6: medicinal chemistry
1/145
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
146 Terms
describe the most common method in the drug discovery process
identify a ‘small’ molecule to interact with a biological target
small = relative to target molecule = large discrete collection of atoms
what are the 2 main biological targets for drug action
proteins:
enzymes
cell-surface receptors
describe proteins
polymers of amino acids joined by peptide/amide bonds = polypeptides
3D structure = governs biological function
describe enzymes
= proteins that catalyse chemical reactions
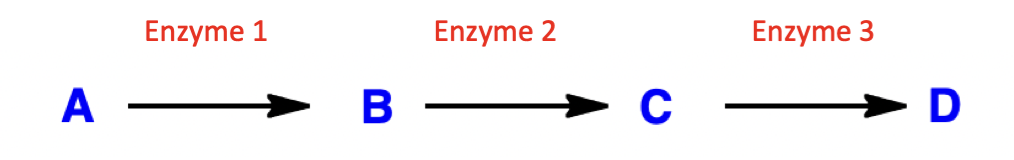
what effect does inhibition of enzyme 2 have on enzymatic pathway?
inhibition of enzyme 2:
build up of compound B
decrease in compounds C and D (downstream)
applications:
treat disease caused by build-up of compound C/D
treat disease caused by deficiency of compound B
kill cell that depend on compound C/D
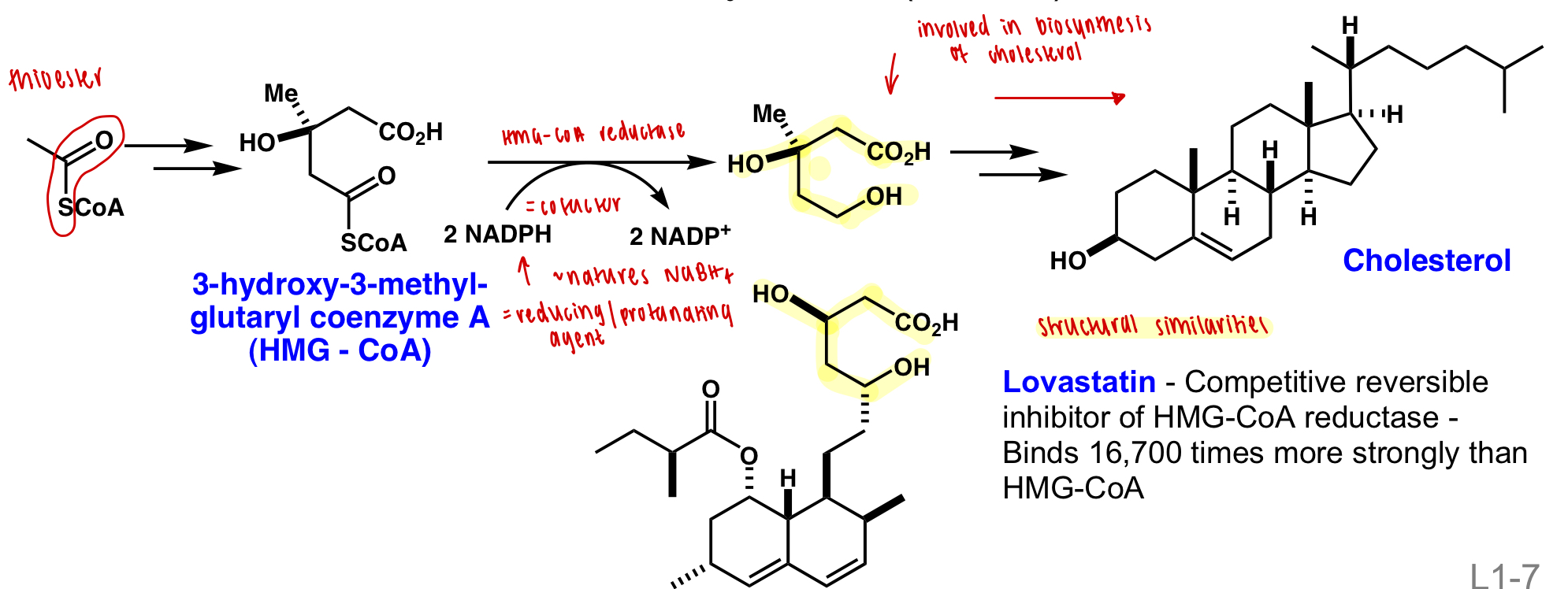
describe a treatment of atherosclerosis
= build up of cholesterol from diet or biosynthesis in the liver
control diet
inhibit biosynthesis:
lovastatin = competitive reversible inhibitor of HMG-CoA-reductase = upstream enzyme of cholesterol biosynthesis
describe cell surface receptors
= proteins that transmit signals in response to ligands
describe the types of drugs that can interact with cell surface receptors
agonists
= mimic the function of natural substrate and activate pathway
antagonists
= bocks the binding site for the natural substrate and deactivates pathways
agonists are more difficult to generate than antagonists
describe methods to find lead compounds (=small molecules)
modification of known structures
systematic screening
rational design
describe modification of known structures
natural products
existing pharmaceuticals:
pros:
well known properties
good profits
cons:
legal and ethical concerns
describe systematic screening
testing “libraries” of molecules against ≥1 biological targets
high-throughput screening (HTS) techniques = >10,000 tests/day
cons:
obtaining structural diversity = libraries have varying side chains but often same functional groups
ensuring ‘drug-like’ properties = screening compounds which could be incorporated into a drug
describe the combinatorial approach to systematic screening
systematically generating compound libraries by combining sets of building blocks (i.e. functional groups) in all possible combinations
describe Lipinski's rules (rule of 5/Pfizer rules)
describe oral bioavailability (not activity)
H bond donors ≤ 5
H bond acceptors ≤ 10
logP: -5 → 5
MW ≤ 500
define H bond donors vs acceptors
donors = H bonded to electronegative atom (O/N)
acceptors = electronegative atom with lone pair/s (O/N)
an atom can be both H bond acceptor/donor = always singly accepting/donating
describe rational design
design of new substrate for biological targets (often enzyme)
pros:
synthesis of fewer compounds
increased potency and specificity
cons:
requires knowledge of 3D structure of target site
describe IC(50) value
= concentration of a compound that inhibits the activity of its target by 50%
= want a minimal IC(50)
measure of potency (not action)
describe pharmacodynamics
= study of interactions of a compound with its target
measure of action
what model is used to model enzyme kinetics?
Michaelis-Menten kinetics
describe Michaelis-Menten kinetics
equilibrium exists between enzyme + substrate and intermediate compex

describe the steady state approximation
the concentration of ES is fixed when reaction is ongoing
rate of formation of ES = rate of breakdown of ES
what is the rate equation in Michealis-Menten kinetics?
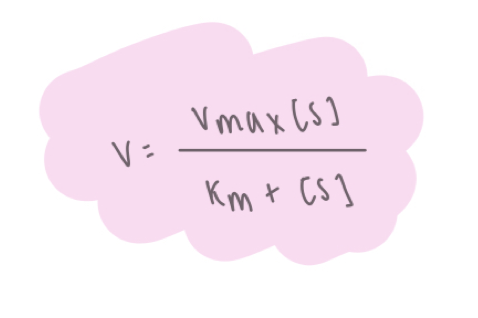
what is Michaelis-Menten plot
y = V
x = [S]
how can Km be found from Michealis-Menten plot?
