Inherited Retinal Diseases (IRDs)
1/18
Earn XP
Description and Tags
Lecture 4
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
19 Terms
What are IRDs?
They are monogenic conditions, caused by over 320 gene mutations
What ocular co-morbidites do they usually have?
High myopia
PSC
ERMs
Macular holes
What 2 things that are part of our job is essential to do well for them?
good refractive correction
offering low vision management
Why are inherited retinal diseases (IRDs) difficult to treat with gene therapy, and why can they be challenging clinically?
IRDs are caused by mutations in over 300 different genes and affect about 1 in 3000–4000 people.
Gene therapies usually target only one specific gene or even one variant, so each therapy can help only a small number of patients (sometimes just 5–10 people in a country).
Clinically, IRDs are also challenging because presentation is highly variable—patients may first show symptoms in childhood or even as late as 80 years old.
They also progress very quickly & therefore we can see sig vision loss in short time, while others are slow
How can you determine the likely inheritance pattern in inherited retinal diseases (IRDs) using pedigree features, and why do IRDs show such varied clinical presentations?
IRDs follow Mendelian inheritance patterns (autosomal dominant, autosomal recessive, X-linked, mitochondrial).
pedigree clues you can use:
Skips generations → Autosomal recessive
Doesn’t skip generations —> autosomal dominant
Sex bias → X-linked
No sex bias → Autosomal
There are 300+ genes involved, each affecting different retinal tissues (e.g., RPE, photoreceptors, mitochondria), leading to variable symptoms and prognosis.
What are the diff ways we can classify IRDs (6 ways)?
1. Age of onset: paediatric/ late onset
2. Region affected first: macular/ cones/ rods affected first, so is central or peripheral vision being lost first
3. Stationary vs progressive: imp to know if disease will get worse, or it stays the same
4. Inheritance pattern: AD, AR, XL, Mt
5. Genes: classify based on genes affected
6. Syndromic vs non-syndromic: syndromic= systemic involvement so not just eye affected but more broad, e.g. Usher syndrome
How do we classify IRDs in optometry?
Panretinal pigmentary retinopathies (e.g., RP, cone-rod dystrophy, chloridaemia)
Affect the whole retina (diff symptoms based on what is affected first)
Rod-cone dystrophy (RP) is most common → loss of night vision + peripheral vision first, progressing to central loss.
Macular dystrophies:
Primarily cone dysfunction → central vision loss, may resemble AMD.
Can occur with or without flecks (e.g., Stargardts = with flecks).
Stationary diseases (e.g., achromatopsia)
Non-progressive (so imp to diagnose & comfort pxs), but baseline vision is poor
example is achromatopsia: VA <6/60, complete colour blindness, severe glare sensitivity).
Hereditary vitreoretinopathies (e.g., X-linked retinoschisis, Stickler syndrome)
Involve inherited diseases affecting vitreous + retina.
May have systemic features (e.g., hearing loss and joint issues in Stickler).
High risk of retinal detachment.
What are the more common IRDs that we should know the signs & symptoms of?
