Mitochondrial quality control and Parkinson's
1/89
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
90 Terms
Mitochondrial defects
the principal drivers/cause of Parkinson’s
Ex. defects in Ca2+ buffering
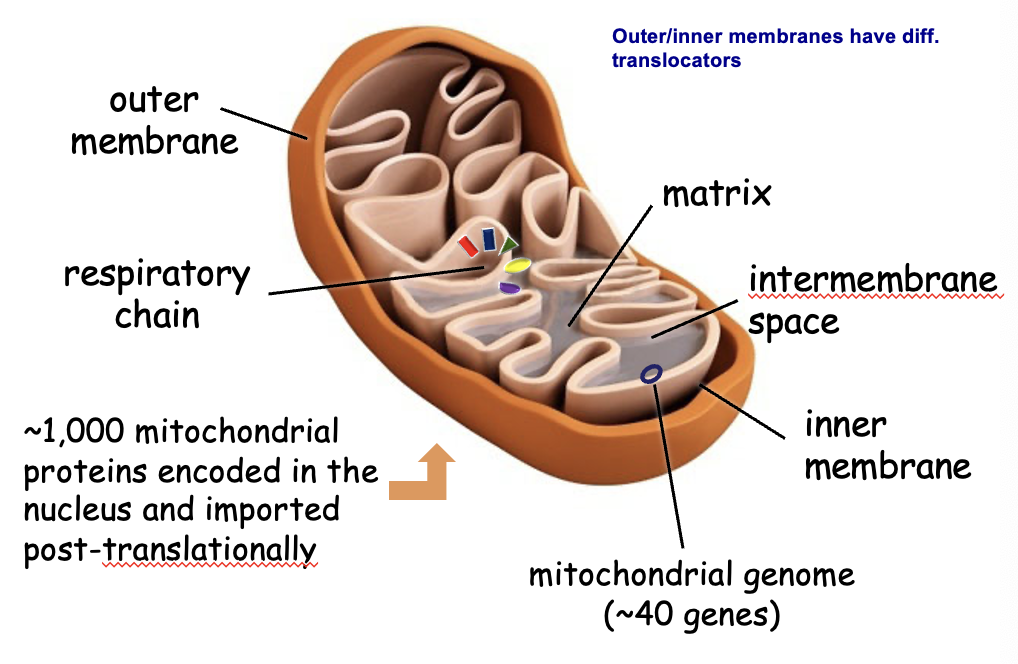
mitochondrial anatomy
Mitochondrial genome
~40 genes → synthesis, trafficking, etc. happens inside (prokaryotic characteristic)
*majority of genes are not mitochondrial
Inner mitochondrial membrane
oxidative phosphorylation → only mitochondria needs O2 to make ATP
Mitochondria (functions)
ATP synthesis, Ca2+ buffering, apoptosis, metabolite synthesis
Warburg effect
tumor cells can survive in the absence of oxidative phosphorylation (no O2) → survives off of 2 ATPs
Mitochondrial dynamics
mitochondria are not static → networks of fusing/dividing mitochondria (dynamically moving b/w 2 states)
combine = fusion
separation = fission
*divide like ancient bacteria
Why is dynamic behavior important?
maintaining dynamic shift b/w fusion and fission is critical for healthy/normal cells → if disrupted, it can cause a harmful disease like Parkinson’s
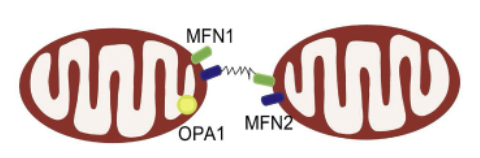
fusion → outer 1st, then inner
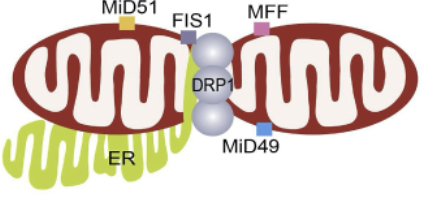
fission → both inner/outer changed simultaneously
*can’t start unless there is a cue (Drp1) from the cytoplasm
Fission proteins
Drp1, Fis1, MFF, MID49, MID51, DNM2
regulated by Drp1 and Fis1 → compress/separate mitochondrial tubules
OMM and IMM fission occurs simultaneously
Fusion proteins
MFN1, MFN2, OPA1
*regulators
Fis1
located in OMM and prevents fusion by inhibiting Mfn2/Opa1 (activity)
Drp1
in the cytoplasm and translocates to the mitochondria
MFN1 and MFN2
regulate fusion (OMM fusion)
Opa1
regulates inner membrane fusion
How are fission and fusion regulated?