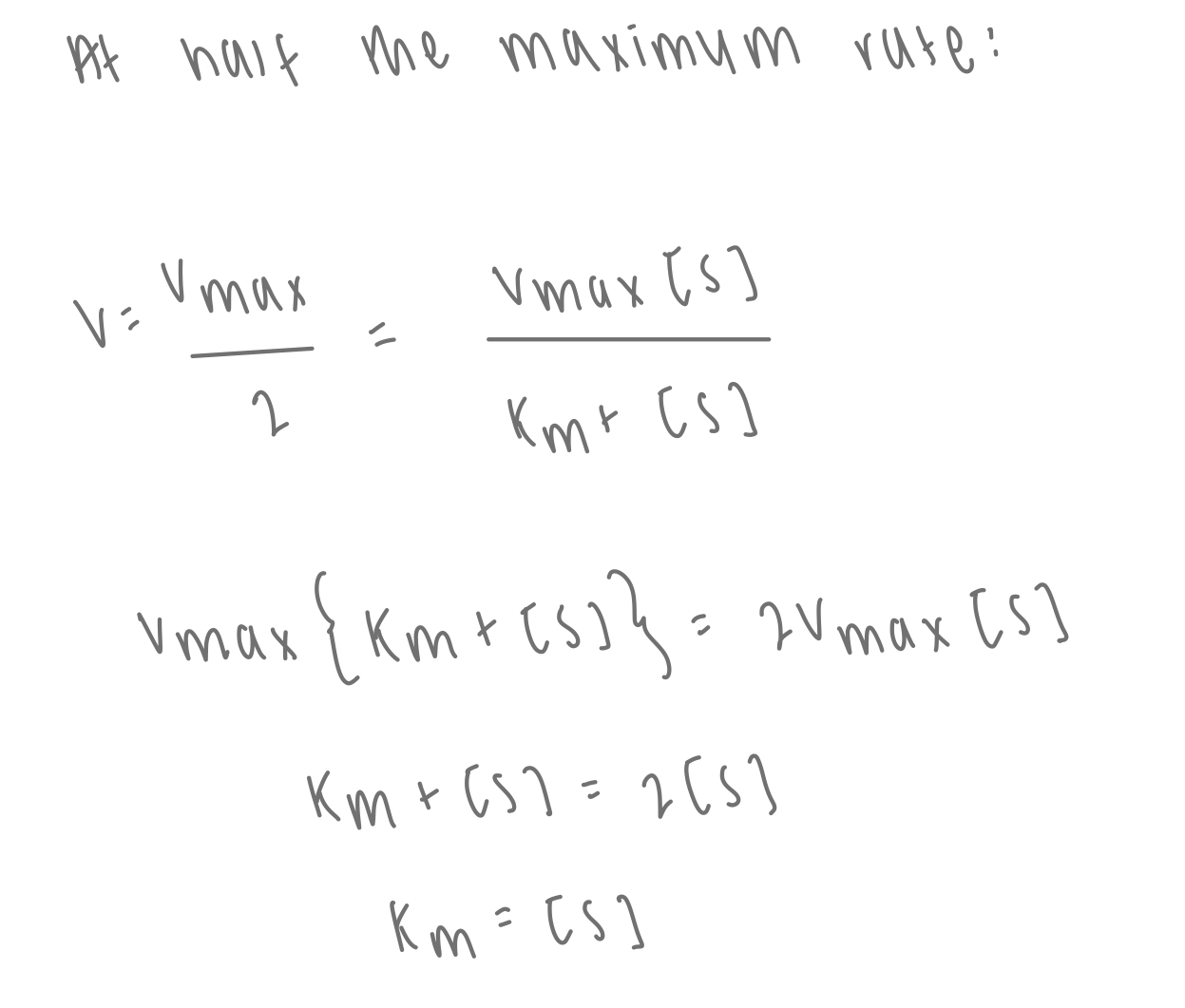
describe the Lineweaver-Burk plot
rearranging the reciprocal Michaelis-Menten equation
y = 1/V
x = 1/[S]
![<p>rearranging the reciprocal Michaelis-Menten equation </p><p>y = 1/V</p><p>x = 1/[S]</p>](https://knowt-user-attachments.s3.amazonaws.com/2f2f0b5c-1e6b-4b25-9be8-937bf7f4839d.png)
what are the 2 types of enzyme inhibition
reversible inhibition
= non-covalently bind target
irreversible inhibition
= covalently bind target
describe types of reversible inhibition
competitive
= bind to the same active site as the natural substrate
non-competitive
= bind to a different (allosteric) site to the natural substrate
describe the effect of increasing [S] on competitive and non-competitive inhibitors
competitive = reverses inhibition
non-competitive = no effect
describe the effect of competitive inhibitors on MIchaelis-Menten kinetics
equilibrium between E + I and enzyme-inhibitor complex
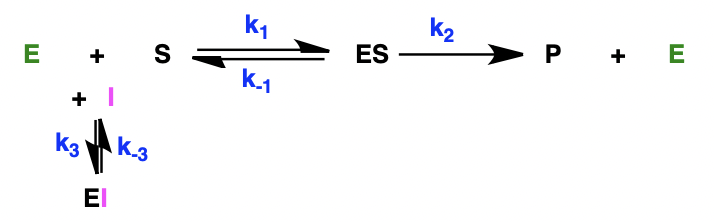
describe the effect of competitive inhibitors on the Lineweaver-Burk plot
V’max = Vmax: y intercept is the same
Km is changed: x intercept is different
(V’max = max rate in presence of inhibitor)
competitive inhibition can be overwhelmed by increasing [S] = Vmax unaffected
![<ul><li><p>V’max = Vmax: y intercept is the same</p></li><li><p>Km is changed: x intercept is different</p></li></ul><p>(V’max = max rate in presence of inhibitor)</p><p>competitive inhibition can be overwhelmed by increasing [S] = Vmax unaffected</p>](https://knowt-user-attachments.s3.amazonaws.com/6084c5b4-6f18-43fd-a6bb-fdc6b633ae0c.png)
describe the effect of non-competitive inhibitors on MIchaelis-Menten kinetics
formation of equilibrium between enzyme-inhibitor complex and enzyme-inhibitor-substrate complex
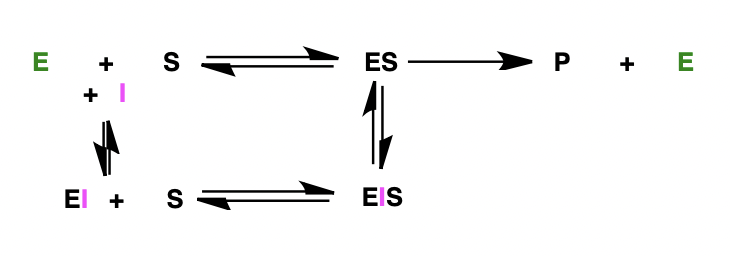
describe the effect of non-competitive inhibitors on the Lineweaver-Burk plot
V’max is changed: y intercept is different
Km = Km: x intercept is the same
(V’max = max rate in presence of inhibitor)
non-competitive inhibition changes the shape of the active site = no effect in increasing [S] = Vmax affected
![<ul><li><p>V’max is changed: y intercept is different</p></li><li><p>Km = Km: x intercept is the same</p></li></ul><p>(V’max = max rate in presence of inhibitor)</p><p>non-competitive inhibition changes the shape of the active site = no effect in increasing [S] = Vmax affected </p>](https://knowt-user-attachments.s3.amazonaws.com/e2958c45-393f-44c0-b2e6-1c9d7cca4ea7.png)
describe irreversible inhibition
‘suicide inhibitors’
= forms covalent bond
= permanently deactivates target
= often electrophilic reacting with nucleophilic residues in target
= often highly toxic = requires selectivity
why can IC(50) not be used to describe irreversible inhibition
equilibrium of non-covalent interaction often precedes covalent deactivation
irreversible inhibition is time dependent since depends on the rate of covalent deactivation = different potencies are observed over time