I think these are listed in order of commonness:
Panretinal pigmentary retinopathies- Retinitis pigmentosa (Rod-cone dystrophy)
Syndromic conditions: Usher' syndrome
Macular dystrophy with flecks- Stargardts disease
Panretinal pigmentary retinopathies- Chloridaemia
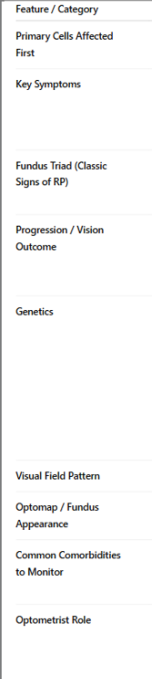
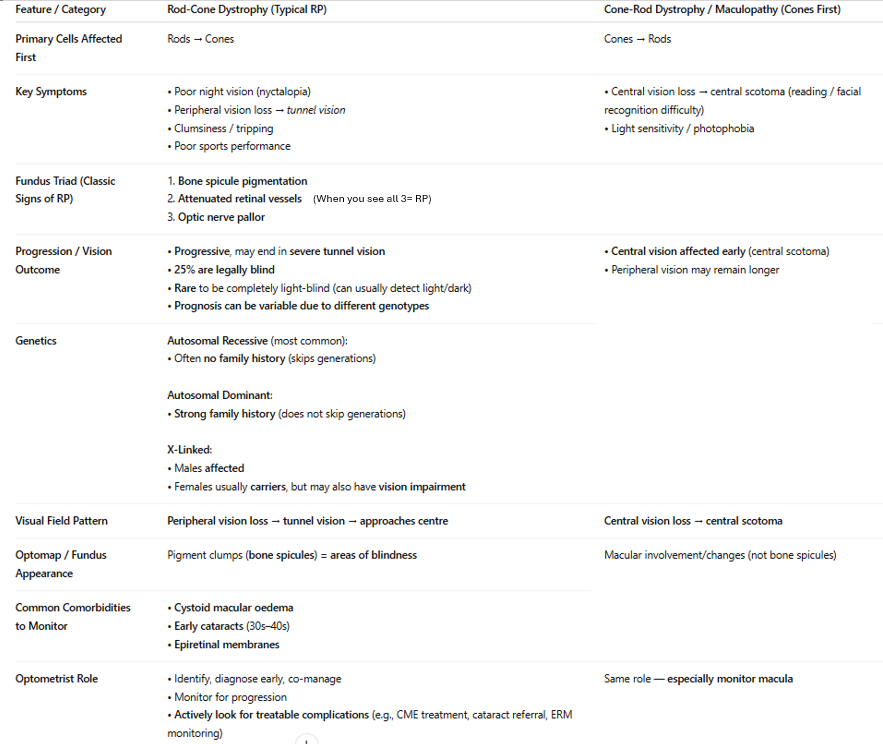
Describe retinitis pigmentosa. How is it diff to cone-rod dystrophy in terms of symptoms & signs?
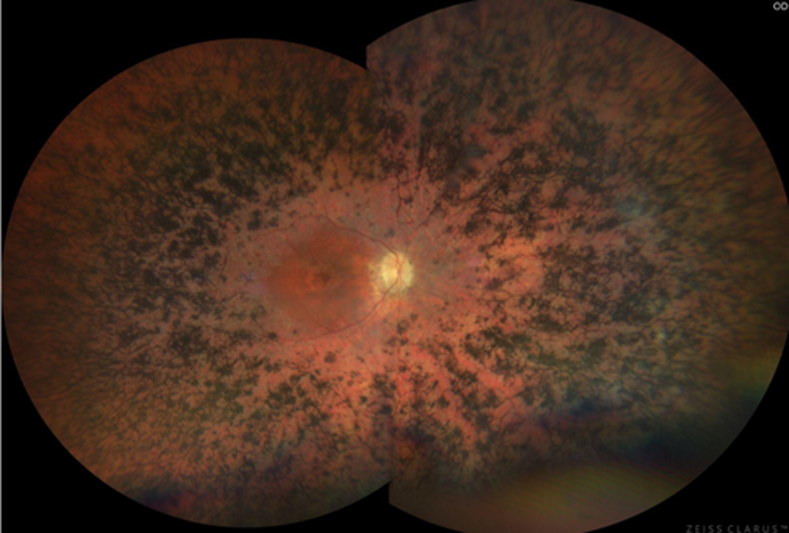
note: Bone spicule pigmentation (called that bc look same as bones when forming in embryo)
What are syndromic IRDs? Give 3 examples (BUS) and what systemic associations they have:
Syndromic IRDs are retinal dystrophies with additional systemic features.
clinical approach: when you identify an IRD look for systemic clues
examples:
Bardet-Biedl syndrome: if someone has a retinal dystrophy, but also extra digits (fingers/ toes) & obese
Usher’s syndrome: retinitis pigmentosa with hearing loss
Stickler syndrome: If with the retinal dystrophy they have a cleft palate & flat face
What is Usher’s syndrome and why is early genetic testing important?
