Pharm Tox Exam 1
1/117
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
118 Terms
pharmacology
the study of substances that interact with living systems through chemical processes
drug
chemical agent that affects living protoplasm
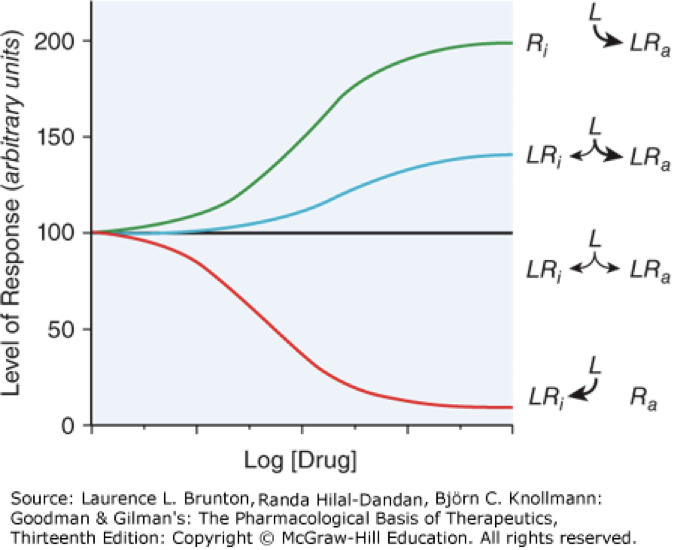
agonist
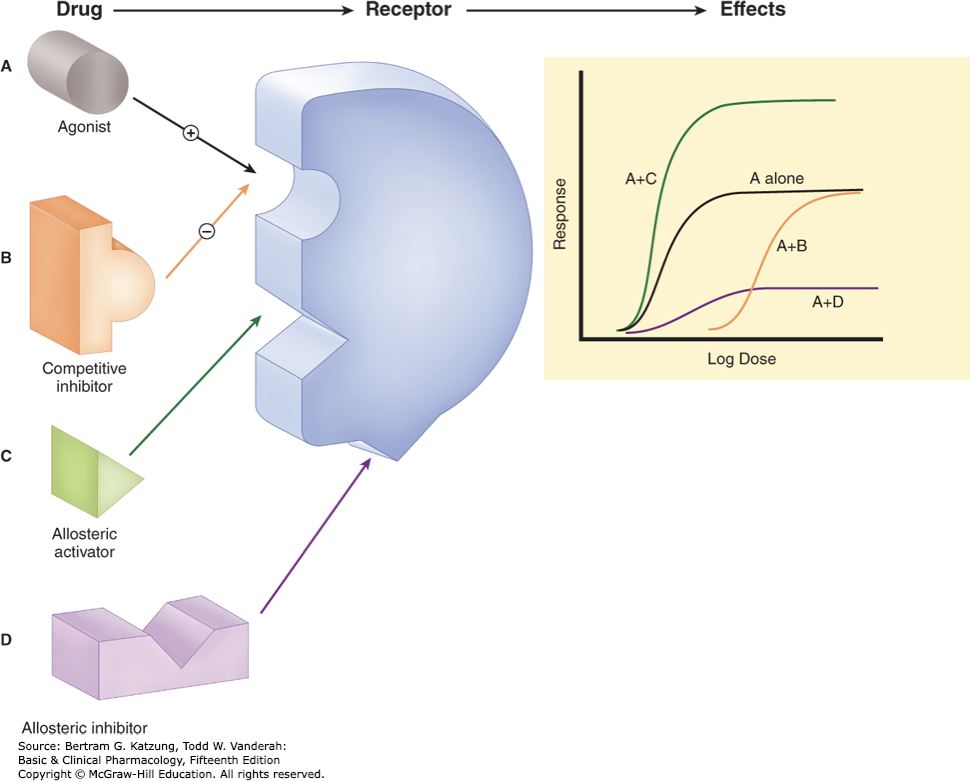
activator, drugs bind and activate a receptor, mimic endogenous ligands, green and blue lines
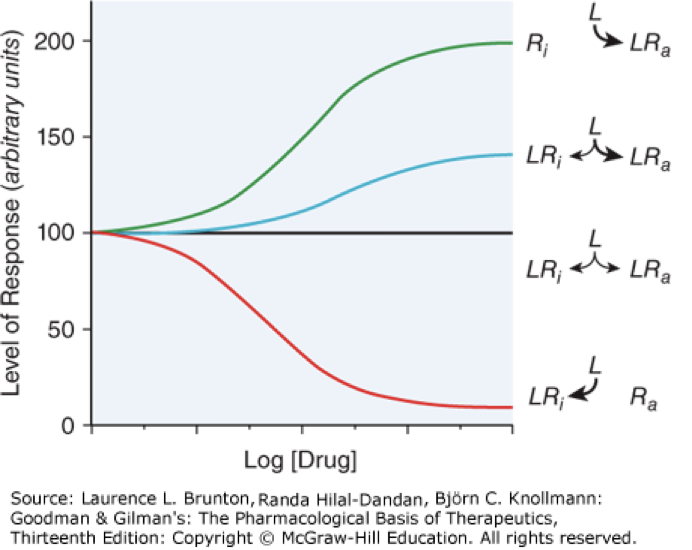
full agonist
activate receptor to the max that is capable, green line
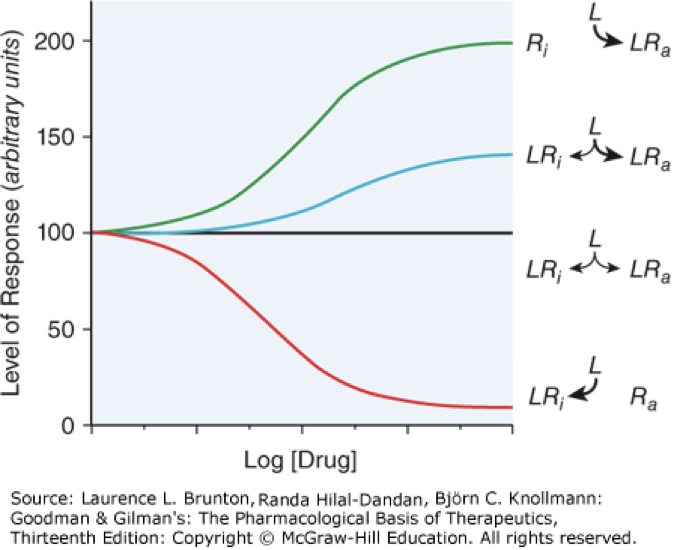
partial agonist
activates but does not reach max activity, can compete with full agonist to reduce activation of a receptor, blue line
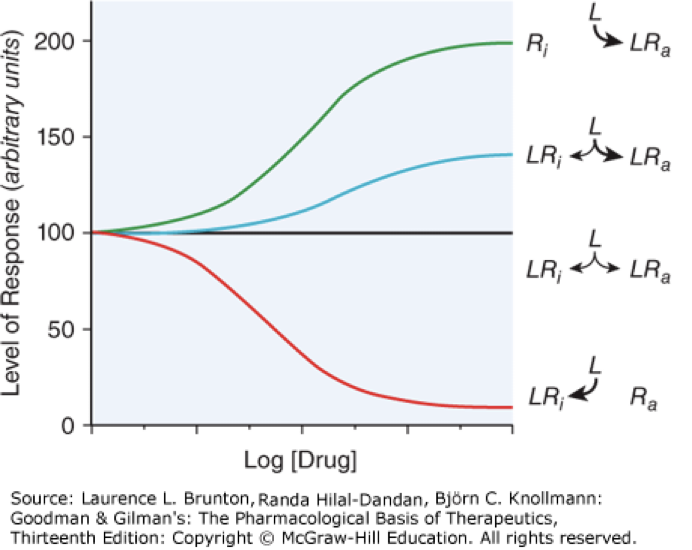
inverse agonist
decreases constitutive (intrinsic) activity, not an antagonist but the graph can look similar to a partial antagonist, red line
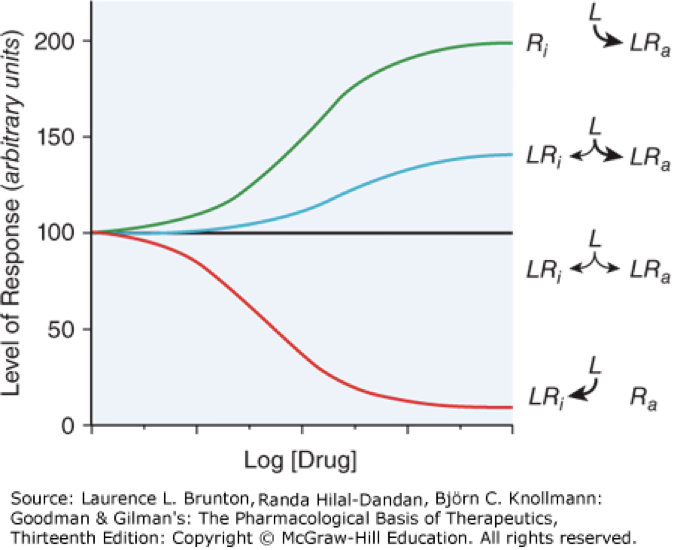
antagonist
inhibitor of agonist, binds and inhibits receptor from binding with agonist, does not change constitutive activity, black line