define a protease
an enzyme that cleaves peptide bonds and degrades proteins
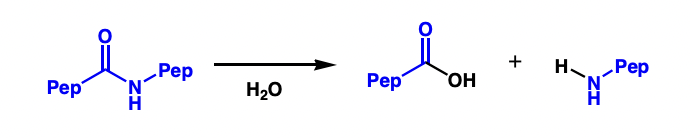
describe X proteases
X = residues in protease active site
i.e. serine protease
= serine residue in active site
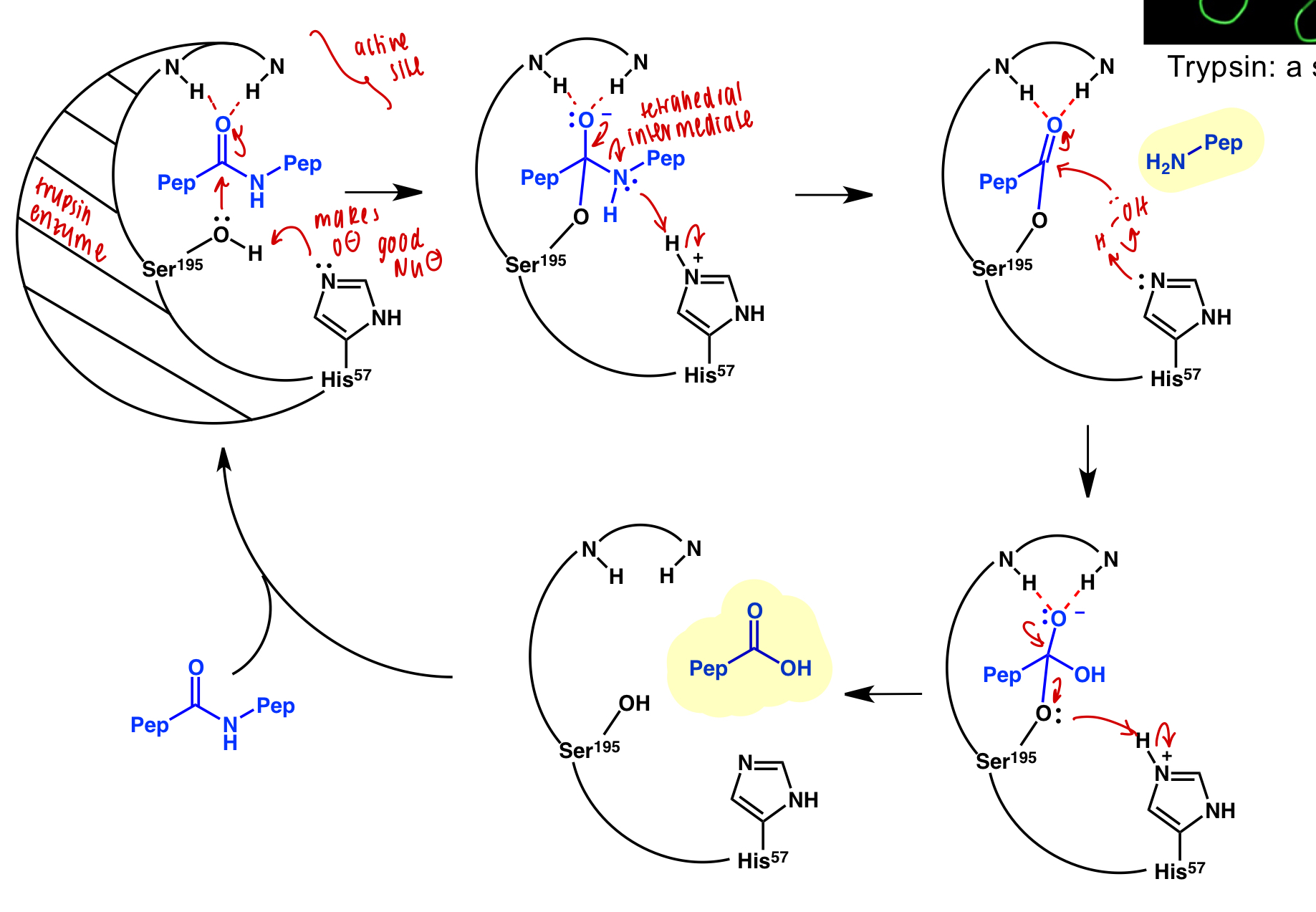
describe inhibition of serine protease trypsin
trypsin active site binds amine residues and cleaves bond
inhibitor = amide residue that will not dissociate from active site
affinity-labelled inhibitor = amide residue with additional function groups participating in secondary interactions that will not dissociate from active site
describe pharmacodynamics vs pharmacokinetics
pharmacodynamics = study of interactions of a compound with its target
pharmacokinetics = study of fate of compound in vivo
what is used to study pharmacokinetics?
‘ADME’/’LADMET’ profile
liberation
absorption
distribution
metabolism
excretion
toxicity
describe liberation
release of drug from the form in which they are delivered
= dependent on route of administration
define exipients
= inactive compounds of a drug formulation used to:
improve absorption
improve stability
serve as carriers
define polymorphs
different crystalline forms of the same substance which may have different physiological properties
describe absorption and distribution
orally administered = absorption into bloodstream occurs in the small intestine
travels in bloodstream to target tissue
must cross two cellular membranes:
outer = hydrophilic
inner = hydrophobic
compound must have a balance of hydrophobic and hydrophilic properties
what is used to access hydrophobic and hydrophilic properties
partition constant, P
describe the partition constant, P
compound is dissolved in equal mixture of octanol and water
octanol = non-polar
water = polar
high P = non-polar
low P = polar

describe logP
high logP = very non-polar = > +6
low logP = very polar = < -6
describe metabolism and excretion
metabolism: broken down (usually to more polar components)
begins in digestive tract; majorly in the liver
first step typically protonation
metabolism too fast = not effective
metabolism too slow = remain in the system longer than desired
generally two stages:
Phase I = biotransformation
Phase II = conjugate formation
excretion: components excreted by kidneys/large intestine
metabolism should be minimised
describe phase I biotransformation
many different processes:
oxidation: most common - installation of polar functional groups
hydrolysis
reduction
describe cytochrome P450
= family of enzymes in the liver
= catalyse oxidation of many functional groups in phase I biotransformation
describe phase II conjugate formation
attachment of ionised group to increase water solubility and ease of excretion
i.e.
addition of sulfate group (SO3-)
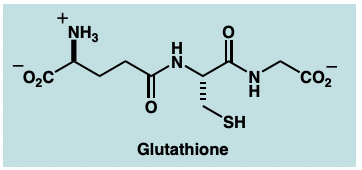
addition of glutathione conjugates:
SNAR or nucleophilic substitution
how can metabolism by prevented/pharmocokinetics improved?