Usher’s Syndrome causes dual deaf-blindness because it damages inner ear hair cells (hearing/balance) and RPE & photoreceptors in the eye (bc as RP)
Children are typically born deaf and then develop vision loss around age 8–10.
Early genetic testing in deaf infants allows families to prepare for expected vision decline rather than being caught off-guard later (or teach them sign language)
cochlear implants can now assist children in hearing, but no solution for vison :(
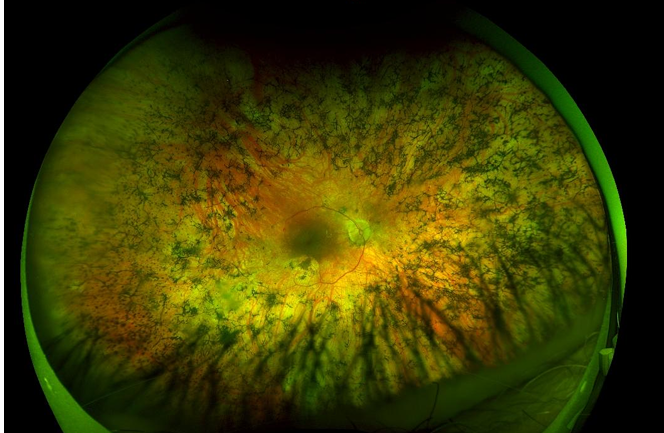
What do you see here? What would change if i told you the person in this photo is deaf?
I see retinitis pigmentosa (bone spicule pigmentation spread around that starts in rods and comes in towards cones/ centre