allosteric modulators and example
bind secondary site to enhance or inhibit action of an agonist, ex. benzos
biologics
larger molecule with receptor (antibody) that binds to endogenous molecules
chemical antagonists
inhibits drug by directly (chemically) interacting with them
osmotic agents
interacts (almost exclusively) with water
hormones
synthesized within the body, chemical messengers
xenobiotics
chemicals not synthesized in the body
poisons
any substance that has the capacity to cause harm, depends on the dose
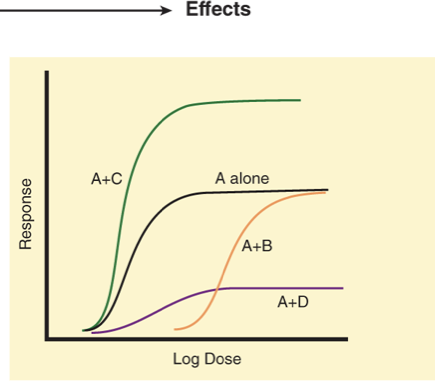
what is A, B, C and D?
pharmacokinetics
ADME, the effect the body has on the drug
pharmacodynamics
the effect the drug has on the body, mechanisms of action
drugs usually alter rate or magnitude of intrinsic responses, not creating new
rational drug design
predicting drug structure based on knowledge of the receptor or target, then optimizing
personalized medicine
fit drug to individual patients based on genetic screening and biomarkers
absorption
movement of drug from site of admin to central compartment (blood)
bioavailability
how much of the drug reaches the active site or systemic circulation, after GI absorption and liver metabolism
bioequivalence
drugs that contain the same active ingredients and are identical in strength or concentration, dosage form, and route of administration
Which of the following best defines a prodrug in pharmacology?
a. exhibits antagonistic effects on multiple receptors simultaneously
b. is administered through intravenous injection for rapid onset of action
c. biologically inactive drug metabolized into an active drug within the body
d. a drug that directly stimulates neurotransmitter release
c. biologically inactive drug metabolized into an active drug within the body
acidic and basic drugs bind to what proteins?
acidic drugs bind to albumin, basic drugs bind to alpha1-acid glycoprotein
drug accumulation in BBB, bone and fat
more lipophilic drugs are able to cross the BBB
tetracyclines and heavy metals accumulate in bone, osteoporosis releases them later
fat stores lipid soluble drugs
what common pain reliever is ionizable?