bioisosteres
prodrugs
describe bioisosteres
structurally distinct compounds that are recognised similarly by biological systems
= less susceptible to metabolism i.e. oxidation, hydrolysis, reduction, (phase I) conjugate formation (phase II)
= chosen depending on which property is key for function
describe carboxylic acid bioisosteres
physiological pH = deprotonated = too polar
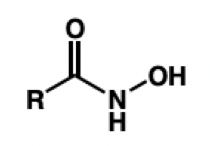
hydroxamic acid = metal chelation
tetrazole = lipophilicity
acylsulfonamide = more H-bond acceptors
draw hydroxamic acid
draw tetrazole
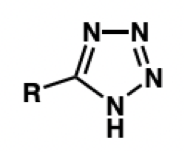
still deprotonated and forms anion but resonance stabilised = lipophilicity
draw acylsulfonamide
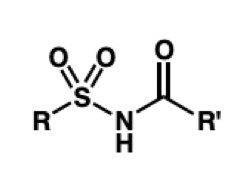
what is a bioisostere for an ester
an amide
what is a bioisostere for a hydroxyl group
an amide
what is a bioisostere for a ketone
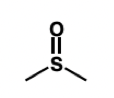
thiocarbonyl group
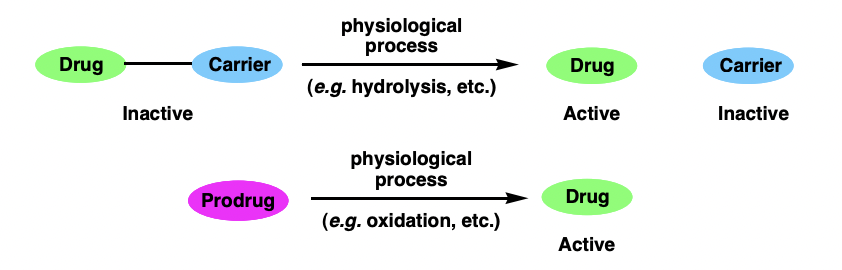
describe prodrugs
therapeutic agents that are converted to their active form in vivo
= converted by early metabolism (phase I processes)
= increased bioavailability
define LD(50)
= measure of toxicity
= drug dosing that is lethal in 50% of a population
= want maximum
define ED(50)
= measure of efficacy
= drug dosing that produces maximum therapeutic response in 50% of a population
= want minimum
define therapeutic index
ratio of toxicity to efficacy
= want a large TI

describe the course of clinical trials
phase I: healthy volunteers ~50
= testing safety of drug candidate and establish maximum dosage
phase II: ill patients ~200
= testing efficacy in ill patients
= must include control group who are treated with best available treament
phase III: larger group ~2000
= determine ideal dosage in broad patient population
describe hERG activity
hERG = K+ ion channel = regulating hearts electrical activity
many drugs show ‘hERG activity’ = hERG inhibition
hERG activity must be minimised = monitored in clinical trials
what is an indole
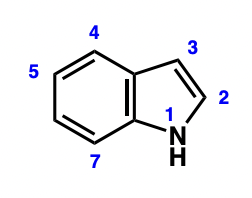
what is a key indole containing molecule?