if also deaf means likely to be Usher’s syndrome (deafness & RP)
What is Stargardts disease?
due to ABCA4 gene mutations
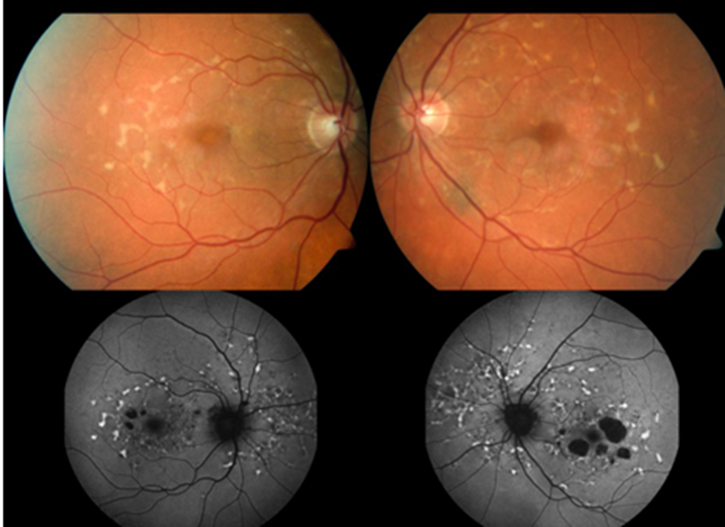
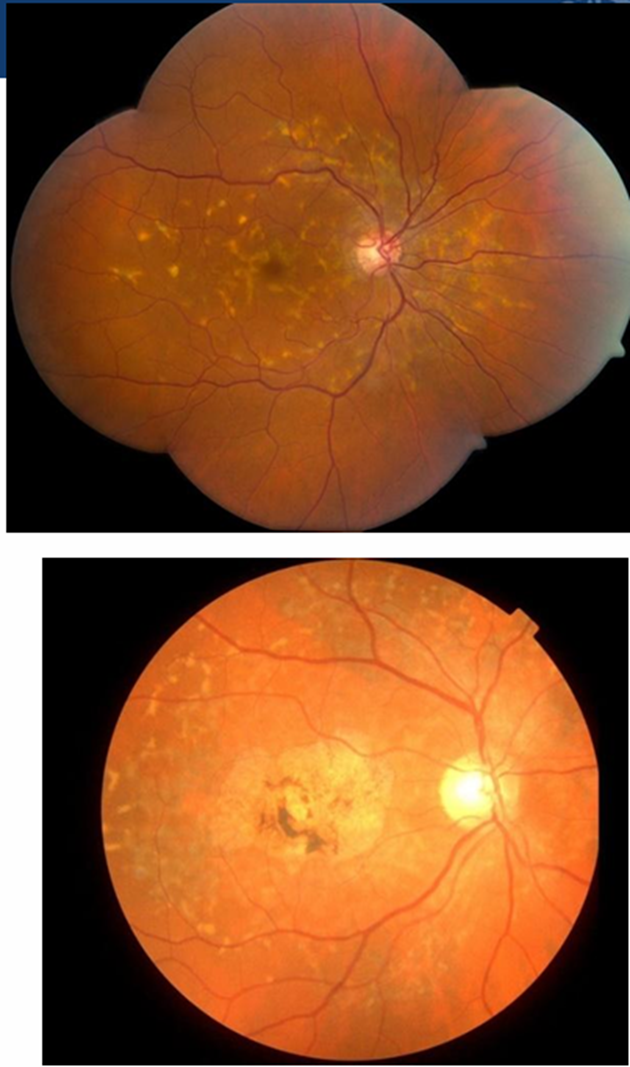
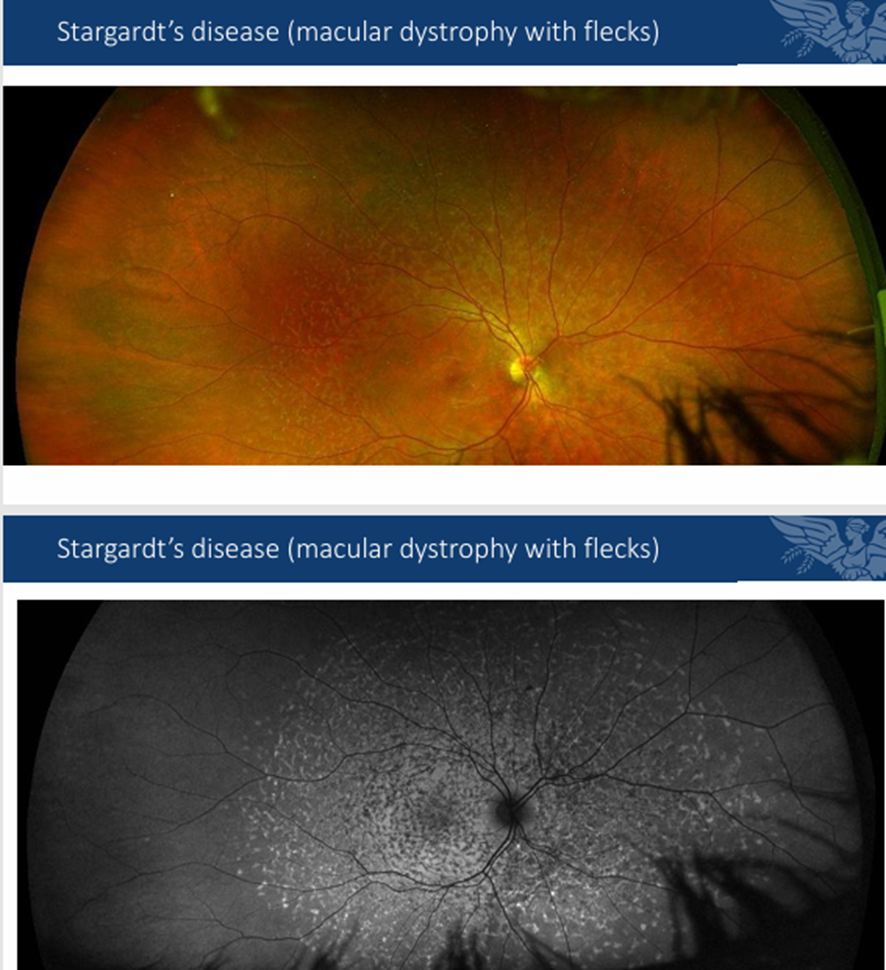
Diff bw normal imaging and FAF imaging when showing flecks:
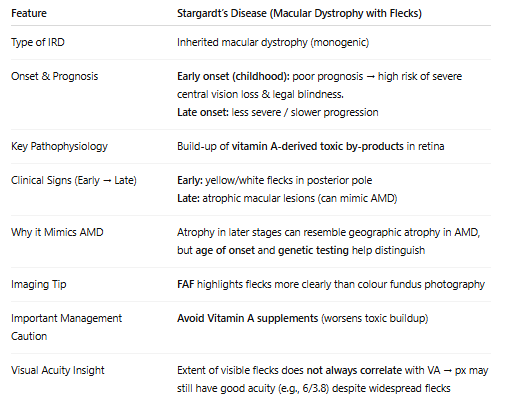
What is chloridaemia? Why is it so easily diagnosed just based on retina alone? What are its symptoms? What type of inheritance pattern is it & what does it imply?
Here you are not just losing PRs and RPE, but also choroid, so you get a retina that looks very brought & white bc you are looking at sclera —> easily diagnosed