tightly and dynamically (happening all the time)
What happens during fission?
low energy demand → uncoupled respiration
reduced ATP synthesis
mitochondrial degradation
*lazy (only work when they have to
What happens during fusion?
energy demands + stress → upregulation of metabolic competence
repair of damaged mitochondria
*Ex. exercising, stress, starvation
Do fission and fusion always lead to the same consequences/results?
in certain contexts yes, but in others the opposite happens (context-specific)
What happens as mitochondria age?
they collect damage → need fission and fusion
*1/2 life = 14 days (life span ~1 month)
Mitochondrial quality control
damages need to be converted to healthy
damage = fission (depolymerized) → removed
healthy part fuses w/ other mitochondria to become healthy
Surveillance mechanisms
mechanisms in healthy individuals → defects lead to Parkinson’s
1) fusion-mediated complementation
2) mitochondria-derived vesicles
3) mitophagy
What is the first line of defense against mild mitochondroal impairment?
fusion-mediated complementation and MDV
What happens when impairment increases?
damaged compartments are segregated from the mitochondria (fission) and undergo mitophagy
PINK1/Parkin-mediated mitophagy
mediate mitophagy in the face of mitochondrial depolarization (loss of membrane integrity → no repolarization)
positive feedback loop → completion
Receptor-mediated mitophagy
induced by hypoxia/during erythropoiesis → connection of damaged mitochondria to autophagosome is built by mitochondrial receptors
NIX, BNIP3, FUNC1, and BCL2L13 (interaction w/ LC3)
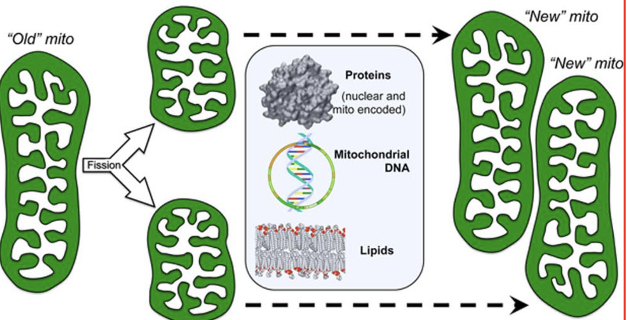
replicative fission and mitochondrial biogenesis
old mitochondria → 2 (each make up new mitochondria)