amino acid tryptophan
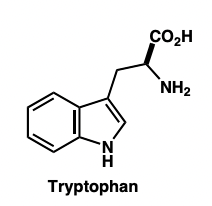
describe tryptophan derivatives
functional group conversion of tryptophan:
tryptamine
serotonin (5-HT)
melatonin
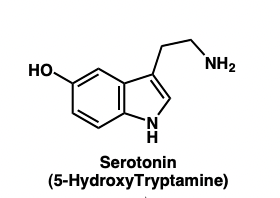
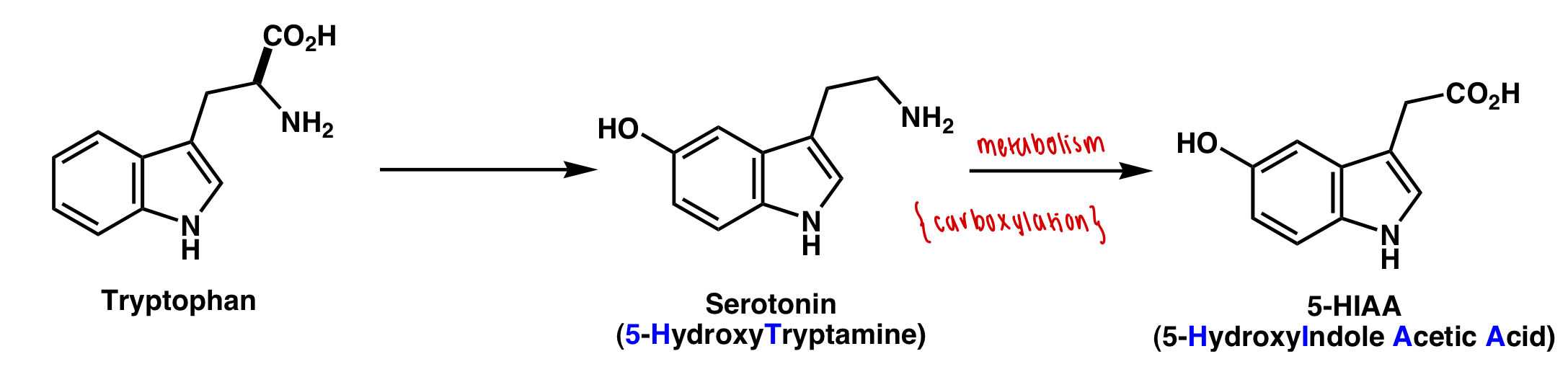
describe the tryptophan to serotonin pathway
serotonin = 5-HT
sumatriptan = 5-HIAA
describe the function of 5-HT
= vasoconstriction
describe the role of tryptophan derivatives in migraine attacks
5-HT levels decrease = vasodilation
5-HIAA levels increase
describe how migraine attacks can be treated
intravenous 5-HT (increase)
drugs activating 5-HT cell surface receptors (vasoconstriction)
what are problems associated with direct use of 5-HT
poor bioavailability
short plasma half life - rapidly metabolised
non-selective action: vasoconstriction elsewhere in body
overall describe Fischer indole synthesis
coupling of an aryl hydrazine with a ketone/aldehyde
hydrazone formation
indole formation
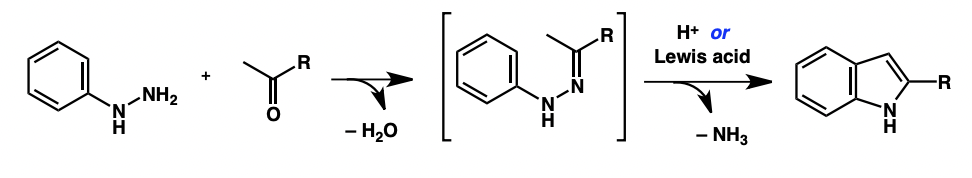
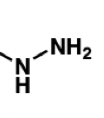
draw a hydrazine
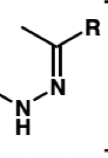
draw a hydrazone
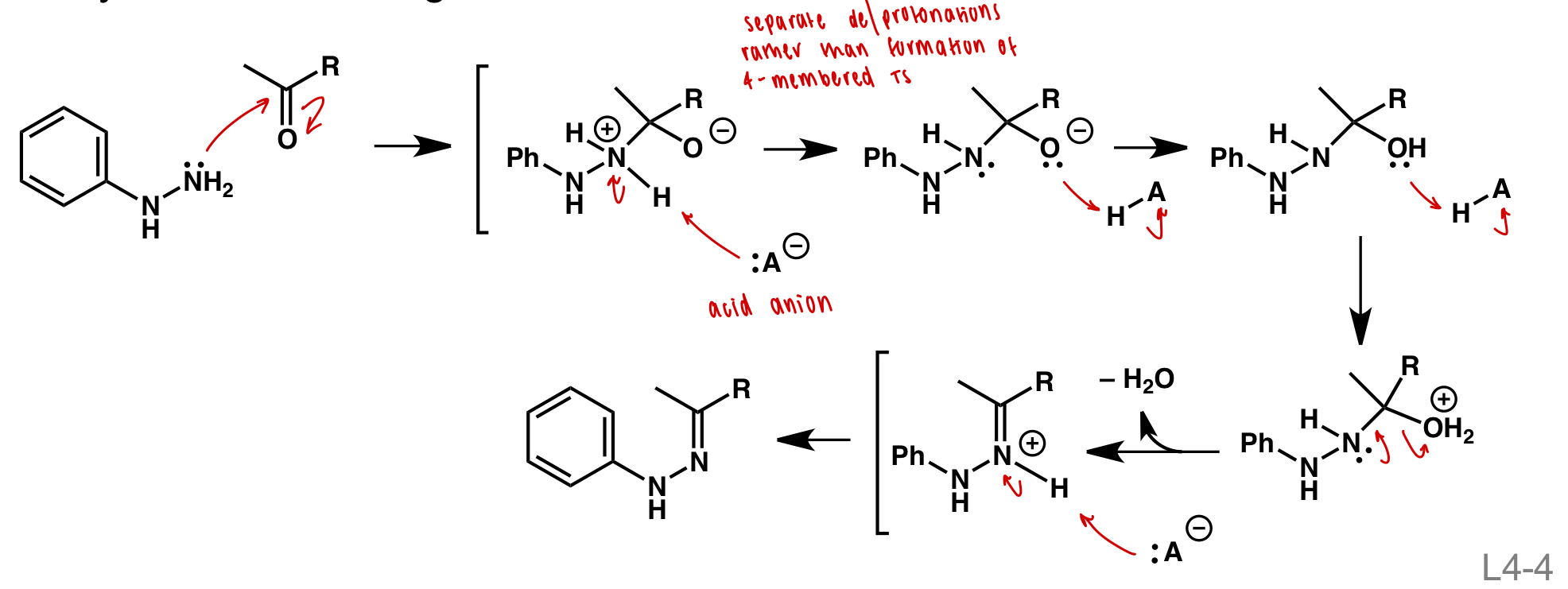
draw hydrazone formation
nucleophilic attack
formation of good LG (H2O+)
deprotonation
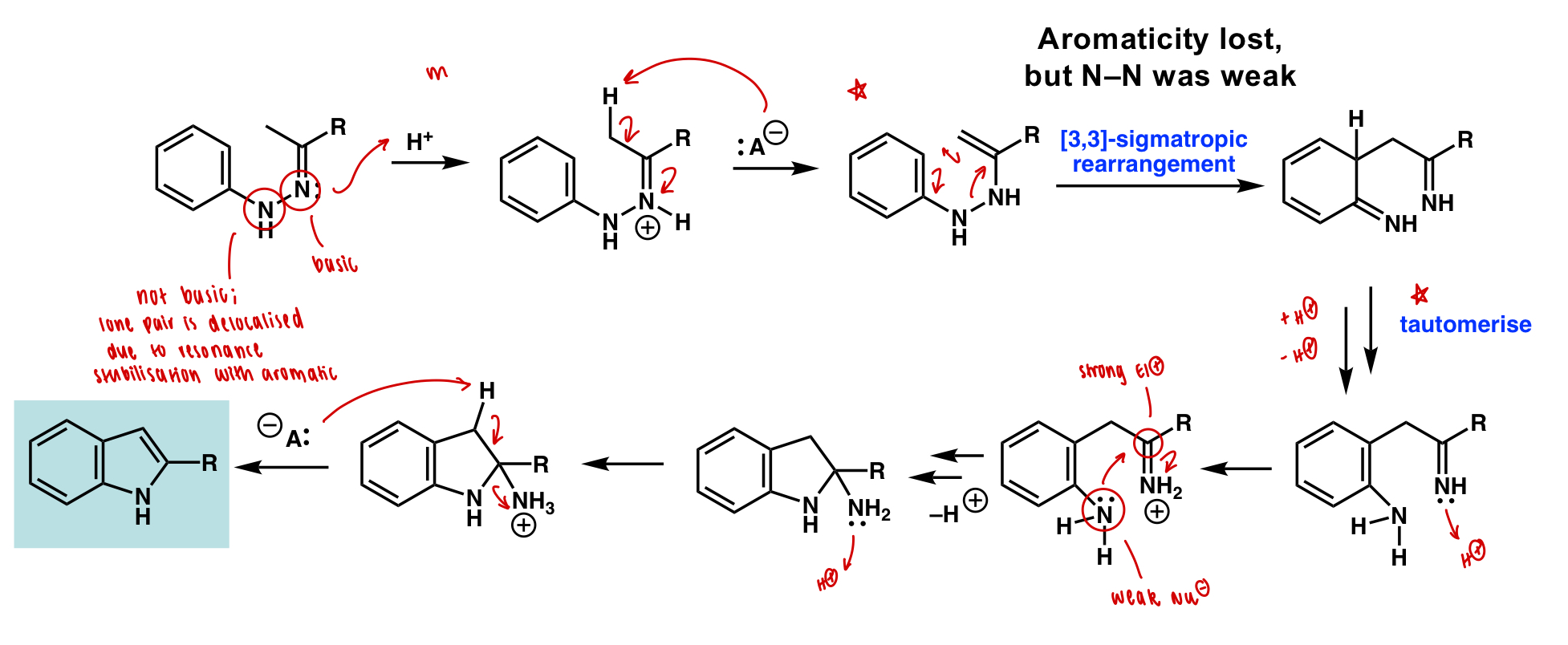
draw indole formation
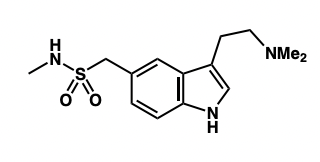
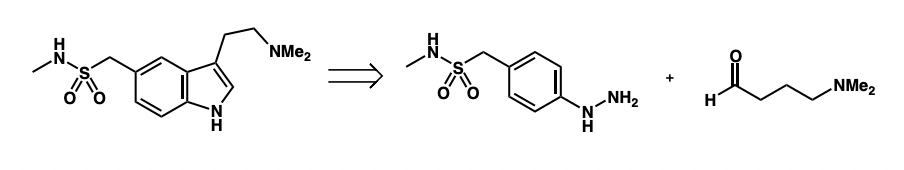
draw the SM required for the retrosynthesis of sumatriptan
describe hypertension
= chronic high blood pressure
= many causes i.e. ACE
describe a bodily response to high blood pressure
= release of bradykanin
= peptide
= vasodilator
describe angiotensin-converting enzyme (ACE)
two forms:
ACE I = inactive = deca(10)-peptide
ACE II = active = octa(8)-peptide
three ways ACE II contributes:
vasoconstrictor = decreases vessel volume
UP-regulates the production of aldosterone = reduces Na(+) and H2O excretion = increases blood volume
degrades bradykanin = inhibits vasodilation = decreases vessel volume
ACE inhibition is target for hypertension treatment

what is a challenge for identifying ACE inhibitors?