symptoms: · Symptoms very similar to RP: Night vision problems, loss of peripheral vision, affects ppl at young age <20, but unlike RP, fundus doesn’t have intraretinal pigmentation & blood vessels are not attenuated
X-linked inheritance therefore affects men more
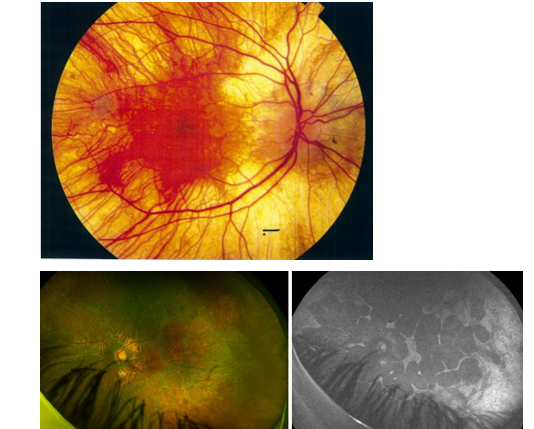
What is your assessment strategy/ what assessments must/ can you do for IRDs?
note: That FAF measures lipofuscin/ metabolic activity and so it can show a lesion hyper fluorescing or hypo fluorescing which is both bad bc if hyper it means there is too much waste (lipofuscin) and if black/ hypo means RPE and PR cells containing the lipofuscin are dead!!