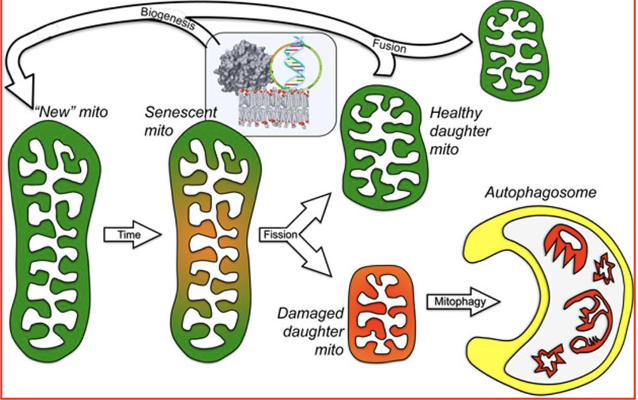
asymmetric fission and mitochondrial renewal/removal
new mitochondria will grow old → separate junk/degrade and keep the good parts → used to create new mitochondria
ER-mitochondria contact
sites mark the location where mitochondria will divide (fission) or merge (fusion)
*ER regulates both states
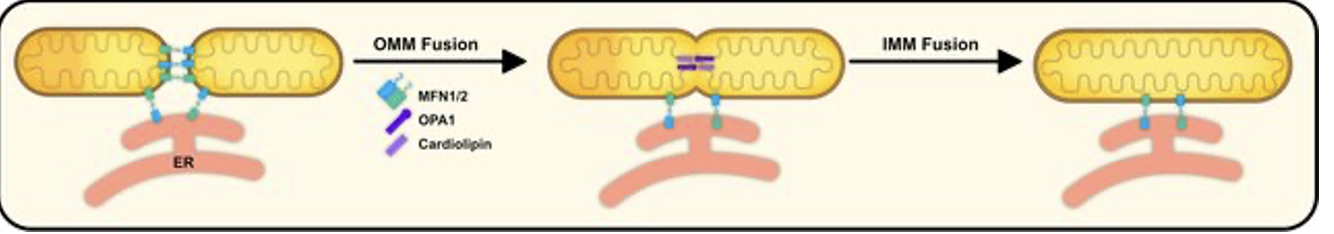
mitochondrial fusion (mechanism)
mediated by interaction of MFN1/2 on OMM
Opa1 interacts w/ cardiolipin on IMM @ ER-mito contact sites
*initiated by ER
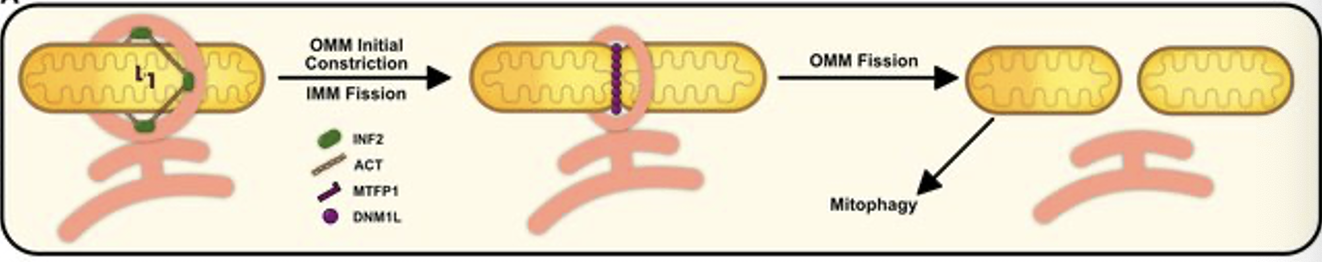
mitochondrial fission (mechanism)
initial OMM constriction via ER protein INF2
mediated actin polymerization
IMM fission by MTFP1 (IMM protein)
DNM1L recruited to OMM adaptor proteins to complete final constriction/scission of mitochondria
mitophagy (mediated by ER)
*initiated by ER
What happens if ER-mito interaction is interupted?
get hyperfusion
What causes cell death?
too much Ca2+ in the cell (mitochondria is a buffer) → can activate all sorts of pathways
Mitochondrial proteins
SIRT5, NRF2, SLP2
purpose - mitigate stress
*upregulated when the cell is under stress → accelerate fusion (hyperfusion)
SIRT5
removes acetyl, succinyl, and malonyl groups from lysine residues in proteins (inhibits Drp1)
NRF2
TF that regulates stress protein
SLP2
mitochondrial protein
Stress responses
mitigation → normal mitochondria
mild → hyperfusion
prolonged → hyperfission (fragmented mitochondria)
*will inhibit fission
Cellular stress response signaling
phosphorylates elF2a → stops all protein synthesis except proteins that help restore homeostasis
Cellular stress
drug induced, starvation, oxidative or proteostatic
What happens during Parkin-induced mitophagy?
