BCH4024 Exam 2 Nitrogen Metabolism
1/132
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
133 Terms
primary metabolites
necessity for normal operation of metabolic pathways and main cellular functions
ie Amino acids, nucleotides, RNA, DNA, B vitamins
secondary metabolites
not needed for cell growth, development, or reproductions
ie alkaloids in plants
amino acid sources
dietary proteins, endogenous protein turnover, and de novo synthesis
dietary proteins
supply essential and non essential amino acids, humans can only make 10
intracellular proteolysis
removes misfolded, old and damaged proteins, regulates cell metabolism and important in cell cycle transitions, also known as endogenous protein turnover
de novo synthesis
needed to adjust AA levels within tissues and for protein synthesis; able to synthesize specialized AAs, controls leval of central pathway metabolites
Provides nutritionally nonessential amino acids that are needed for protein synthesis Adjusts amino acid pools in different tissues Adjusts energy metabolism by controlling concentrations of central pathway metabolitesAllows cells to adapt to metabolic stress Needed to make nucleotides, heme, hormones, as well as neurotransmitters.
essential AAs
histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine
PVTTIMHLL; variety needed in diet!
conditionally essential AAs
arginine in pregnant women/children
tyrosine when Phe is low
Cystine when Met is low
intracellular proteolysis
breaking down and building new proteins in cells; not at a constant rate, Low Nitrogen intake leads to increased of this; 3 pathways are lysosomal, proteasome, and autophagic
lysosome pathway
proteins protonated in lysosome with low acidic pH and undergo partial unfolding making them susceptible to proteolysis
proteasome pathway
protein ubiquitin marks unfolded proteins for breakdown, only ubiquinated proteins can enter proteasomes, barrel like macromolecule strcutruers that use proteases to form small peptides and AAs
autophagic pathway
removes old organelles ie mitochondria/ribosomes via engulfing in vacuole using ubiquitin+ lysosomes
ubiquitination
post translational modification that marks proteins using 3 enzymes, E1/E2/E3, initiating conjugating and ligase,
mono and poly; mono adds to linked molecules and poly adds to N terminus of ubiquitin
digestion
mechanical and enzymatic within GI; HYDROLYTIC process as water cleaves functional groups; biopolymers myst be broken down into monomers so done so via this route;
enzymes of this are produced/stored in pancreas and stomach and neurohormonally controlled
proteolysis
enzymatic cleaves of proteins; 2 steps by breaking protein into fragments and then into AAs and di/tri peptides
protein fragment proteolysis
occurs in small intestine at neutral pH where protein fragments can not refold; done by chymotrypsin, trypsin, carboxypeptidase, and elastase- cleaves in CT
protein proteolysis