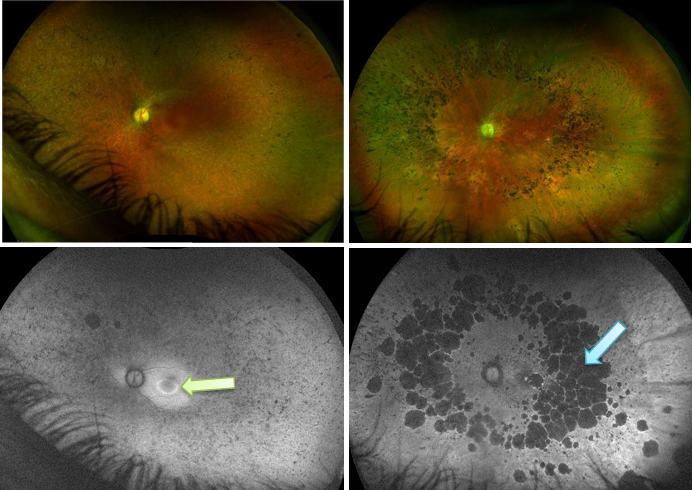
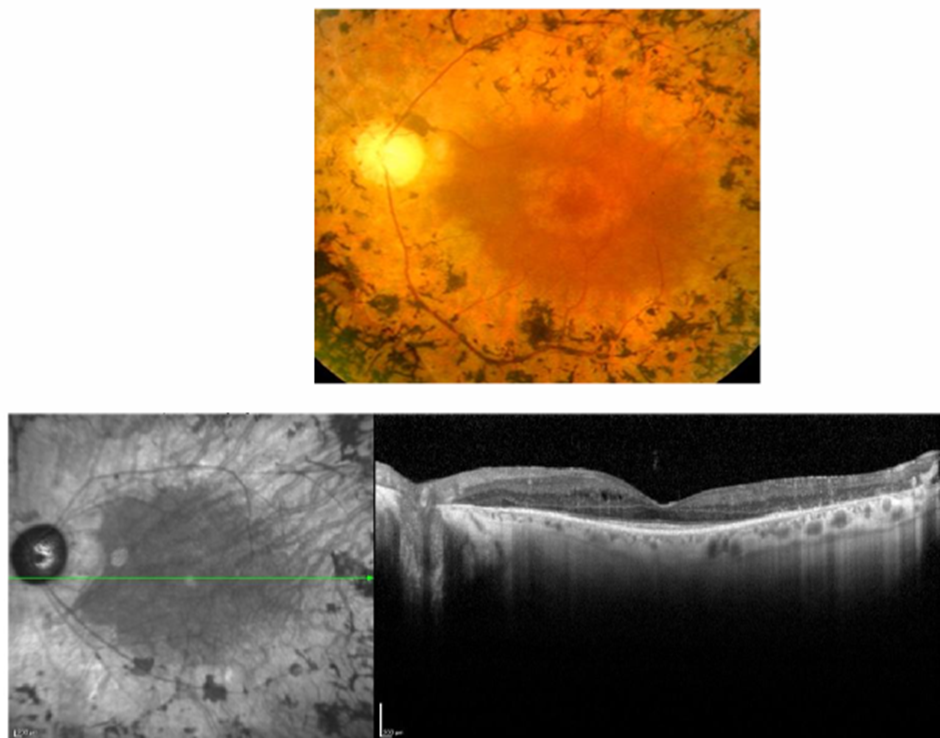
What do you see here? These are 2 diff conditions (one on left has a colour fundus version above and FAF version helping see lesion better, and same for right):
left= cone dystrophy (hyper/ glowy in centre indicating IRD related to cones bc too much lipofuscin/ waste there)
right= retinitis pigmentosa/ rod-cone dystrophy: you see hypo, so cells are dying and its starting peripherally and coming in centrally
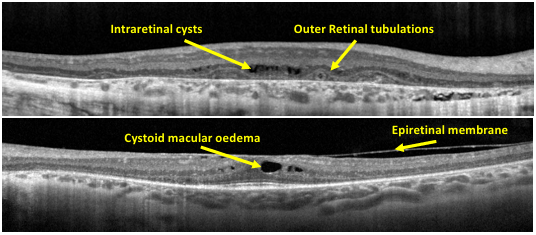
Just look at this image and study the dI ff lesions you can see in OCT.
How should we manage those IRDs (the more common ones)?
What are the different treatment options that are being investigated for IRDs? Make sure you mention what Luxantra is:
Side note/ additional info: Leber’s Congenital disease (mutated RPE65), causes blindness so when they take Luxantra one time only, it prevents blindness later in life from occurring (but it is only helping small amount of ppl bc its rare condition)