1) PINK1 phosphorylates ubiquitin → attracts double membrane → autophagosome
2) parkin recruited to mitochondria and activates UPS → degrade mitochondrial membrane proteins
3) parkin promotes recruitment of autophagy adaptors to damaged mitochondria (OPTN, NDP52)
4) mitochondrial Rab GTPase activating protein
What is the importance of parkin-induced mitophagy?
principally defective process
specific indicator that mitochondria is damaged
depolarization, DNA mutations, increased ROS, misfolded proteins (tau, amyloid beta, alpha-synuclein → Alzheimer’s, Parkinson’s)
What is the role of PINK1 in healthy mitochondria?
used and degraded by the ubiquitin pathway (tight regulation)
What is the role of PINK1 in damaged mitochondria?
more PINK1 tagged to the outer membrane → attaches Parkin → forms complex in damaged mitochondria → mitophagy (autophagy and proteosomal degradation)
What happens if PINK1 is damaged?
stabilize on damaged mitochondria → phosphorylate ubiquitin → inactive parkin transferred to the outer membrane → parkin activated
*PINK1 is ubiquitin kinase
Parkinson’s (symptoms)
stooped posture
back rigidity
flexed elbows and wrists
tremors in the legs
shuffling, short steps (obvious sign)
slightly flexed hips and knees
hand tremor (uncontrolled movements)
reduced arm swing
forward tilt of trunk
masked face
*motor functions affected most significantly (Ex. Michael J Fox)
Parkinson’s (non-motor skill symptoms)
mental/behavioral issues (depression, anxiety, fatigue, personality changes, etc.)
sense of smell
sweating and melanoma
GI issues (sexual, urinary, weight loss)
pain
Parkinson’s (motor skill symptoms)
vocal symptoms
rigidity
tremors
walking difficulties
dystonia (repetitive movements make body parts twist)
bradykinesia (mask-like face)
Parkinson’s (RFs)
toxins
genetic factors
pesticide exposure
water pollutants
air pollutants
aging
family history
male
chemicals
hydrocarbon solvent exposure
*thought as disease of old age, lifetime exposure to these factors increase risk of diagnosis (exact mechanism unknown)
Rotenone
*pesticide → mitochondrial complex I inhibitor (activity decreased in many cases of idiopathic PD)
produces Parkinsonism in rats
What did the epidemiological and animal studies show?
associations w/ altered risk → farming (herbicides, pesticides), welding (Fe, Mg), well water (toxins), smoking, coffee, etc.
if you could reproduce a diseased model w/ some RF, there is an association
*smokers had a lower risk of being diagnosed w/ Parkinson’s
Potential role of environmental factors (Parkinson’s)
MPTP metabolized to MPP+
taken up through dopamine transporter
affected mitochondria = mitochondrial defect
disrupts complex I of ETC
neuronal cell death (RF)
*took an analgesic and tested it in animals → caused Parkinson’s-like disease
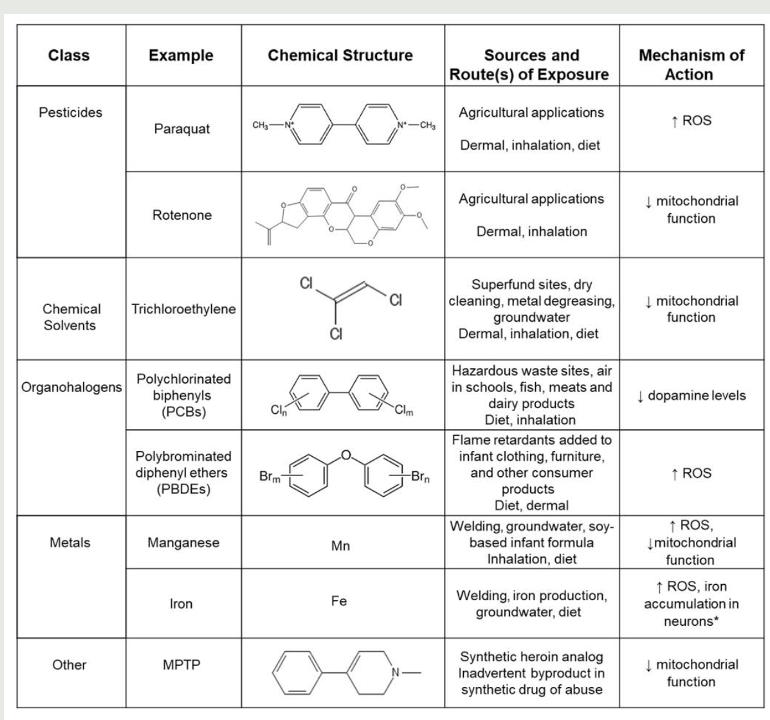
chemicals linked to Parkinson’s → disruption of mitochondrial function
treatment - dopamine agonists (bring levels close to physiological) → manage systems (no cure)
Positive association (Parkinson’s disease risk)
medication
coffee
plasma urate levels
exercise
cigarettes
Negative association (Parkinson’s disease risk)
heavy metals
solvents
TBI
psychological state
dairy consumption
pesticides
medication
Dopamine-producing neurons
cells primarily affected by mitochondrial disfunction (in basal ganglia → substantia nigra)
produce dopamine → neurotransmitter for movement control (motor symptoms of disease)
*maximum loss of function
How do astrocytes relate to PD?