saliva- proteases from bacteria/WBCs
stomach- acidic pepsin
zymogen
protease enzymes as inactive precursors made/stored in stomach/pancreas which secretes into small intestine; safer to store and prevents autophagy and apoptosis
ie pepsinogen, chymotrypinogen, trypsinogen, pro-carboxypeptidase
pepsinogen
pepsin, stomach (gastric chief cells), ph 1-3; autocatalytics, 2 active site aspartyl residues use to mediate low pH catalyssis
gastric chief cells also excrete chymosin, an acid protease that speeds up pepsin cleavage by curdling milk
chymotrypsinogen
active trypsin cleaves this then also pi chymotrypsin+ remaining trypsinogen to activate sequential proteolysis
trypsinogen
made in pancreas and stores in vesicles, TI keeps in inactive form, in small intestine TI is diluted so enterokinase activates this to turn into trypsin
trypsin is used for activation of chymotrypsionogen
enterokinase
ectoprotease on intestinal mucosal wall
pro-carboxypeptidase
cleaved by trypsin
AA transporters
symporter imports AAs/ sodium into intestinal brush cells against AA gradient; driven by transmembrane ion gradient because of high sodium content in intestine lumen and low sodiium on brush border cells
sodium gradient maintained by sodium potassium ATPase; maintains chemiosmotic concentration gradient
chemi= using energy of ATP hydrolysis
osmotic= imbalance of ions on different sides
true nitrogen balance
intake=excretion; de novo synthesis important in maintaining N balance, different across orgnas
positive nitrogen balance
intake exceeds excretions; required for growth in children/pregnancy, wound healing and convalescence
negative nitrogen balance
intake less than excretion; starvation, malnutrition, disease ie burns, trauma surgery
marasmus
Protein energy malfunction ie PEM from inadequate protein and calorie intake, deficency of all nutrients; dry skin and loose folds of skin hanging over butt,
SUPER SKINNY
kwashiorkor
protein malnutrition but getting carbs so fat; irritability, enlarged liver, edema, osmotic imbalance
big stomach, washing of african fruit to lose nutrients
transamination
conversion of keto-acids and AAs through loss of alpha amino group via double displacement by transaminases; alpha keto acids essential for metabolism in right quantiy ie not too much or too little
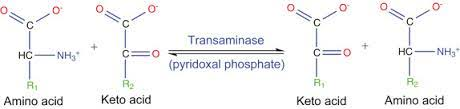
transaminases
requires cofactors PLP derived from vitamin B6 and bind extremely tight; PLP converted to PMP
Pyridoxal phosphate/PLP
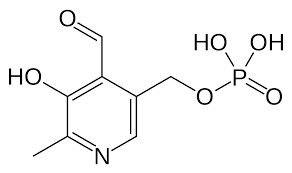
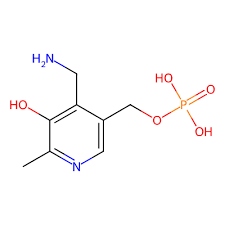
pyridoxamine phosphate/pmp
pyruvate
alpha keto acid of alanine
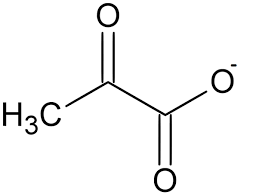
oxaloacetate
alpha keto acid of ASP
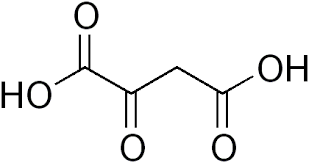
alpha ketoglutarate
alpha keto acid of glutamate
transamination mechanism
2 half reactions, half 1: formation of aldimine, then conversion to ketimine which is hydrolyzed to ketoacid ;half 2: occurs in reverse with R2
