First year physical
1/458
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
459 Terms
Describe how reversible protein folding was discovered
Anfinsen showed in 1957 that proteins fold reversibly
Added 8M urea and mercaptoethanol, this uses complete unfolding of the protein due to denaturation of the protein
If you then added things back in the correct order to reverse these effects, you could fold the protein properly
--> remove urea first to reform H bonds, then added oxidising agents to reform the disulphide bonds. Implied that structural information was inherent in the protein
Describe how insulin goes against the reversible folding experiment
the primary structure is folded and then cut, so would not reform the same structure as previously
Describe how proteins fold
Proteins fold and form compact structures in vivo
Dogma/thermodynamic hypothesis: the native conformation is determined by the totality of interatomic interactions and hence by the amino acid sequence in a given environment
--> primary sequence determines the proteins structure
Proteins are dynamic structures
Proteins fold to a unique tertiary structure under physiological conditions
What is the thermodynamic hypothesis for protein folding?
the native conformation is determined by the totality of interatomic interactions and hence by the amino acid sequence in a given environment
Describe the protein folding problem
Can't determine how proteins fold, only their structure
Homology - looks at sequence similarities to determine structures that may form
How the protein gets to the structure is undetermined, we don’t fully understand the energetics behind it
Describe how free energy changes determine whether a protein folds in a specific way
ΔG = ΔH - TΔS
Depends on enthalpy change (faithful interactions) and entropy (the energy dispersal such as the hydrophobic effect)
They don’t both have to be favourable as long as the free energy is favourable (the favourable term is overcome by the unfavourable term)
What are the 3 electrostatics
Simple electrostatics, dipoles, induced dipoles
Describe the equation for simple electrostatics
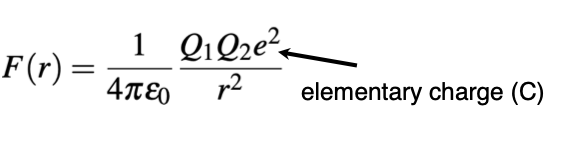
What sign are positive and negative charges as attractions and repulsions?
Attractive - negative
Repulsive - positive
Describe the electrostatic potential energy term for two charges

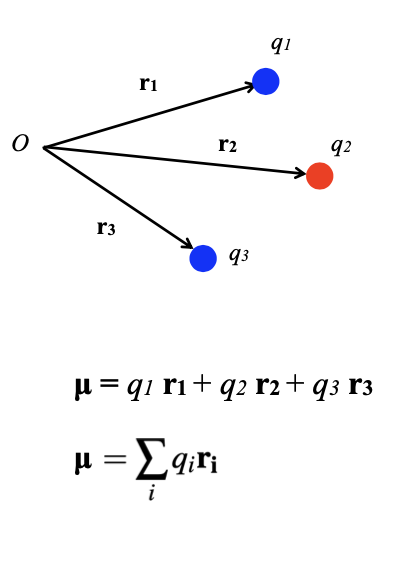
Describe how to deal with multiple charges

Describe what dielectrics are used
Charges are not in a vacuum, they are in a medium
Water can shiueld charges through forming hydration shells and other interactions, this reduces the force between the two charges
Water molecules themselves feel forces due to the two charges and as a result align between the two charged, this has the net effect of reducing the coulombic force that q2 experiences due to q1
--> how much It is reduced depends on: how easily can an applied electric field induce a separation of charge in the molecules of the medium and how easily can the molecules of the medium move to minimise the electrostatic force they experience
--> this is induction effect and orientation effects
What is the dielectric constant?
a dimensionless factor that accounts for the behaviour of the medium in which the charges exist - macroscopic approach
Why may a dielectric constant be high?