mitochondrial disfunction of supporting cells of brain also play a role in PD progression
*glial cells also affected
Familial PD
associated w/ several genetic mutations linked to mitochondrial disfunction
PINK1 and Parkin → important for segregating damaged mitochondria and recreating healthy mitochondria

increased damage

decreased removal
What are key features of PD?
dopamine neuron degeneration in midbrain
lewy bodies (protein aggregates) in neurons
*protein misfolding
What are lewy bodies made of?
alpha-synuclein, ubiquitin, synphilin-1, neurofilaments, amyloid-beta, tau
*alpha-synuclein thought to be primary protein responsible; amyloid-beta and tau used to be unique to Alzheimers
What proteins regulate mitophagy?
LRRK2, PINK1/parkin, alpha-synuclein, DJ-1
Pattern of inheritance
can be autosomal dominant or recessive
*onset is important for cost of treatment (chronic/early = more important) → finding therapeutic targets to prevent initiation of disease
What are the consequences of mitochondrial disfunction in PD?
leads to progressive cellular disfunction = neurodegeneration
impairment of mitochondrial biogenesis
increased ROS
defective mitophagy
compromised trafficking
ETC disfunction
variations to mito dynamics
Ca2+ imbalance
*alone or in combination
Impaired biogenesis (mutations)
alpha-synuclein, CHCHD2, parkin, PINK1
Ca2+ imbalance (mutations)
alpha-synuclein, PINK1
Altered mitochondrial dynamics (mutations)
VPS35, CHCHD2, Parkin, PINK1, ATP13A2
ETC disfunction (mutations)
LRRK2, VPS35, CHCHD2, parkin, PINK1
Impaired mitochondrial trafficking
LRRK2, PINK1, parkin
Defective mitophagy (mutations)
parkin, PINK1, ATP13A2 (lysosomal protein)
Oxidative stress
alpha-synuclein, LRRK2, CHCHD2, VPS35, parkin, PINK1, ATP13A2
What happens in PD?
Ca2+ buffering is defected, metabolism is defected, and inflammation is increased
Astrocyte function
#s and functions most abundant in striatum/caudate putanem → where dopaminergic neurons is highest/loss of astrocytic function has most detrimental effect on PD
How is astrocyte disfunction treated?
