Transcriptomics
1/47
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
48 Terms
Transcriptome definition
Complete set of transcripts in a cell, or a population of cells, for a specific development stage or physiological condition
Transcriptome
RNA content in the cell
Includes IncRNA, miRNA and circular RNA
Includes tRNA and rRNA but not often as they’re housekeeping genes
Genome is the same in every cell in the organism
Transcriptome and Cancer
In cancer, gene expression can be used to identify the evolution of cancer
If a patient is asymptomatic, their transcriptome can be checked for biomarkers of a developing cancer to use as a diagnosis
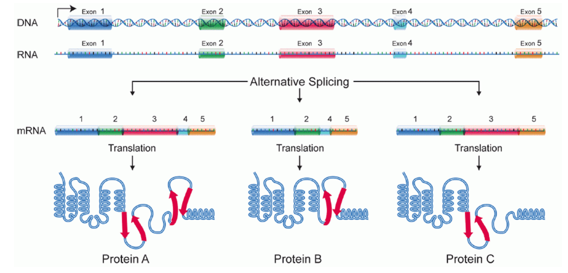
Alternative splicing
A sequence of exons doesn’t always correspond to the final protein made
A few different variations can be formed if the gene comprises of more than one exon
e.g. If exons are emitted from the final sequence or rearranged
The final proteins of different exons can have slightly different functions too
Mechanisms: Spliceosome
Structure made of RNA
Recognise splicing sites
Spliceosome directs which portion of the gene is being considered and spliced out
Two main types - major and minor
These snRNPs have different functions within spliceosome activity
Transesterification
Major type
snRNPs U1, U2, U4, U55, U6
Minor types
snRNPs U11, U12, U4atac, U5, U6atac
snRNPs
Small nuclear ribonucleoproteins
Transesterification
Introns are removed from an immature mRNA (pre-mRNA) by transesterification
Mature mRNA molecule is made
Guanine and adenine base are bonded by transesterification
Hydroxyl (OH) group on the carbon atom of the adenine attacks the bond of the guanine nucleotide at the splice site
Spliceosome example
Peromyscus mice
Agouti gene gives whiter colouration
It binds to melanocortin cell receptors, which is involved in regulating pheomelanin pigment synthesis (gives reddish pigments)
In the belly, there is normal agouti genes
On their back, the cells have a different transcription type that contains the 1D exon, which has a lot of start codons
This reduces the efficiency of translation into a function agouti protein
Lamins example
Class of genes encoding lamins are important for normal development
Exon and intron mutations can alter splicing which causes premature stop codons and frameshifts
Point mutations cause stop codons
These can cause muscular dystrophy diseases as the truncated short protein doesn’t fully perform its function
Spliceosome therapies
Certain therapies develop enhancers and silencers so splicing doesn’t happen int he same way
To get different proteins or administer protein to patient and stop splicing happening so ‘bad’ protein doesn’t appear
Long non-coding RNAs (IncRNA)
Transcribed by polymerase I
Often spliced, sometimes capped and/or polyadenylated
Appear and behave like mRNA
Don’t code for proteins
IncRNA functions
Act as a guide
Interaction with a whole protein complex
Transcription regulation
MALAT-1 interacts with serin-rich proteins
Stabilise mRNA
Act as a decoy
IncRNA function: Act as a guide
Inc-DC stabilises STAT3 protein through phosphorylation
The protein is better suited to enter the nucleus and target specific genes
Which indirectly affects gene expression
IncRNA function: Interaction with a whole protein complex
RNA attaches to it
Changes gene conformation, enhancers and markers
Therefore, changes expression
IncRNA function: Transcription regulation
