3D Structures of Proteins
1/35
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
36 Terms
Structure Hierarchy
primary: chemical structure
secondary: local structural elements
tertiary: long-range interactions overall fold single polypeptide chain
quaternary: interactions between polypeptide chains
Backbone Configuration
peptide bond can be cis or trans with cis being the most common (xxx-Pro)
the plane of each peptide bond shares one alpha carbon with the next peptide bond plane
rotation of the psi and phi angles changes the relative position of the amide planes → some configurations are NOT possible
this causes steric hindrance (BAD)
peptide bonds to gly or pro residues are sometimes found in unallowed regions due to the small size of gly side chain and ring structure of pro
Secondary Structure Elements
alpha helis
beta sheet
beta turn
loops
alpha-Helix Structure by Linus Pauling
right handed helix with 3.6 residues per turn
pitch of 5.4 angstroms per turn
held together by H bonds between C=0 residue n to NH of residue n+4
helix propensity is the tendency of certain amino acids to occur more frequently in alpha-helices
amino acid side chains point outward from the axis
proline, lots of charged residues, and lots of bulky residues are bad
Beta Sheets
two or more separate strands of polypeptide H bonded together through peptide bond groups
could be parallel (not straight H bonds) or antiparallel (straight H bonds) orientations, could be mixed
strands are not flat but puckered and slightly twisted to the right as a beta pleated sheet
beta bulges happen when residues don’t line up
turns can occur between strands
strands in a beta sheet are connected by tight turns (beta-turns) or large loops
beta turns always have pro at position 2 and connect anti-parallel beta strands
type 2 beta turns always have gly at position 3
Sequence and Secondary Structure
pro rarely occurs in secondary structure
different residues have different helical propensities
side chain interactions can disrupt or distort secondary structure
Sequence Affects Helix Stability
not all polypeptide sequences adopt alpha-helical structures
small hydrophobic residues like Ala and Leu are strong helix formers
pro is a helix breaker because can’t rotate around N-Ca bond
gly is a helix breaker because the small R-group supports other conformations (too much free rotation)
Circular Dichroism Analysis
measure molar absorption difference of left and right circularly polarized light
peptide bond in each secondary structure has characteristic CD spectrum
chromophore in chiral environment make characteristic signals
spectrum can be deconvoluted to give a measure of %helix, % sheet, and % coil
Protein Tertiary Structure
overall spatial arrangement of atoms in a polypeptide chain or protein
two major classes:
fibrous proteins: typically insoluble, made from a single secondary structure long extended structures, many polypeptides, often structural
globular proteins: water-soluble, lipid-soluble membrane proteins, roughly spherical in shape, polypeptide folds back on itself, enzymes
Fibrous Proteins
much or most of polypeptide chain is approximately parallel to a single axis
often mechanically strong
usually insoluble in water
structural role
Alpha Keratin
helical rod segments capped with non-helical N and C termini made of coiled coil of 2 alpha helices, left handed twist, 3.5 residues per turn, primary structure of rods has 7-residue repeats
intra and inter-strand ionic interactions stabilize coiled coil
coiled coils are frequently seen in the dimerization domains of transcription factors
Silk Fibroin
main protein in silk, antiparallel beta sheet structure with small side chains of ala and gly allowing close packing
stabilized by H bonding within sheets, LDFs between sheets, hydrophobic interactions between ala sidechains
Collagen
made of 15-30% gly, pro, hydroxypro, and hydroxylys
strands crosslinked at N and C termini
many repeats of Gly - X - Y where X is pro and Y is 4hydroxypro - increases stability of collagen by increasing electrostatic interactions
gly occurs where chains cross by its NH H bonding to X carbonyl in adjacent strand
L-handed helix with 3 residues per turn
no intra-chain H bonds
R-handed superhelix
Globular Tertiary Structure
form wherever possible since big numbers of H bonds
helices and sheets often pack close together
backbone links between elements of secondary structure are usually short and direct
proteins fold to make the most stable structures, make H bonds, minimize solvent contact of hydrophobic residues
secondary elements themselves not usually active → must fold to become active
Globular Proteins
design principles:
most polar residues face outside of protein + interact with solve