reversal of mitochondrial dysfunction in the striatum → significant therapeutic benefit
GSDMD pores
protein-formed channels in cell membranes involved in inflammation and cell death
release pro-inflammatory cytokines → release of IL-1B
Microglia
involved in PD
skewed towards fission → activate inflammasomes (cause inflammatory disease)
activate innate response and increase neuronal inflammation
Inflammasome
accumulation of proteins that form a super complex of proteins
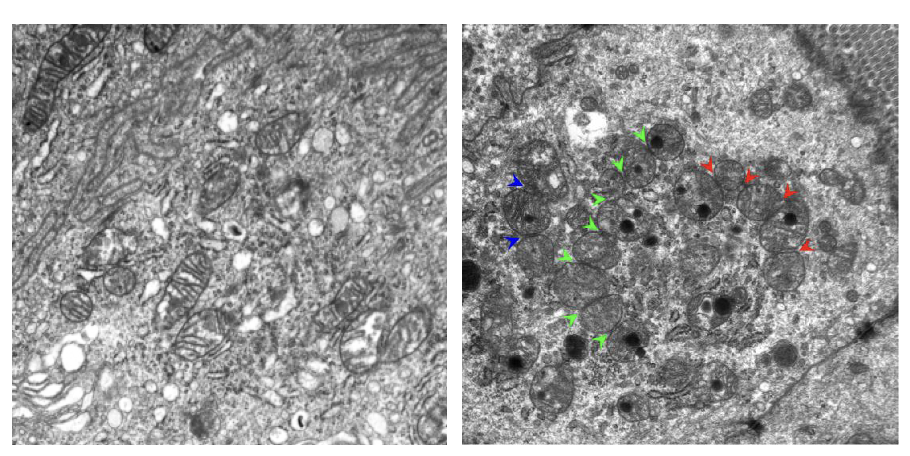
multiple mito fission events in enterocytes from small bowel of DKO mice
2 genes (mitochondrial defects) involved in etiology of Chron’s → increased fission
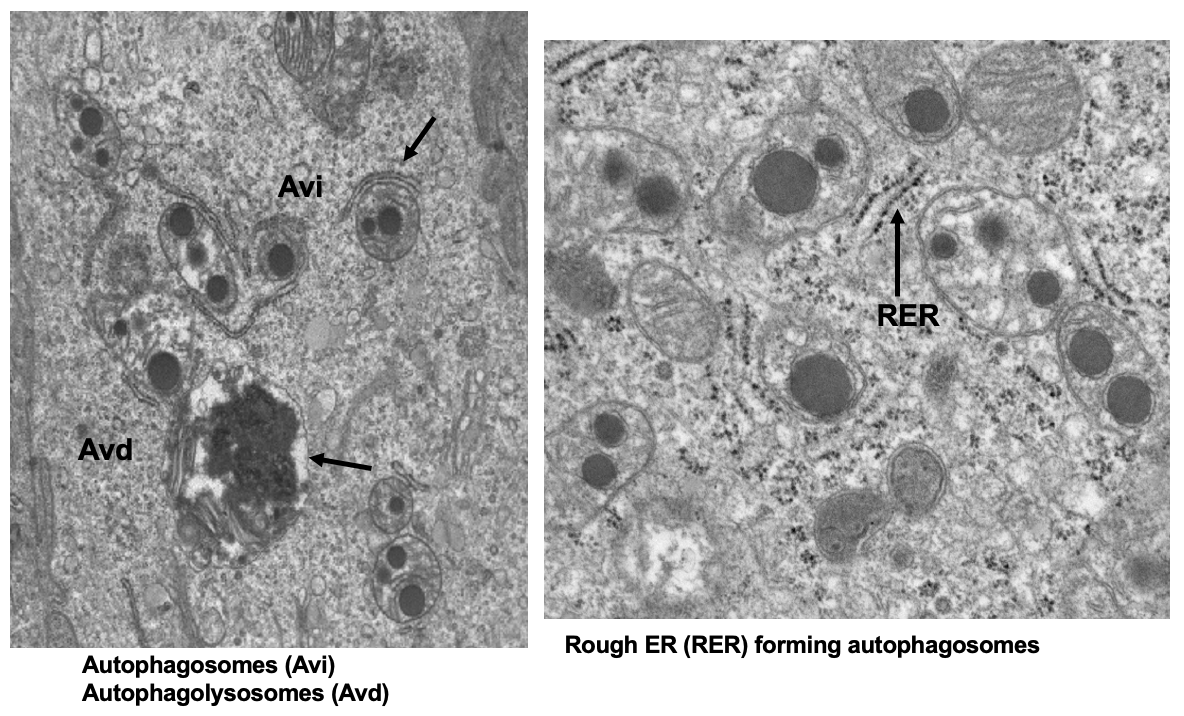
abnormal mitochondria in autophagosomes (Avi) and autophagolysosomes (Avd) in SI of DKO mice
double membrane shows formation of phagophore
lipids aggregate inside of mitochondria
isolation membrane forms around mitochondria → form Avi → fuses w/ lysosome (Avd)
*process of autophagy but it doesn’t get cleared out
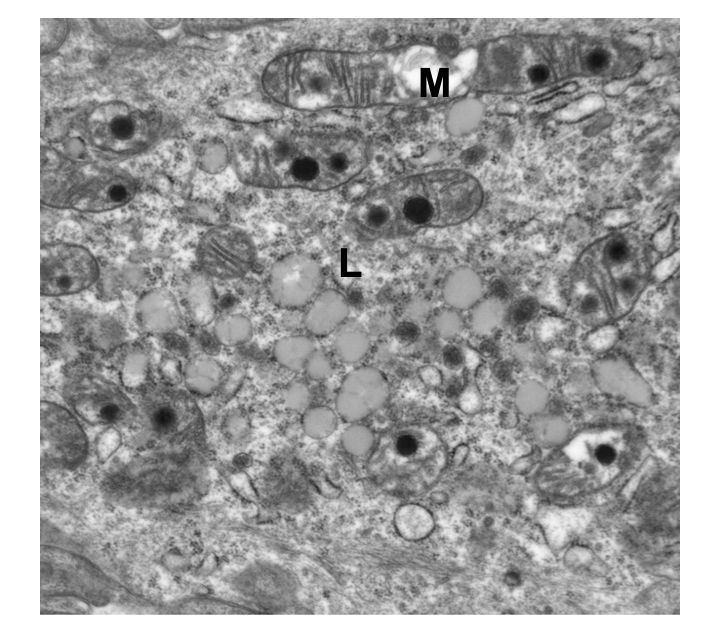
DKO mice show abnormal mitochondria (M) and accumulation of lipofuscin (ceroid; L)