fullreversible
AAs that do not undergo transamination
proline/hydroxyproline: secondary amines no transamination
lysine: would cyclize into toxic non metabolite
threonine: dimerize into toxic non metabolite
transaminases cannot bind lysine or threonine
oxidative deamination
done by glutamate dehydrogenase GDH to regenerate alpha-ketoglutarate, the amino acceptor and prodduces ammonia for reutilization or disposal; located in mitochondrial matrix
GDH mechanism
amine oxidized to imine with NAD+ reduction, imine then hydrolyzed to keto acid; NADH formed as well
transaminase and deamination can be coupled to oxidative degrade 14 AAs
NAD in GDH
converted to NADH; creates large energy storage release if C-N bond broke, enough energy to make 3 ATP; nature’s batteries; eaction runs on dependence of availability of NAD/NADPH, NO NADH OR NADP+
when high levels of NAD+ mitochondria favor oxidative deamination and GDH catalyzes oxidation; makes atp
when high levels of NADPH, favor reductive amination, GDH catalyzes reduction; used to make fatty acids and sterols
GDH products
a-KA enters citric acid cycle, NH4+ to urea cycle, NADH reoxidized to NAD by oxidative phosphorylation ie making ATP;
GDH activity
dependent on Energy state of cell and available AAs ie allosteric control; when
HIGH ATP/GTP/NADH; GDH IS INHBITED, so low rate of AA degradation and high protein synthesis, deamination favored at lower pH
HIGH ADP/ GDP/ FREE AAs, GDH is highly active, a-KA stimulates TCA to make ATP, fuels ETS and oxidative phosphorylation restoring energy and amination favored at HIGH pH
routes of Deamination
GDH main system for removing ammonia from AAs
can also be hydrolytic or eliminative
L/D AA oxidases
similar to GDH ie converts AA to a-KA; H2O2 produced and flavin mononucleotides used as cofactors; humans make both L and D
D AAs found in damaged protein and old, dry food
D- AA oxidase has antibacterial effect by preventing accumulation of D AAs
hydrolytic deamination
glutaminase- widely dsitributed in body in mitochondria; ammonia enters urea cycle, also done by
asparaginase
eliminative deamination
histidinase or histidine ammonia lysae
ammonia assimilation
incorporating of ammonia into metabolic form, toxic at high concentration so must be kept at 5-10 micromolar; free ammonia can cross membranes and dissipate proton gradient
free ammonia depletes a-Kglutatarate inhibiting TCA activity
high ammonia in mitochondria of liver because urea is produced here
ammonia assimilation pathways
GDH, Glutamine synthetase, carbamoyl synthetase, glycine synthetase
GDH in ammonia
reductive amination, reverse of oxidative amination reaction, no effect on NAD/NADH
NADPH + NH3 + α-KG → Glutamate + H2 O + NADP+
glutamine synthetase
main way of trapping ammonia, GLN serves as major N donor in biosynthetic reactions and is major inter-organ N shuttle; doesnt alter blood pH; allows for production of ammonia in situ which is then used as Nuc