ACE = membrane bound protein
= structural information is difficult to obtain
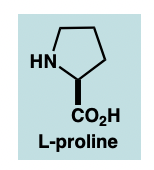
what moiety has activity as an ACE inhibitor
L-proline at C terminus (with free COOH)
describe the interactions of these ACE inhibitors at the ACE active site
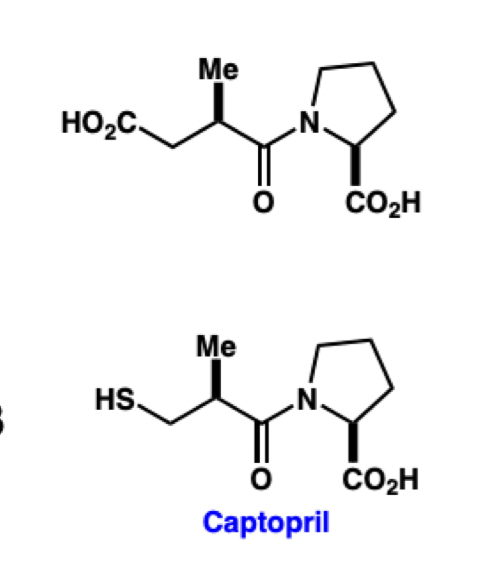
COOH = COO(-) in vivo = coordinates to Zn2+ ion
Me = fits into hydrophobic pocket
SH = S(-) in vivo = improved coordination to Zn2+ ion
IC(50): captopril « COOH-containg compound
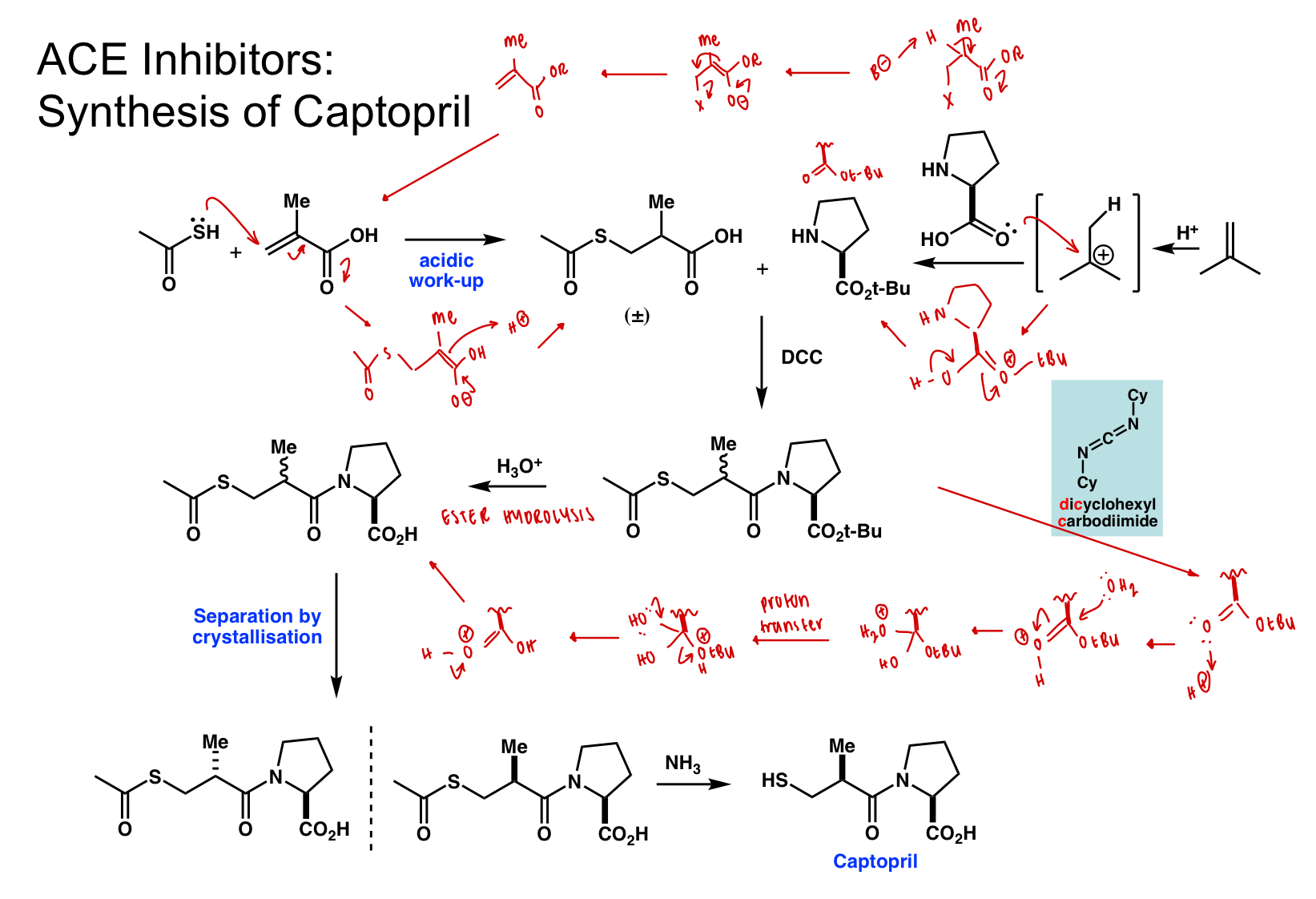
describe retrosynthesis of captopril
amide bond formation
= coupling reaction
thiol group installation
= a,B unsaturated carbonyl (Michael acceptor) = nucleophilic addition of SH(-)
= a,B unsaturated carbonyl from carbonyl with B LG
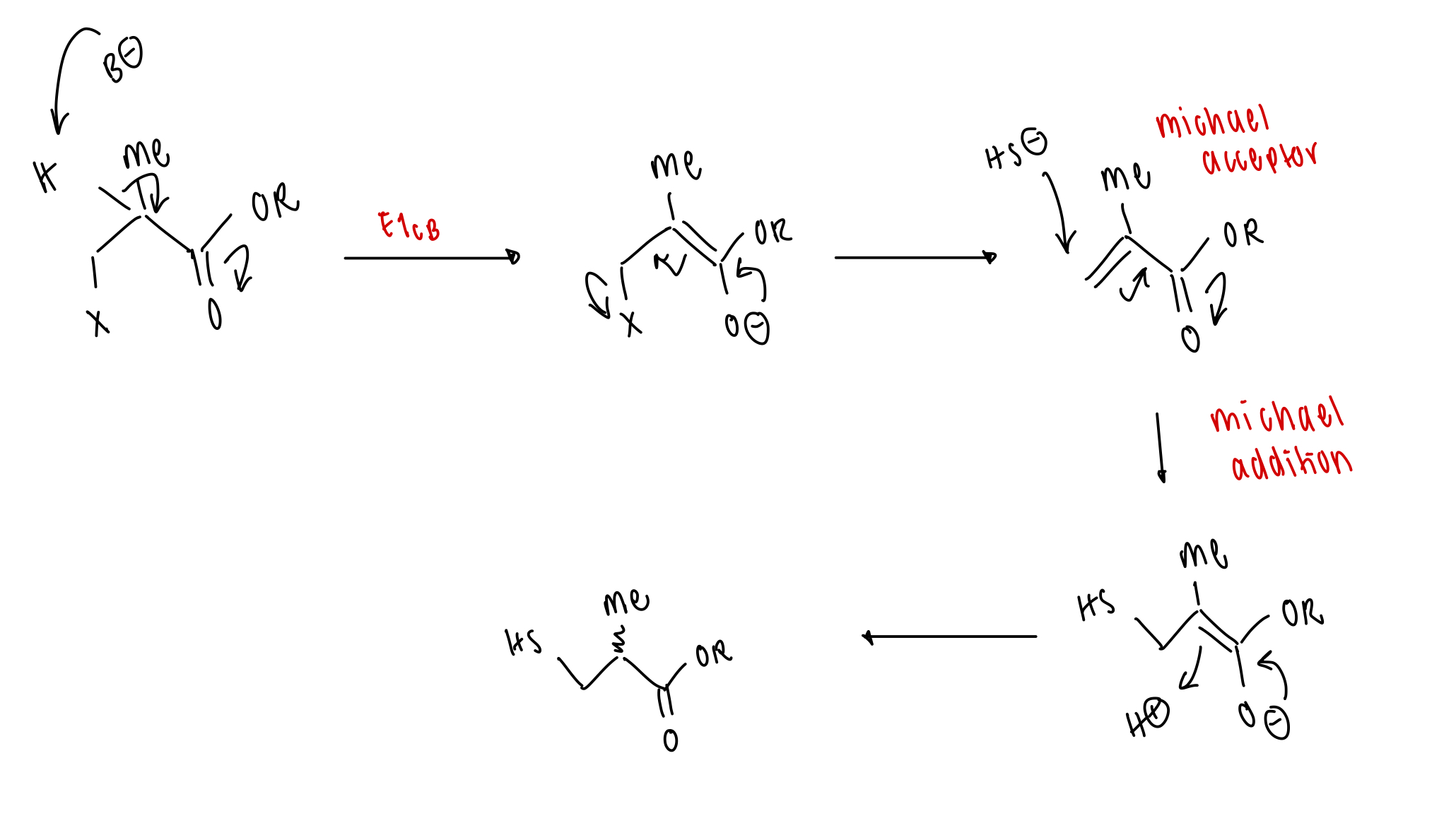
how can amide bonds be made?
= requires catalysis
old method = SOCl2