aspirin
redistribution
termination of drug effect after withdrawal of a drug, lipid soluble drug are not eliminated as fast
placental transfer of drugs depends on
lipid solubility, extent of PPB, degree of ionization
first order kinetics
the amount of drug metabolized per unit time is proportional to the plasma concentration of the drug (Cp) and the fraction of drug removed by metabolism is constant
zero order kinetics
metabolic capacity is saturated at the concentrations usually employed, so a constant amount of drug is metabolized per unit time, does not depend on concentration, occurs at toxically high drug concentrations
inducible biotransforming enzymes
broad spectrum enzymes with predictable genetic variation, CYPs
phase I reactions
what is the purpose?
oxidation (CYP), reduction, hydrolysis
to deactivate (or active if prodrug) and slightly increase water solubility
phase II reactions
what is the purpose?
conjugations
add polar group that greatly increases water solubility to allow elimination
where does metabolism happen?
primarily in the liver, some in GI, kidneys, lungs
clinical pharmacokinetics
used to find a quantitative relationship between dose and effect, framework to interpret drug concentration and how to adjust dose as needed
where does excretion happen?
kidneys (30% of unchanged drugs), feces (unabsorbed oral drugs or metabolites in bile), lung (volatile)
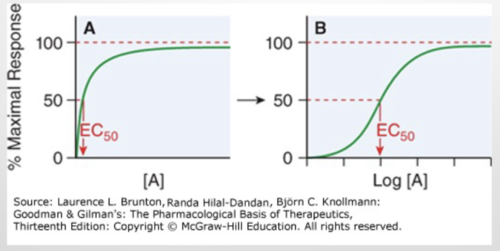
dose-response curve EC50
effective concentration of agonist for 50% response
dose-response curve LD50
median lethal effective dose, 50% of animal participants die
hormesis
low dose stimulates receptor, high dose inhibits receptor
pharmacophore
simplest form of structure needed to interact with target or receptor, can be identified by computer modeling and optimized
affinity
measured binding of ligand to receptor, determined by chemical structure, represented by dissociation constant
specificity
how promiscuous the drug is, non-specificity can come from racemic mixtures and leads to side effects
potency
drugs have the same response but one is at a lower concentration, when the EC50 is less or the dose-response curve more to the left
a mix of affinity and efficacy
efficacy
the capacity of a drug to activate a receptor and generate a response
radioligand assays
measures functional response to determine affinity of ligand for receptor
positive synergism
super-additive effects of two drugs used together
negative synergism
less then additive effect of two drugs used together
what factors modify drug action?
medication errors
patient compliance
rate and extent of absorption
body size and composition
distribution in body fluids
binding in plasma and tissues
rate of metabolism and excretion
physiological variables
pathological factors
genetic factors
interaction with other drugs
development of tolerance and desensitization
drug-receptor interaction
functional state of targeted system
selectivity of drug, propensity to produce unwanted effects
placebo effects
resistance to antimicrobial or antineoplastic agents
pharmacogenetics
study of genetic bases for variation in drug response, large effects from small number of DNA variants
pharmacogenomics
study of larger number of variants, genetic component of variable drug responses, can be across populations
how genes affect PK, PD, efficacy and ADE of drugs
difference between gene and allele
a gene is a DNA sequence that codes for a protein, alleles are different versions of one gene
different between mutation and polymorphism
a mutation is a change in the gene’s DNA sequence, a polymorphism is a mutation that is present in 1% or more of a population
relationship between genotype and phenotype
genotype is the DNA sequence itself, phenotype is how the genes physically present (different based on regulation and expression of genes)
haplotype
group of alleles that are inherited together from one parent, the closer genes are on a chromosome the more likely they are to be inherited, set of single nucleotide polymorphisms (SNPs) found to be statistically associated on one chromatid
frequency of polymorphisms
important when deciding what is generally best for the population you are treating
non synonymous coding SNPs
codes for a different amino acid
synonymous coding SNPs
changes gene sequence but codes for the same amino acid
noncoding SNPs
can be in the promoter region and affect the transcription of the gene
can be intronic and affect splicing
gene duplications