Glutamine synthetase mechanism
double displacement via 2 SN2 rxns;
GLE phosphorylated by ATP to give gamma glutamyl P intermediate which reacts with NH3 to produce GLN
pH less than pka so mostly NH4+; deprotonation reaction driven by free energy of ATP ie Delta G = +10
Futile cycle prevention
keeping enzymes in separate cell compartments ie GLN synthetase and glutaminase; synthetase only in cytoplasm where glutaminase is in mitochondria
Carbamoyl Phosphatase Synthetase-I/CPS-1
main reaction for ammonia assimilation in mitochondria; bicarbonate phosphorylated by ATP to give C-Pi intermediate which reacts with NH3 to produce to Carbamate which is then phosphorylated
requires 2 ATP; associated with urea cycle
Carbamoyl Phosphatase Synthetase-II/CPS-2
GLN dependent enzymel GLN used as N source; occurs in cytosol using GLN and aprotic transfer tunnel allows for free NH3 with no energetic cost to deprotonate
CPS-2 mechanism
GLN- hydrolyzing subunit initially filled with water which is displaced by GLN, water is w5 so too big; hydrolyzes GLN to release NH3 into tunnel while a gamma thioester is formed and acts as lid to block proton entry
ammonia moved unprotonated through tunnel between active sites where it goes to react with intermediate
GLN dependent enzyme reactions have low michaelins constant for GLN, high affinity
liver cells
show division of labor ie carry out different metabolic tasks in different organelles or subcellular locations
metabolite pool- cells make and store metabolites where needed, pool is reservoir of metabolite; carbamoyl P has 2 destinies in liver
urea cycle basics
form in which N is excreted, varies between species ie
fish excrete NH3- ammonotelic
Birds excrete uric acid- uricotelic
mammals excrete urea- ureotelic
90-95% of N excreted in urea form; excretion depends on daily N intake in dietl some excretion via creatinine and uric acid; gout= excess uric acid
urea cycle
occurs almmost exclusively in liver ie principal NH3 detoxification site, N transported between organs as ALN, GLE, GLN, urea; enzymes in cytosol and mitochondria; this cycle liberates considerable acid helping control body pH
urea cycle mechanism
ornithine+CP1→ citruline
citruline+ aspartate→ arginosuccinate
arginosuccinate→ arginine+ fumarate
arginine→ ornithine +urea
step 1 urea cycle
ornithine nuc amine group attacks carbamoyl- P to make citruline; requires ortnithine transcarbamoylase OTC, ornithine comes from GLN semi aldehyde
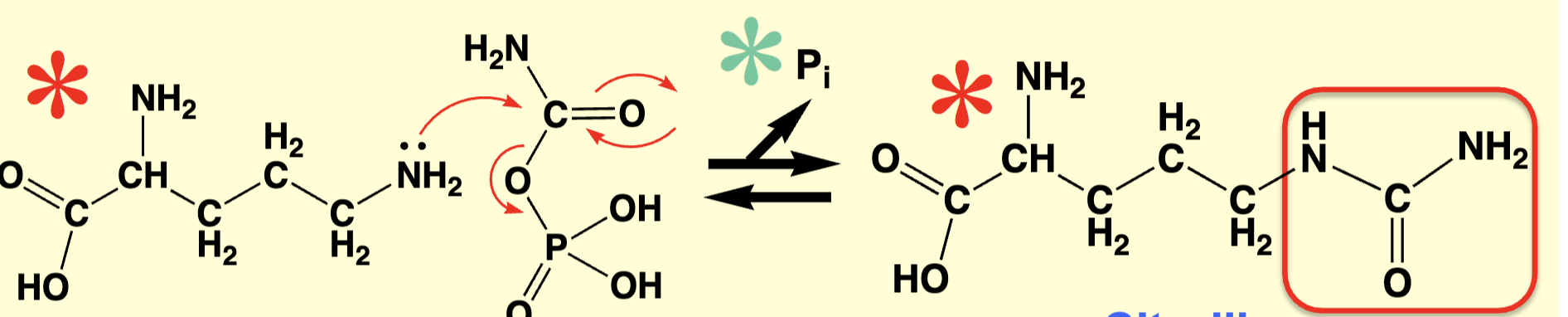
step 2 urea cycle
citruline + asp make arginosuccinate;
REQUIRES ATP AND ARGINOSUCCINATE SYNTHASE
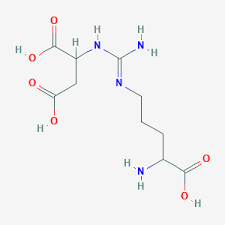
step 3 urea cycle
breakdown of argininosuccinate into arginine and fumarate using argininosuccinate lyase
fumarate can be converted to oxaloacetate or ASP and enter TCA
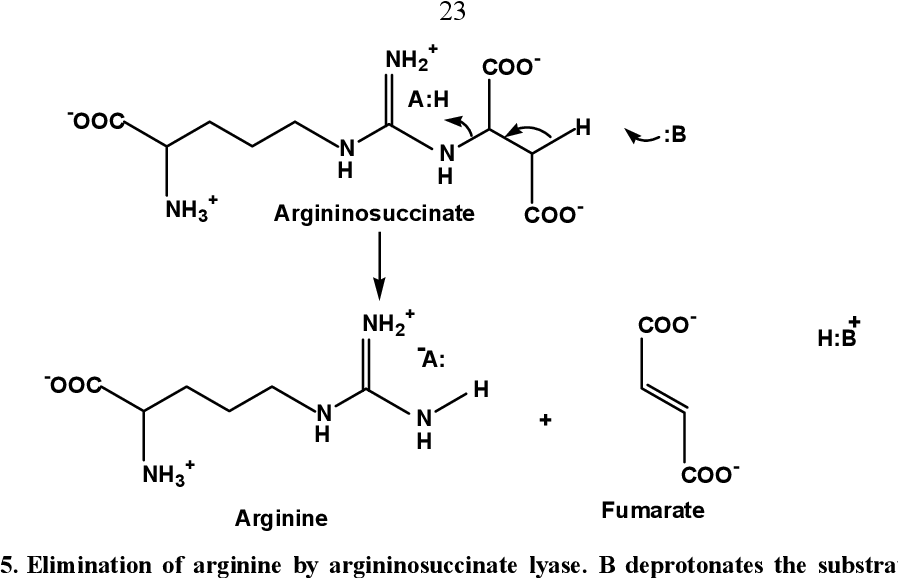
step 4 urea cycle
