Functional Genomics- Ch
1/82
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
83 Terms
Homozygote
An individual with two identical alleles for a particular gene, resulting in uniform expression of that trait. (BB)
Heterozygote
An individual with two different alleles for a particular gene, leading to mixed expression of that trait. (Bb)
Segregation
The separation of homologous chromosomes (aka when paternal and maternal chromosomes pair together) after recombination during meiosis. Or, seperation of corresponding alleles during the reproductive process.
Recombination
Chromatids from each homologous chromosome exchanges segments of alleles, which results in different gene combinations. Results in genetic diversity.
Chromosomes pair up with their homologous partners (same genes), and recombination shuffles the alleles between them.
Alelles
An allele is a different version (or variant) of the same gene, and each version can lead to a different trait or form of a trait.
Ex: B and b are on/from the same gene (like coding for eye color), but different alleles (B for brown eyes and b for blue eyes).
Linkage
Refers to the physical proximity of genes on the same chromosome.
Genes that are close together tend to be inherited together because there’s less chance of recombination separating them.
Ex: If Gene A and Gene B are close on chromosome 1, a crossover is less likely to occur between them. So they “travel together” during meiosis = linked.
Linkage Disequilibrium
Refers to non-random association of alleles at two or more loci in a population.
Aka …some combinations of alleles appear together more often (or less often) than expected by chance even though they are far apart.
Ex: Locus 1 has alleles A and a. Locus 2 has alleles B and b.
Even if these two loci are far apart on the chromosome, they might be in LD if you find that:
AB combination appears more frequently than Ab or aB, even though they are physically far apart.
Genetic Map
A diagram that shows the arrangement of genes and their relative distances on a chromosome, based on the frequency of recombination events. b
Higher % = alleles are further apart, higher likelihood of crossing over
Lower % = alleles are closer together, lower likelihood of crossing over
centiMorgan (cM): 1cM = recombination frequency of 0.01
“closely linked” = they are more likely to be inherited together (aka linkage disequilibrium) because it is more than expected
Physical Maps (Mapping Genomes)
An assembly of long, continuous pieces of DNA (contigs), and on these contigs, scientists mark known DNA sequences (landmarks) and measure the physical distance between them in kilobases.
Provide information about the arrangement and location of genes on chromosomes to help study genomes.
Methods of Assembly Contig: Hybridization Techniques
chromosome walking
sequence-tagged sites (STS): single occurance in the genome
Chromosome Walking
It’s a method to find and connect overlapping DNA fragments to gradually move along a chromosome, starting from a known DNA sequence. Start from a known DNA sequence (probe), connect it to another fragment that contains the same DNA sequence (overlapping sequence), sequence the unknown parts, create another DNA probe at the end of the sequence, repeat.
Sequence tagged sites
An STS is a short, unique DNA sequence that occurs only once in the genome. Because it's unique, if a clone contains an STS, scientists know exactly where that clone came from in the genome. STSs mark precise locations on the DNA. STSs help scientists match up and organize DNA fragments by anchoring them to a known position in the genome.
Cytological Map
A cytological map is a chromosome map created by analyzing the physical structure of chromosomes using microscopy techniques. Provide a way to visualize and locate specific regions on chromosomes based on banding patterns that appear when chromosomes are stained.
Synteny
The conservation of blocks of genes or genetic sequences on chromosomes between different species. In simpler terms, it means that genes are found in the same relative positions on the chromosomes of different organisms.
Homologs
Genes with very similar sequences, evolved from a common ancestral DNA sequences
1) Orthologs
2) Paralogs
Orthologs
homologs in different species (evolution)
Genes in different species that evolved from a common ancestral gene by speciation.
Usually retain the same or similar function.
Paralogs
homologs within the same species (gene duplication)
Genes that are related by duplication within the same genome.
Often evolve new functions, even if they’re related.
Sanger Sequencing
Denature DNA- Split strands
Bind primer (small section of DNA)
DNA polymerase lengthens strand according to the template strand
DNA Polymerase randomly places dideoxy nucleotide, which terminate chain
Chain terminated
Process repeated above until sample contains fragments of all different lengths (all terminated by differently clored flourescently labeled dideoxynucleotides)
Fragments separated by size, and the color of dideoxy nucleotdes can tell us the template stand nucleotide sequence
Dideoxy Nucleotide
Missing an OH group, which is what allows the next nucleotide group to bind, randomly placed by DNA Polymerase, dyed according to the base last placed.