exceptions: membrane-spanning portion of membrane proteins, subunit interfaces (hydrophobic residues face interior and interact with each other)
random coil is not random
structures are not static (various elements and domains of protein move to different degrees)
some parts are flexible and disordered
empty space exists in form of small cavities
Structural Families
all alpha
all beta
a + b secondary structure elements separate in sequence, antiparallel beta strands
a/b mixed in the sequences, parallel strands in beta sheet
Motion in Globular Proteins
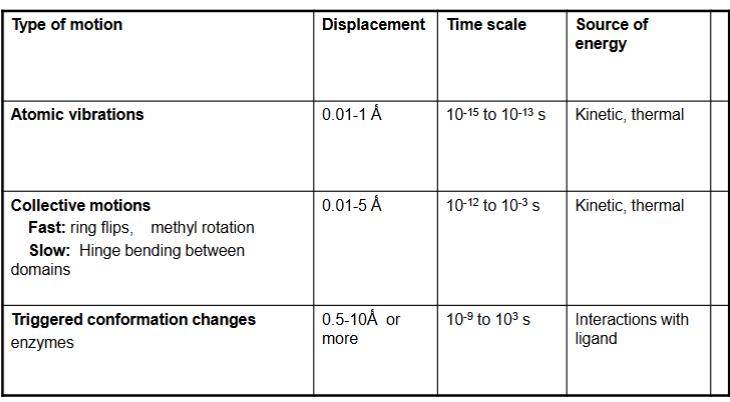
Repeated Structural Units in Proteins
motifs, small parts of proteins
Rossman fold - nucleotide binding domain
domains, independently folded regions of structure that are functional when removed from larger protein
Rossman Fold
motif found in nucleotide binding domains
gly-x-gly-x-x-gly in ADP, NAD
gly-x-gly-x-x-ala in NADP
Other Structural Motifs
greek key
helix turn helix
beta turns
beta bulges
Domains
SH2 domains are found in proteins of the tyr kinase signaling cascade - binds phosphorylated tyr residues
Kringle Domains
around 80 amino acid looped domains with 3 disulfide bonds per domain
found in proteases of the blood clotting cascade and growth factors
2 families, one without 3 disulfides in ECM
Proteins with similar function…
may not have similar sequences but they will have similar structures
Quaternary Structure
interactions are specific and reproducible and increase stability of subunit
active site is at subunit interface
cooperativity of substrate improves binding efficiency and regulates activity
distal binding site for activators and enhancers increase sensitivity to such molecules
each chain in a homo oligomer is coded for by a different gene, if one is wrong other compensates
one gene has many subunits (genetic efficiency)
Protein Stability tertiary and quaternary Structure
electrostatic forces (ion-ion, close range forces, salt bridges)
van der waals (between permanent and induced dipoles, LDFs)
H bonds
hydrophobic interactions
What holds proteins together?
mostly the hydrophobic effect - the greasy stuff stays on the inside
hydrophobic side chains get buried in layers like a cake
H bonds that are buried in the protein (30-40%)
buried salt bridges (smaller contribution)
Interior of the Protein
microenvironments affect properties of amino acids
charged side chains buried in hydrophobic pockets stay uncharged - increase pKa for (-) and decrease for (+)
buried salt bridge residues remain charged - decrease pKa for (-) and increase for (+)
Mutations
conservative mutations cause smaller changes except when at a critical location
mutations in surface residues cause smaller changes except if amino acid is part of interactions
nonconservative mutations cause more drastic changes except hydrophilic on outside
Methods for Determining Protein Structure
X-ray crystallography
NMR spectroscopy
CD
cryo electron microscopy
modelling
Protein Denaturation
native structure = biologically relevant 3D structure
denaturation = disrupting forces that hold native structure
CD spec
activity assays
NMR spec
can be denatured by:
heat or cold
pH extremes
organic solvents
chaotropic agents: urea and guanidinium hydrochloride
Ribonuclease Refolding Experiment
ribonuclease has 8 cysteines linked via disulfide bonds
sequence alone determines native conformation when urea and 2-mercaptoethanol are removed slowly
Protein Folding
levinthal’s paradox: if a polypeptide searches all possible conformational space to find the lowest E state it will take a very long time
protein retains partially folded intermediates called nucleation condensation model
folding is hierarchical
hydrophobic collapse → secondary struc → tertiary struc → final conformation
protein disulfide isomerase: enzye reduces disulfides then helps reform new disulfide bonds
molecular chaperones: heat shock proteins, groEL, groES
GroEL and GroES - protein folding cages
each chamber has 7 subunits of groEL protein
cap is made of 7 subunits of groES protein
ATP binds groEL → misfolded portein binds to opening of chamber → chamber is capped up, ATP is hydrolyzed → ATP and a new substrate protein bind to other side → first protein and ADP are released
Alzheimer’s
normal folded peptide is in brain as helical bundle
one unfolding → others get beta sheet conformation → plaques in the brain
p53 Tumour Supressor
mutations in core of protein destabilize structure and make it unfold
mutants may be unable to bind to DNA/get fully unfolded
new target is small molecules that will stabilize the structure of the protein
Cystic Fibrosis
deletion of phe 508
destabilizes overall protein structure → misfolded protein degrades on ER
phe residues is important in stabilizing interdomain interactions via hydrophobic interactions