argninine breakdown into ornithine and urea via ARGINASE and a hydrolysis recation; ornithine returns to mitochondria to restart urea cycle; urea excreted
overall reaction
(HCO3-)+ (NH4+)+(3ATP)+(ASP)+(H2O)→ Urea +(2ADP)+(2Pi)+ AMP+ PPi+ Fumarate+ (5H+)
the big picture
lots of protons are released helping to control body pH, carbonate comes from TCA, NH3 from GDH, ATP from oxidative phosphorylation
Urea cycle control
high protein diet stimulates urea cycle enzymes, AAs caused glucagon release inducing enzyme biosynthesis, low proein diet not a lot of urea, certain AAsa re effective in stimulating glucagon release ie casein, decreases NH3 thus increasing OAA and a-KG for gluconeogenesis
glucagon
pancreatic alpha cell hormone that stimulates glucose synthesis
indirect activation of urea cycle
ARG + GLE important regulators due to role in NAG production (N-acetylglutamate); R allosterically stimulates conversion and NAG synthase, E is substrate for synthesis
NAG
allosertic activator of CPS1, the first step of the urea cycle; deficient leads to hyperammonemia, primary deficiency is mutation in NAG gene,
secondary deficiency= mitochondrial changes affecting NAG function
NAG anolgue N-carbamoylglutmate (carbaglu) is used when deficiencies exist AND activates CPS1
OTC deficiency
ornithine transcarbamoylsae, most common inherited urea cycle disorder, X-linked in adult of any age or sex, rapid NH3 increase leading to lethargy and potential death
arginase deficiency
episodic hyperammonia if untreated, not life threatening, plasma R levels 3-4x normal, birth and early childhood levels are normal because R required for growth
liver acinus
averting ammonia toxcity by prevents NH3 re-entry into blood through differential use of NH3 , hepatocytes in direct contact with blood to permit rapid metabolite exchange, early cells in first segment of acinar space are rich in glutaminase and urea cycle enzymes, high km=low affinity for NH3, removes majority of NH3, perivenous scavenger cells are rich in GLN synthetase, low km, high affinity nH3, mops up leftover NH3 and supplies GLN to many organs
endothelial lining of liver is fenestrated exposing to hepatocytes permitting rapid metabolite exchange between liver and blood
biosynthesis of arginine
key urea cycle intermediate, diet, cell protein turnover, and ornithine; can be converted to nitric oxide and citrulline; can be used to synthesize creatine; nitric oxide helps protect tissues from damage due to low blood supply
creatinine synthesis
creatine is converted to a phosphorylated form in muscle using creatine kinase and then creatinine without enzyme; produced at fairly constant rate bc no enzyme; production amount dependent on muscle amount so higher in men; clearance rate is indication of how well kidney functions
biosynthesis of glutatamate
transamination; reductive amination ie GDH, reduction amination GLN synthetase, GLN hydrolysis with glutaminase