IncRNACONCR activates DDX1
This phosphorylates cohesion
Cohesion is important for DNA, as it keeps strands together during cell cycle
IncRNA function: MALAT-1 interacts with serin-rich proteins
Enhances splicing
Facilitates it for the particular isoform of exon the cell requires
IncRNA function: Can stabilise mRNA
Leads to amyloid-peptides accumulation which can cause Alzheimer’s disease
IncRNA function: Act as a decoy
GAS5 IncRNA acts as a decoy for GR transcription factor, inhibiting apoptosis
miRNAs
Short RNAs that regulate gene expression
Original transcript is pri-miRNA
Association between miRNA expression and cardio-vascular diseases finds cancer is quite frequent
However, the exact role is not known – possible that miRNAs are biomarkers
miRNAs can be used in gene therapy by targeting transcripts associated with disease
pri-miRNA
73 bases long
pri-miRNA is cleaved twice to get rid of the loop structure and forms miRNA (20-25 bp)
In this process, the passenger strand is unimportant and degraded
The complex of proteins binds to active miRNA and form a RISC complex
The RISC complex binds to mRNA to repress translation or degradation of that mRNA as it may no longer need to be degraded
Repress gene expression
Dependent on how well it matches mRNA - perfect match for miRNA/mRNA or partial match for miRNA/mRNA
Perfect match for miRNA/mRNA
Deadenylation - followed by decapping and degradation
Proteolysis - degradation of nascent peptide
Partial match for miRNA/mRNA
Initiation block - repressed cap recognition or 60S joining
Elongation block - slowed elongation or ribosome drop-off
Circular RNAs
No longer functional RNA
Formed by back-splicing
Act as decoys more efficiently than incRNA
miRNA can degrade RNA when it shouldn’t be so it needs to quickly be soaked up so it doesn’t degrade useful RNA
Circular RNAs decoy example
The regulator never reaches the nucleus (and therefore, the target) as it becomes bound to circular RNA
Epitranscriptomics
Emerging field that combines epigenetics and transcriptomics because RNA can be modified the same way DNA can be modified by markers, readers and writers
Most common modification is methylation of 6 adenosine that happens after transcription, and causes a writer
Methylation can cause alternative splicing, which can be involved in structural changes (e.g. formation of loop where there wasn’t one before)
m6A methylation
Associated with myD88 alternative splicing
MyD88 interacts with Toll-like receptors
MyD88S inhibits inflammatory response through NF-KB response pathway
A decrease of m6A methylation to MyD88 will cause enhanced inhibition of signalling pathway
m6A methylation in celiac disease
Excessive inflammation
It was found that individuals with the allele associated with celiac disease have more m6A methylation
cDNA microarrays
First high-throughput method for transcriptomics
Tag cDNA with fluorescent tags and it will emit light
Brightness and duration - approximate how much in sample
High-throughput sequencing: Illumina
Dominant type of next-generation sequencing and most accurate sequencing
Limit of sequencing length so new ones have been made e.g. PacBio, oxford nanopore
Transcriptome is easy and cost-effective as genes there are less genes and they are closer/easier to find
After DNA purification in Illumina, they need to be fragmented because Illumina has a limit to how many bases it can read
Paired-end sequencing gives more accuracy by sequencing the forward and reverse strands at the sample amplified spot