increases number of enzymes and can increase how quickly a drug is metabolized
large deletion
if a gene has redundancy this shows how important it is and will not be as affected by deletions
consequence of SNPs
nonsynonymous substitutions
splice site mutations
early stop codon, degradation of mRNA, reduced amount of protein
increased proteolysis
changed promoter function
intronic SNPs can lead to truncated protein, from early stop codon
codeine and mutations
prodrug that is metabolized by CYP2D6, individuals missing a copy of the gene don't get full effects of drug
warfarin and mutations
warfarin inhibits VKORC1 to stop clotting and is metabolized by CYP2C9, mutations can affect how effective warfarin is or how quickly it is metabolized
important drug characteristics for drug development
drugs ability to interact with receptor: size, charge, shape, atomic composition of drug, drug shape or chirality
transportation to site of action
appropriate duration through ability of the body to inactivate or excrete drugs
signal transduction pathways
activated by receptors on the surface of cells
two major functions: ligand binding domain (LBD), message propagation (effector domain)
regulatory action may be exerted on effector proteins or on transducers (intermediary signaling molecules)
one receptor can create a large effect because of the ability to amplify the signal, often through enzyme cascades
excellent drug targets
what are the four types of receptor structures?
nuclear or intracellular receptors
ligand-gated ion channels
G coupled protein receptors
enzyme-linked receptors
G protein coupled receptors (GPCR)
span plasma membrane as bundle of 7 alpha helices
target for many drugs due to large quantity and physiological importance
mediated by secondary messengers
signal transducer with one or more effector proteins (AC, PLC, cGMP, PDE6, Ca and K)
GPCR alpha subunit types
alpha subunit types: Gs, Gi, Gq and G12/13
Gi decreases cAMP
Gs increases cAMP
Gq increases IP3 and Ca
can be homo or heterodimers of subtype
Gq-PLC-DAG/IP3 Ca Pathway
Gq or Gi recruits PLC which generates DAG and IP3, releases Ca
Ca regulates metabolic processes, secretion, contraction, gene expression and electrical activity
types of ion channels
voltage gated: Na+, K+, Ca2+
ligand gated: glutamate (excitatory), GABA (inhibitory), ACh
transient receptor potential channels: nociception, hot and cold sensation, mechanosensation, sensation of chemicals
enzyme linked receptors
receptor tyrosine kinases: receptors for hormones like insulin, GF, ephrins
Jak-STAT receptors: regulates transcription, no intrinsic enzymatic activity
receptors that stimulate cGMP synthesis
nuclear (intracellular) hormone receptors
transcription factors that regulate gene expression
receptors for steroid hormones like androgens, estrogens, glucocorticoids, thyroid, vitamin D
Jak-STAT receptors
binds cytokines
dimerization induced by ligand (cytokine) binding, JAK proteins bind, JAK phosphorylates other STAT proteins, phosphorylated STAT proteins translocate to the nucleus and regulate transcription
apoptosis signaling pathway
highly regulated set of reactions
lead to cell rounding and shrinking of cytoplasm, nucleus condenses
cell membrane presents phosphatidylserine on outer surface
recognized by macrophages so they phagocytize the dying cell
two inducing pathways for apoptosis
external signals
internal signals, triggered by DNA damage or improperly folded proteins
autophagy
catabolic pathway that sequesters damaged cellular contents in vesicles then delivers to lysosomes and contents are degraded
can also provide cells with substrates for energy and biosynthesis
testicular feminization syndrome
deficiency of androgen receptor
myasthenia gravis
autoimmune disruption of nicotinic cholinergic receptor function
insulin-resistant diabetes (type I)
autoimmune disruption of insulin receptor function
cancer
many forms come from mutations in growth factor receptors
xenobiotics
foreign chemicals in the body
plants are dietary xenobiotics
can be beneficial or toxic
enzymes metabolize these and eliminate them from the body
most are lipophilic to cross lipid membranes
downsides of drug metabolism
different between species, animal models are limited in predicts effects in humans
inter-individual variations in capacity to metabolize drugs
drug interactions with metabolizing enzymes
metabolic activation of chemicals to toxic and carcinogenic derivatives
what is metabolism for?