biosynthesis of Glutamine
only GNL synthetase, cytosolic enzyme
biosynthesis of Aspartate
transamination, hydrolysis ie asparaginase
biosynthesis of asparagine
asp hydrolysis, uses 2 active sites and transfer tunnel
biosynthesis of alanine
transamination only; used for inter-organ transport btwn liver and organs of ammonia nitrogen to prevent toxicity, “alanine shuttle”, alanine→circulation→ alanine
biosynthesis of proline
reduction of E to E semi-aldehyde, cyclase, then reduced again, note that this semi aldehyde can undergo transamination to make ornithine
biosynthesis of serine
2 pathways beginning with 3-Pi-glycerate; made from glycolytic intermediate
biosynthesis of glycine
4 pathways, simplest structure yet hardest to make
biosynthesis of tyrosine
conditionally essential, requires Phe; 2 step catalytic proc
biosynthesis of hydroxyproline
collagen fibril maturation- conversion within collagen, stabilizes collagen by forming key hydrogen bonds within tropocollagen triple helix, Prolyl hydroxylase is easily deactivated, forming bound Fe(III)–O – ,which must be reduced by Vitamin C to restore enzyme:; iron dependents and associated with scurvy
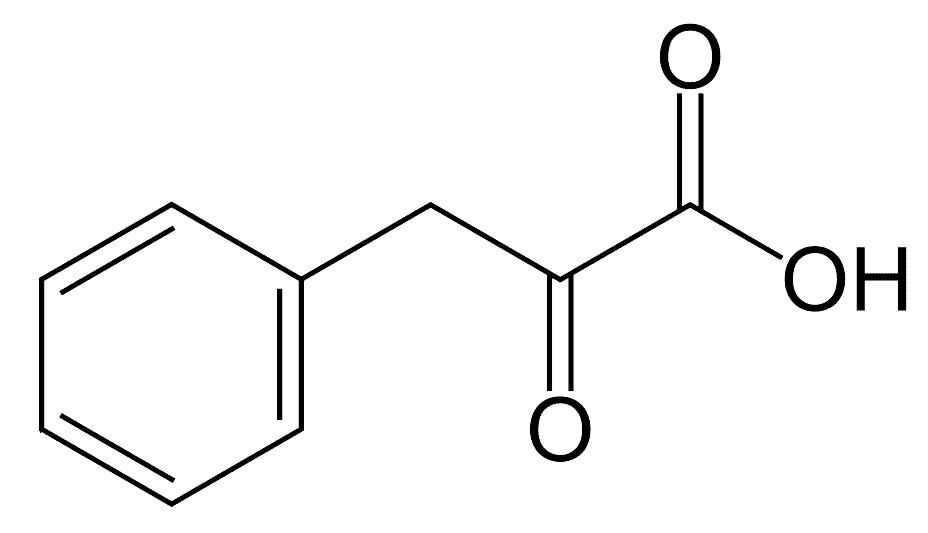
phenylketonuria (PKU)
deficiency in PHE hydroxylase; high serum PHE (20x normal) + high urinary phenylpyruvate upon transamination; very rare, damages nerve function but is treatable with low PHE diet that is maintained into adulthood
phenyl-pyruvate
autosomal recessive
two copies of abnormal gene must be present for the disease or trait to develop; PKU is an example
biosynthesis of cysteine
conditionally essential, requires MET, SAM is involved, a methylating agent for 300 metabolites
s-adenosylmethionine
thyroxin biosynthesis
formed on thyroglobulin; protein synthesized within thyroid cells but stores outside, iodide is actively transported into thyrocytes and oxidized to reactive iodine in ERl occurs thru EAS, forming thyronine ring, reabsorbed after CNS stimulationl protelyzed to form T4 endocrine
iodination of free tyrosine does not occur
thyronine
specialized fused ring AA, T3 is active form (10x over T4 (thyroxin))
metabolic actions of T3
t3 formed by t4 peripheral deiodinase; enters cells through active transporter binding to mobile receptors/transcription factors to stimulate or inhibit gene expression, increase basal metabolic rate
thyroid hormones improve development, growth, and metabolism but not necessity for life
triiodothyronine (t3)
AA neurotransmitters
E and N are excitatory, D-ser, gly; some are converted to GABA
GABA
gamma- aminobutyric acid; formed by decarboxylation of L- E
important neurotransmitters
D-Ser from racemization of L-Ser
serotonin from tryptophan
dihydroxyphenilalanine from PHE
pyrimidine biosynthesis
fewer AAs/steps than purine with simpler structures, ring formed first then added to PRPP, UMP initially formed which is phosphorylated to UTP
cytosine
thymine