Cyclic Sequencing
Cyclic sequencing refers to a DNA sequencing method where the process happens in repeated cycles, each cycle revealing one base (A, T, C, or G) of the DNA sequence at a time. (concept behind many NGS)
Capillary electrophoresis (done after sanger sequencing)
loaded into capillary tube filled with gel
electric current is applied
fragments migrate through the gel/ capillary tube
smaller fragments move faster through the gel and exit the capillary tube faster
Laser and detector at the exit of the tube
Laser: excitrd the dye
Detector: reads the color of the dye
Base Calling
Software records the order of the colors and translates it into a DNA sequence
Each peak is a detected base
Chromatogram: this is what the color peaks are called
Phred Scores
(q) Number assigned to each base in a DNA sequence to represent how confident the software is that the correct abse was identified
Phred Score formula
Phred Score (Q) = -10 log10(P)
p= probability that the base call is incorrect
q= quality score
Interpretation of Phred Score
Q20 score or higher is considered high quality base calling
Good: sharp clear peaks (Q20 and higher)
Bad: messy/overlapping peaks (lower than Q20)
Bad: no phred score could be calculated, sequencer could not determine which base was present (N designated for base)

Heirarchal Sequencing
Break the entire genome (~3 billion bp) into large chunks (150bp) using BACs
Build a map: align and organize these large clones (chunks) into a scaffold using…
chromosome walking
finger printing
Subdivide each BAC clone into smaller pieces (3kb)
Sequence smaller fragments (Sanger Sequencing)
Assemble fragments into contigs, then super-contigs using overlapping regions
Fill gaps using cDNA or mate pair reads
BAC
Bacterial Artificial Chromosome, used to clone large DNA fragments.
Tiny, circular piece of DNA (plasmid)
Use to clone large pieces of DNA
Cut up BACs to get smaller fragments for sequencing.
Fingerprinting
enzymes slice DNA at specific recognition sites to create fragments of known specific lengths
Measure fragment size
Size of the fragment is the “fingerprint”
Fragment size/pattern tells you which DNA seqeunces go together by comparing their sizes and patterns
You want to line up the BACs before you sequence them
cDNA
complementary DNA
Can onyl help fill gaps inside genes
Can help locate genes → since you already know what that gene sequence looks like
Mate Pairs
Come from ends of large DNA fragments — before it is cut up into smaller pieces
So you know which contigs/ sequence on the contigs are on both ends of the long DNA fragment (as well as how many bp are between them) and if they are near each other
Whole Genome Shotgun Sequencing
Break entire genome into large DNA chunks
Skip initial scaffolding step! Go straight to…
Cut up large DNA chunks into smaller chunks
Sequence smaller chunks
Assemble all reads into contigs, unitigs, and eventually scaffolds
Use mate pairs to help assemble unitigs into contigs/scaffolding
Problems with whole genome sequencing
Repetitve sequences because there is no intiial scaffold/map to go off of
You can’t tell which unitigs go on which end of repeats (ATATAT…)
Mate pairs will tell you which unitig will go on each end of the repeat sequence, as well as how many bp are supposed to be between each unitig
Unitig
made up of multiple smaller fragments
perfectly ovrelapping, no ambiguity (no gaps, etc.)
100% confident
Contigs
may include multiple contigs, contains gaps etc. made up of multiple small DNA chunks
chrUn
unlocalized contigs → These are sequences from a genome that haven't been assigned a specific location on a chromosome yet.