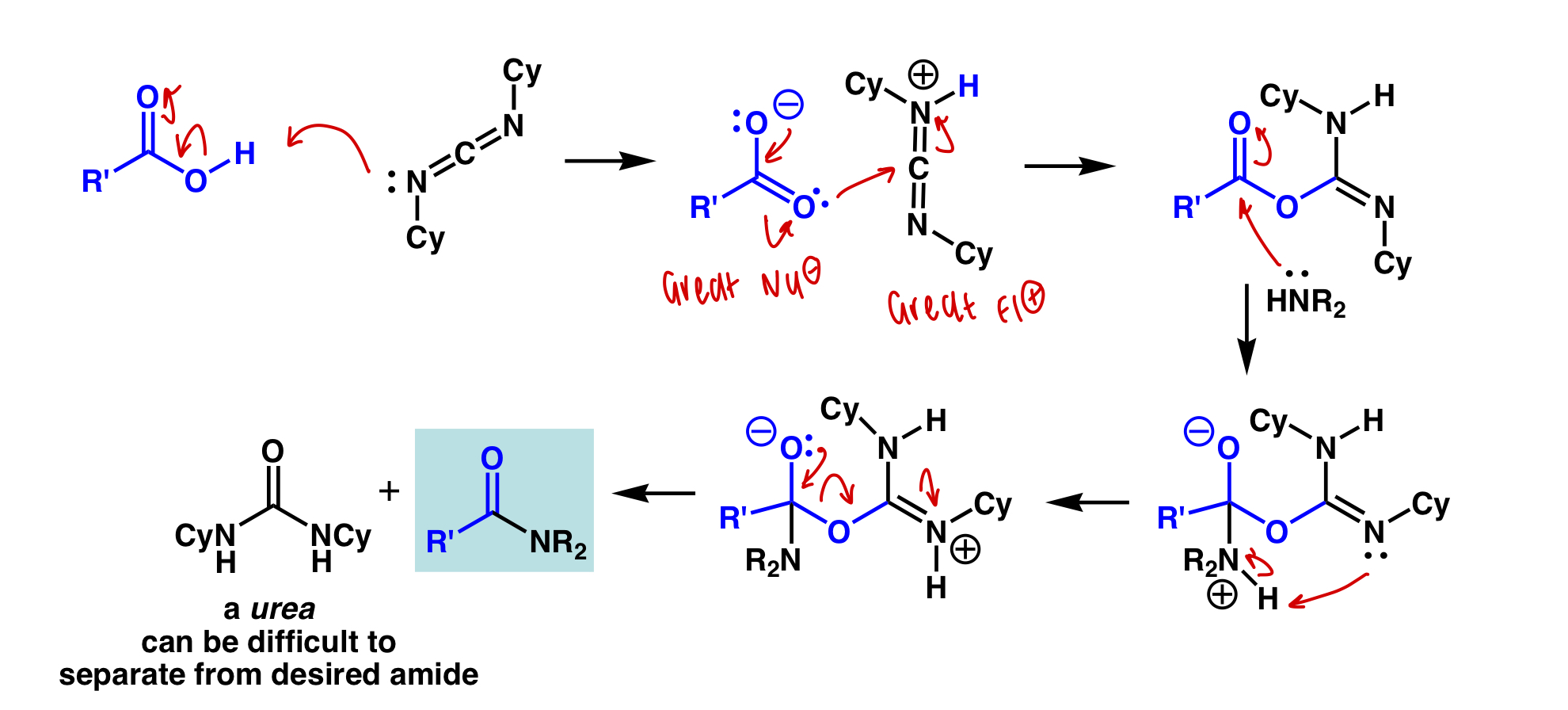
new method = peptide coupling reagent
draw the mechanism of peptide coupling reagent DCC
draw the full synthesis of captopril
last step = nucleophilic substitution
define bacteriostatic
inhibit cell growth, allowing natural defences time to respond to infection
define bactericidal
actively kill bacterial cells
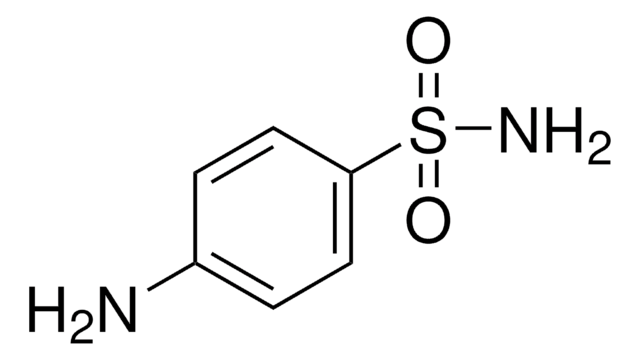
draw sulfanilamide
= anti-bacterial
draw sulfanilamide prodrug (prontosil)
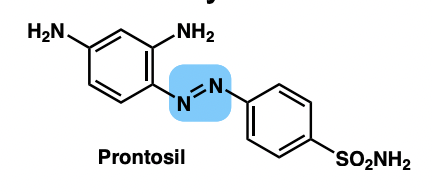
describe sulfa drugs
= sulfanilamide analogues
= inhibit folic acid biosynthesis in bacteria
describe the action of sulfa drugs
structure ~ PABA
PABA = upstream substrate of folid acid biosynthesis = substrate of dihydropteroate synthase
sulfa drugs are competitive (reversible) inhibitors of dihydropteroate
folic acid = essential for DNA and amino acid biosynthesis