converts hydrophobic xenobiotics to be hydrophilic so it can easily be eliminated from the body
prevents accumulation and toxicity
rates of metabolism of drugs
if metabolized too quickly, reduced therapeutic effect
if metabolized too slowly, accumulates and causes toxicity
first round of metabolism
bacteria in the gut, varies in each person, changes drug disposition by different gene expression of the microbiome
phase I enzymes
Cytochrome P450 (CYP): oxidation, has a heme to use oxygen during reactions
Flavin-containing monooxygenase (FMO): adding an oxygen atom to substrates, high levels in the liver and bound to ER
Epoxide hydrolase (EH): hydrolysis of epoxide rings
phase II enzymes
glutathione s transferase (GST)
glucuronosyltransferase (UGT)
sulfotransferase (SULT)
N-acetyltransferase (NAT)
methyltransferase (MT)
role of CYPs
phase I oxidation
one compound can be metabolized by different CYPs, one CYP can metabolize many drugs or the same drug at different sites, this is the cause of many drug-drug interactions
CYP inducers
increase their own rate of metabolism or metabolism of other drugs
sites of drug metabolism
GI tract, liver, nasal and lung mucosa, ER
toxicity
physiological response to a drug that is an adverse effect, can depend on dose
ED50
concentration of drug where 50% of population will have desired response
LD50
dose that is lethal in 50% of the population (in animal models)
therapeutic index
LD50/ED50, the higher the ratio (larger difference) the safer the drug
graded dose-response curve
normal curve, response increases as dose increases
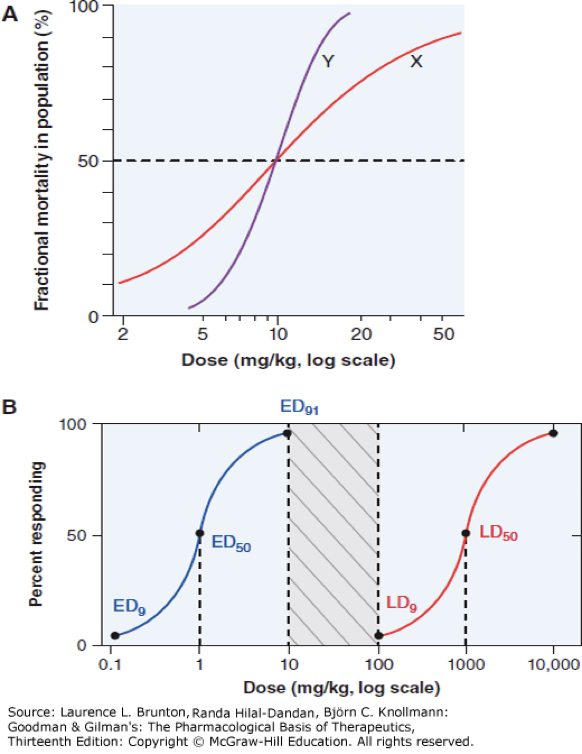
quantal dose relationship
percentage of the population that responds increases as the dose increases
U-shaped dose-response curve

usually for endocrine disruptors, hormones, essential metals and vitamins
initial high adverse response due to deficiency, then dips when homeostasis is achieved, then beyond required amount toxicity occurs
hockey stick dose-response curve

for toxicants with saturable removal process, no response until reaching a threshold where the removal process is saturated then zero-order increase in adverse response
inverted U-shaped dose-response curve

for ligands that down-regulate receptors, or there is an additional negative effect beyond the concentration that produces the primary positive effect
five rights of safe medication dispensing
drug, patient, dose, route, time