Because of repeat sequences and lack of information
Chr1 xxxx random
Unplaced contig names consist of the chromosome number, followed by the NCBI accession number, followed by "random“
Chr# = chromosome #
NCBI number = unique identifier for the contig in the GenBank database
Random = indicates the contig is not placed at a specific location
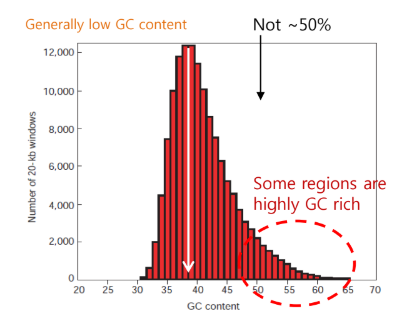
GC contents in human genome
generally low GC content, but some regions are highly GC rich (CpG island)
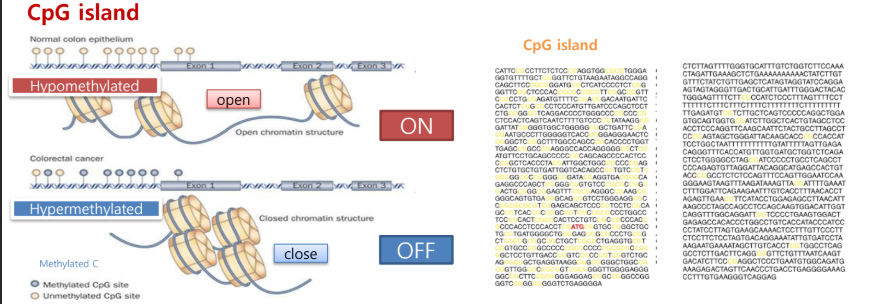
CpG Islands
CpG islands are regions with a high frequency of CG dinucleotides, often near gene promoters.
Their methylation status influences gene expression:
Unmethylated CpGs (hypomethylated) → chromatin open → genes ON
Methylated CpGs (hypermethylated) → chromatin closed → genes OFF
Segmental Duplication
Segmental duplications are long stretches of DNA that are nearly identical copies of each other — with greater than or equal to 90–95% sequence identity.
Intrachromosomal
Interchromosomal
Intrachromosomal Duplication
Both copies are on the same chromosome.
Tend to be less similar (less % identity).
Can be longer.
Interchromosomal Duplication
Tend to be more similar (higher % identity).
Usually shorter
One copy is on one chromosome, the other on a different chromosome
Fragment
a small piecce of genomic DNA - typically several hundred bp in length - subject to an individual partial sequence determination, or read
Single-end read
technique in which sequence is reported from only one end of a fragment
Pair-end read
technique in which sequence is reported from both ends of a fragment (with a number of undetermined bases between the reads that is known only approximately)
Read length
the number of bases repoorted from a single experiment ona single fragment
Assembly
the inderence of the complete sequence of a region from the data on individual fragments from the region, by piecing together overlaps
De novo sequencing
determination of a full-genome sequence without using a known reference sequence from an individual of the species to avoid the assembly step
Resequencing
determination of the sequence of an individual of a species for which a reference genome sequence is known. The assemble process is replaced by mapping the fragments onto the reference genome.
DNA sequencing by NGS
start with extracted DNA
DNA is broken into small pieces using: sonication, dnase (enzymes)
Short artifiical DNA sequences (adapters) are added to bothh ends of each DNA fragment.
These help the fragments bind to the sequencing platform and get read
PCR Amplification: fragments are amplified (copied) to increase the amount of DNA
Sequencing library: a collection of amplfiied DNA fragmentes with adaptors, ready to be sequenced.
NGS sequencing platform → put in libaray into NGS machine
Amplification methods
emulsion pcr
Bridge Amplification
NO AMPLIFICATION (single molecule)
Emulsion PCR
start with DNA that has been ligated
Attqach DNA to beads - each fragment gets attached to a small bead of water
Form an emulsion
Bead +PCR reagent are mixed with oil to create an emulsion
each droplet acts like tiny test tube
PCR amplification inside droplets
PCR performed
within each bead, DNA is copied many times and coats bead
Bridge Amplification
Prepare DNA with adapter
fragments have special adaptor sequences ligated to both ends
complementary to oligos stuck on the surface of the illumina flow cell
Bind DNA to flow cell
each DNA fragment sticks to the surface via base pairing with attached oligos which is now anchored at one end
Bridge Formation
Free end bends over like “bridge” and binds to second oligo on surface
DNA Polymerase codes second strand
Denature both strands → now two complementary strands
Repeat
DNA Polymerase Reaction
DNA polymerase reads the template strand
Adds complemetary strand
When it adds a base to the new strand…
the base is added to the strand
A byproduct (phosphate/ H+) released → that release is what gets detected in sequencing
Pyrosequencing, reversble termination, chain termination
SBS (Sequencing by synthesis) method
pyrosequencing, reversible termination
Pyrosequencing
when nucleotide is being added by DNA polymerase, pyrophosphate (PPi) is released
Enzymes convert PPi into ATP, which produces light
How much light → how many bases were placed
One nucleotide added at a time → light tels you if it was added or not
Detection through light (monocolor)
Reversible Termination
same as chain termination EXCEPT:
chain termiantion reversible through chemically removing dideoxynucleotide, allows for continuation of synthesis
after every base, machine snaps picture of fluorescent label
Detection through fluorescence (4 colors)
Alternate Sequencing methods
ligation, translocation
Translocation (channel)
DNA strand is threaded thorugh nanopore (tiny biological or synthetic pore embedded in a membrane
As each base passes through the pore, it disrupts an electric current in a base specific way
Sequencer measures the current changes to identify bases in real time
Electric detection
Ligation
uses fluorescently labeled probes (oligonucletodies)
Each broke contains known bases and a dye tag
Binds to matching sequence, and DNA ligase attaches it
Fluorescence detected → sequence decoded based on color pattern
Multiple rounds of probing give full sequence
Illumina
Bridge Amplification → Reversible Termination → Fluorescence (image/color)
Pacific Bioscience
Single molecule (no amplification) → pyrosequencing → Fluorescence
Oxford Nanopore
single molecule (no amplification) → nanopore → H+ (pH)
H+ (pH) detection
nucleotide added (ATCG)
H+ is released, changes pH of solution
pH change detected
Tells you how many of each nucleotide is added
Sequence Alignment
The process of arranging DNA or protein sequences to identify regions of similarity that may indicate functional, structural, or evolutionary relationships.
Think of it like comparing two strings of letters to see how closely they match
Common Ancestor: ACGCTGA ←→ ACTGT
Sequence Alignment 1
ACGCTGA
A - - CTGT
Sequence Alignment 2
ACGCTGA
ACTGT - -
You want to maximize the amount of matches you have with minimal gaps or mismatches.
Identity
nucleotide sequences are identical, likely to have similar functions or origins
Substitution
One base swapped for another.
Insertion
extra base added
deletion
base removed
Pairwise Sequence Alignment
Aligns two sequences and compares them to find matches/mismatches.
Match = same letter in both sequences.
Mismatch = different letters.
Optimal Alignment Involves
Scoring: How similar are the sequences? Use:
Distance (like Hamming or Edit distance).
Score (based on matches, mismatches, gaps).
Dynamic Programming: Algorithm that finds the best alignment.
Global Alignment
Aligns entire sequences end to end (useful if sequences are similar).
High sequence similarity, homolog, same functiom
Local Alignment
Finds regions of highest similarity (useful if sequences only share some regions).
Conserved region of sequence > functional domain / element
Hamming distance/ edit distance
Hamming distance: use when there are no caps and count the number of mismatches (or if there are gaps needed, still line them up with no gaps)
Edit distance: how many editing operations there are to transform x to y (how many changes you need to use in order for the sequences to match)

How to measure sequence similarity: score
Score = (match or mismatch penalty) - gap penalty
Score values:
Match =3
Mismatch = -1
Gap = -2
Local Alignment
finds the best matching region(s) between two sequences (better if sequences vary in size or contain different domains).
Global Alignment
aligns sequences from start to finish (useful when they’re similar in length)
Haplotypes
group of genes (DNA regions) in the chromosome that are inherited (segregated) together from a single parent during recombination
A collection of specific alleles (SNPs) in a cluster of tightly linked genes on a chromosome — likely to be continually passed down unchanged (low rate of mutation)
SNP
An SNP is a single base-pair change in the DNA sequence at a specific position in the genome.
SNPs differ between people (one person might have A at that specific location, while another person might have G at that specific location)
However, SNPs in biological families are passed down unchanged
Polymorphism
Tagged SNPs
Specific representative SNPs within a haplotype block (that include other SNPs) that act like “markers” or shortcuts to identify the whole region.
help in tracking inheritance of traits and diseases.
Three major types of mutagen
gamma rays- strong mutations, disrupt multiple genes
Chemical- full spectrum of mutations, random distribution, mutation detection difficult
Insertion- nonrandom distribution
Structural variations
Large-scale alterations in the genome, including deletions, duplications, inversions, and translocations of DNA segments. They can affect gene function and contribute to genetic diversity and disease.