lipid droplets (beta-oxidation disrupted) → mitochondrial pathway can’t clear out fat (mitophagy is disrupted)
mitochondria are rounded = dividing more
*translucent
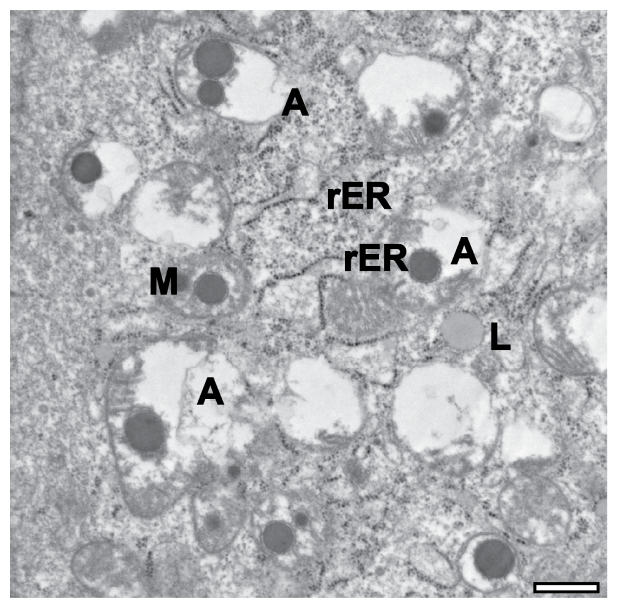
mitochondria undergoing autophagy
seeing a lot of vacuoles → the cell has died
phagophore (rER), autophagic vacuoles (A), lipofuscin (L)
What pathways coordinate metabolism and regulate lifespan?
AMPK, insulin/IGF signaling, mTOR signaling
*responses are highly context-dependent/not uniform (Ex. during exercise, increased energy needs → fission)
What happens during starvation?
AMPK activated
insulin/IGF, mTOR inhibited
favors fusion → longevity, insulin sensitivity, glucose tolerance
What happens during nutrient excess?
activates insulin/IGF, mTOR
AMPK repressed
favors fission → premature aging, cardiomyopathy, obesity, diabetes
Fusion and fission in cancer
regulates metastasis, migration, apoptosis, autophagy, proliferation, metabolism
fission increases metastasis/proliferation in cancer cells
decreased fission inhibits apoptosis
increased fission increases OXPHOS
What are mitochondrial defects associated with?
Alzheimer’s, NAFLD, cardiac ischemia/myopathy, PD
*not unique to PD (fission/fusion)