sulfa drugs = bacteriostatic
define a structure-activity relationship (SAR) study
study which seeks to improve pharmacokinetic (body) /pharmacodynamic (function) properties of an active compound by modifying structure
draw the general structure of sulfa drugs
majorly NH2 at para position
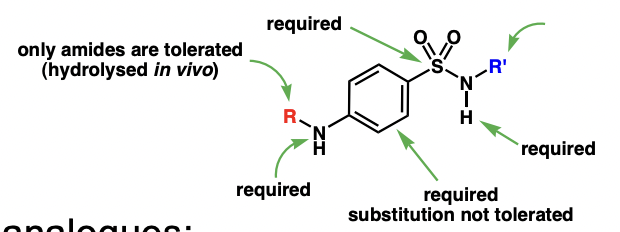
describe sulfa synthesis
(protected) sufonyl chloride backbone
variable amine
coupling + deprotection
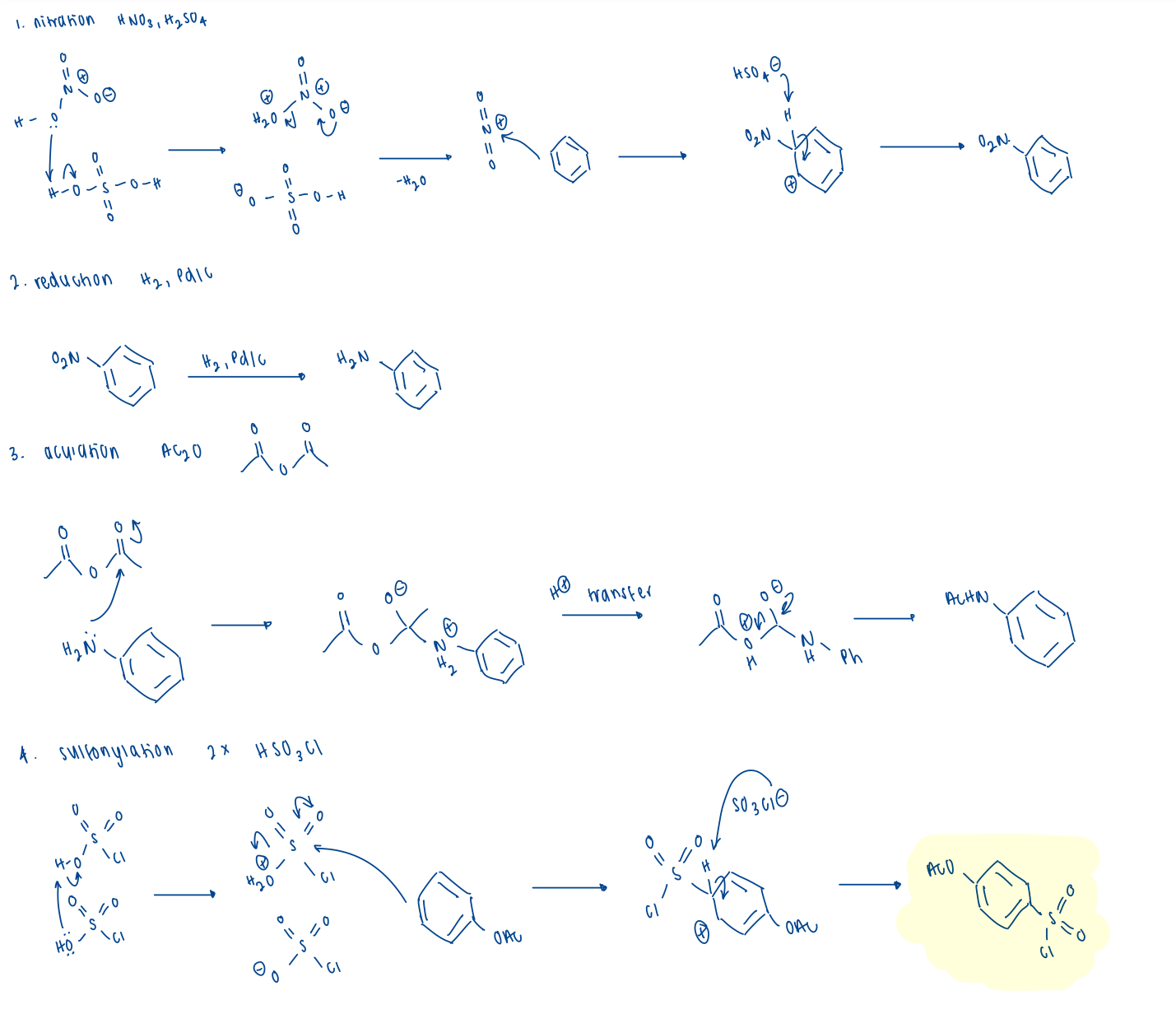
draw the sulfonyl chloride backbone synthesis
4 steps: benzene SM
nitration (NO2)
reduction (NH2)
acylation (NHAc)
sulfonylation (SO2Cl)
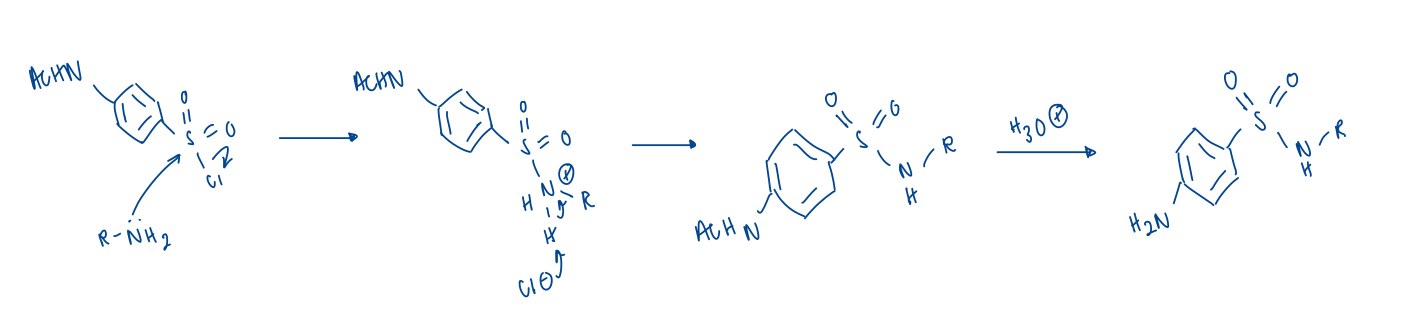
draw the coupling and deprotecting mechansim
what are methods of bacterial resistance to sulfa drugs?
mutated dihydropteroate synthase no longer binds = mutation at active site
overproduction of PABA = competitive inhibitor
efflux of drugs