Parallel sequencing
Assembling a transcriptome
For humans, there will be a reference genome
If there is no reference genome, it will be assembled de novo
The shorter reads will be assembled and overlayed into a consensus sequence
Once genes have been mapped to reference, gene expression can be compared between different treatments
However, RNA is not stable and needs to undergo library preparation
They’re mapped onto exons
Need at least 3 samples to draw conclusions about transcriptome
Library preparation
cDNA is shattered into fragments using an ultrasound
The ends are sequenced and onto map reads
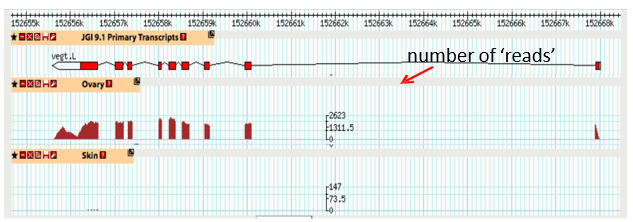
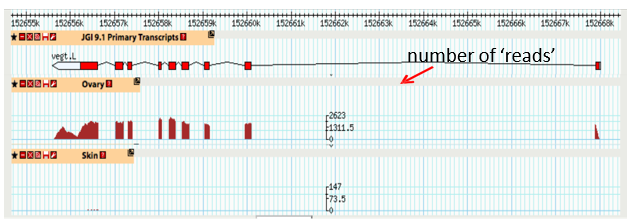
RNAseq output for a single gene
When comparing tissue types, must consider the amount of genes that may be expressed as it can change between tissue types
Two types of graphs can be made - volcano plot and heat map
In the graph, Xenopus VegT is expressed in the ovary, but not the skin, as it is mainly expressed in the egg
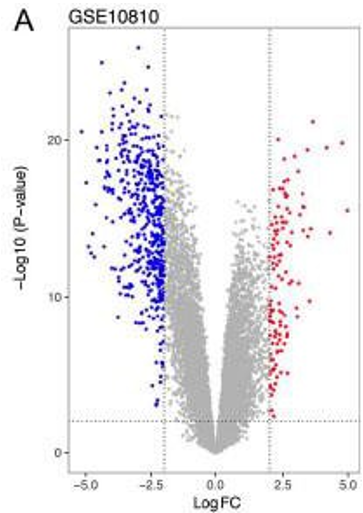
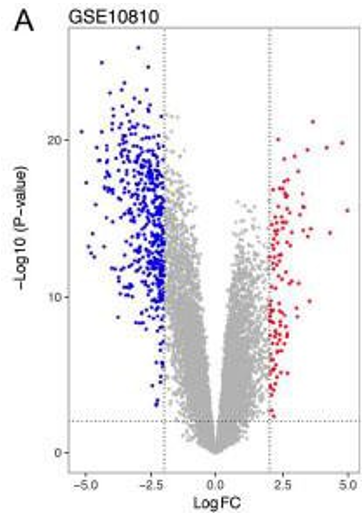
Volcano plot
The x-axis of LogFC shows how much more of the gene is expressed than a base level expression
The y-axis shows how significant it is
Blue is under expressed
Red is overexpressed
The highest gene on the graph may be picked as a candidate gene
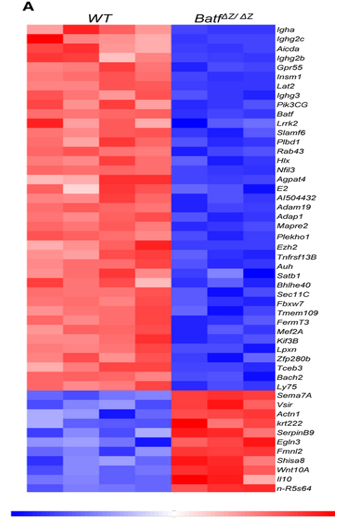
Heat map
Used to compare two different conditions – wild type and individuals with the condition
Intensity of colour symbolises how much they’re expressed
There are several columns to show the different times the experiment has been replicated
ENCODE project
DNA elements encyclopaedia for human genome and mouse genome
To make a very comprehensive catalogue of transcripts
Map sequence reads to the genome sequence to identify the genes, exons and transcriptional start sites
ENCODE project problem
Cell specific expression
ENCODE project solution
Repeat for 15 different cell types
ENCODE project techniques
CAGE
RNA-PET
CAGE
Selects RNAs for the methylated 5’ cap which defines the start of transcription
RNA-PET
Selects 5’ cap and 3’ polyA together which gives full length RNA
Deriving transcriptomes for different cell types
Must prep as the transcriptome is only as specific as the cell preparation
Single cell transcriptomics - brain, different brain regions and cells
Individual dissociated cells can be sorted by microfluidics
Can examine cell differentiation dynamics
RNA-seq can identify genes that have just started to be expressed, is in stable expression and are turning off
Single cell transcriptomic: Brain
Extract RNA and sequence, then you get the average transcriptome of the brain
Single cell transcriptomic: Different brain regiond
Isolate specific brain regions
Extract RNA
Sequence
Get average temporal lobe transcriptome
Single cell transcriptomic: Cells
Isolate single cells from a specific region
Extract RNA
Sequence
Get the precise temporal lobe cell transcriptome
Individual dissociated cells can be sorted by microfluidics
Dissociate cells
Place into individual aqueous droplets containing sequencing reagents in an oil medium
Add a bead containing sequencing primers with a barcode that is unique to that droplet
Anneal and make cDNA
High-throughput sequence all the droplets together
Sort the sequences to each cell by barcode
Devise mathematic processes to analyse the data