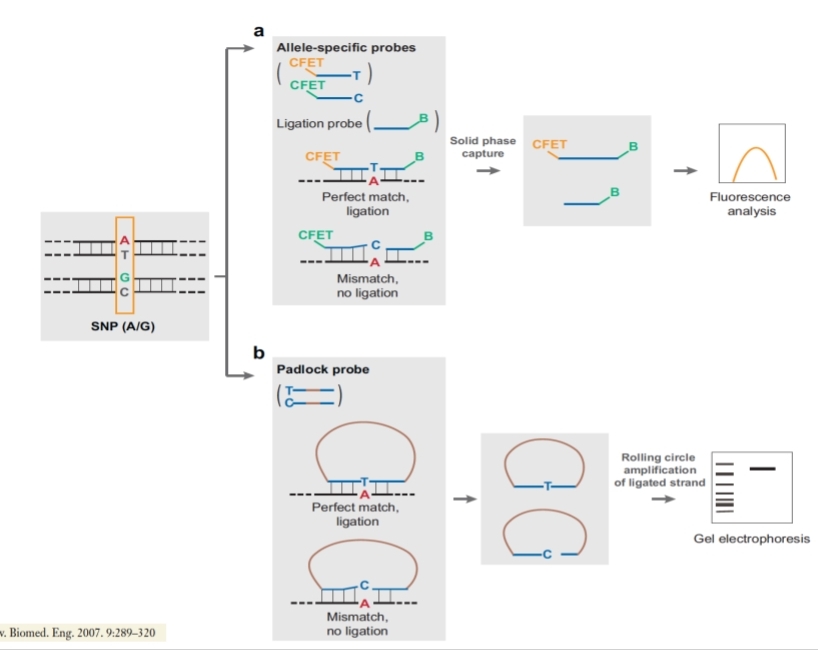
DNA Ligase Oligonucleotide ligation Assay (OLA).
We have to design 3 different probes. One probe is complementary to the wild type allele, the 2nd is complementary to the wild type allele but with a mutation (to cause mismatch), the 3rd is designed to link after the mutation and is universal. We can have the usual situation so probe 1 and 3 create a perfect match and bind. If we use 2nd and 3rd, the 2nd doesn't bind well, so we use DNA ligase to bind the probes only if they perfectly match. We can visualize fluorescence if we bind a fluorophore or we can do electrophoresis so the result of the link is going to be just one fragment given by the sum of their bp.