Medium is polar and readily moves in response to the presence of electric charges, it is therefore very effective at shielediing electrostatic charges
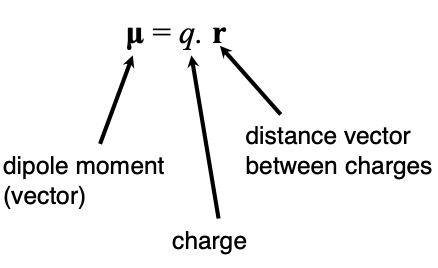
Define an electric dipole
a dipole of two electrical charges with opposite signs ±q, separated by a small distance
Describe the equation for dipole moment
How can properties of dipoles be understood?
Think of ir as 2 charges separated by a small distance
Can look at the different orientations and look at the doifferent energies produced
How can we produce one dipole from many?
Combine all to give a net dipole moment
Describe the consequences of the a-helix dipole moment
The C=O and N-H groups are generally aligned with the axis of the helix
The net dipole moments can stabilise ion binding sites in proteins
Each a-helix has a net dipole moment pointing towards its N-terminus
Describe antiparallel helix packing in proteins
To understand this, remember each dipole can be represented as two charges separated by a small distance
Can orientate different alpha helices to determine which will be more or less stable
Antiparallel are more stable due to the opposite directions of the dipoles
Describe polar molecules and how their dipole magnitude can e calculated
Elements differ in their ability to attract electrons towards themselves when forming covalent bonds
Can make predictions about electronegativity from the periodic table, this gives you a qay of calculting the magnitdue of the dipole without doing extensive calculations
This is electronegativity, can be characterised using the pauling scale
Electrons ith a high electronegativity have a high tendency to attract electrons
Homonuclear: electrons evenly shared between atoms
Heteronucelar: electrons not evenly shared between atoms, separation of centres of charge, net permanent dipole moment
Describe induced temporary dipoles
An uncharged molecule comes into contact with a permanent dipole, this will change the charge of the uncharged molecule - inducing a dipole on it
The induced dipole depends on the polarizability - how we can more the electrons around in that molecule
What does the magnitude of an induced dipole depend on?
Polarizability of the molecule
How can iyou calculate the magnitude of an induced dipole
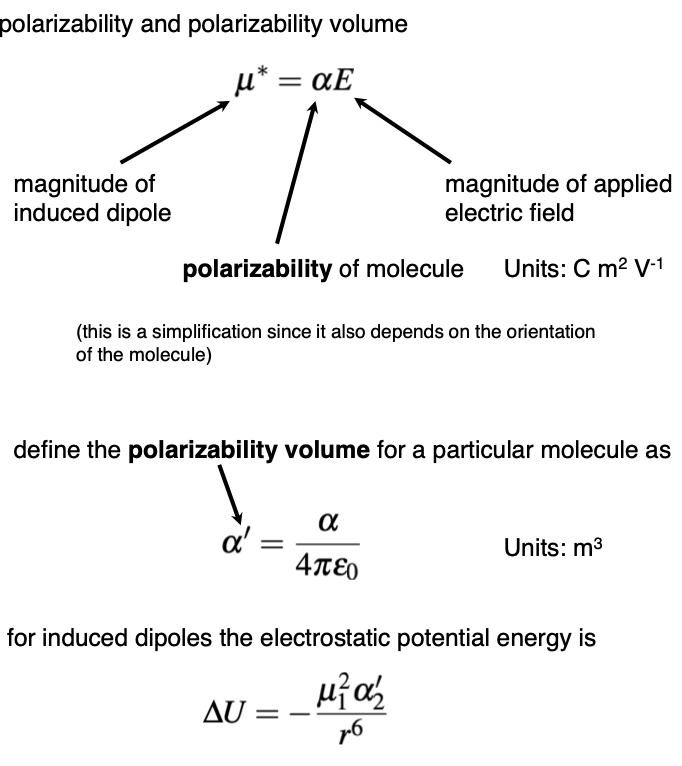
What is a polarisability volume?
Can average all of the orienatations of the molecule and their different dipole magnitudes
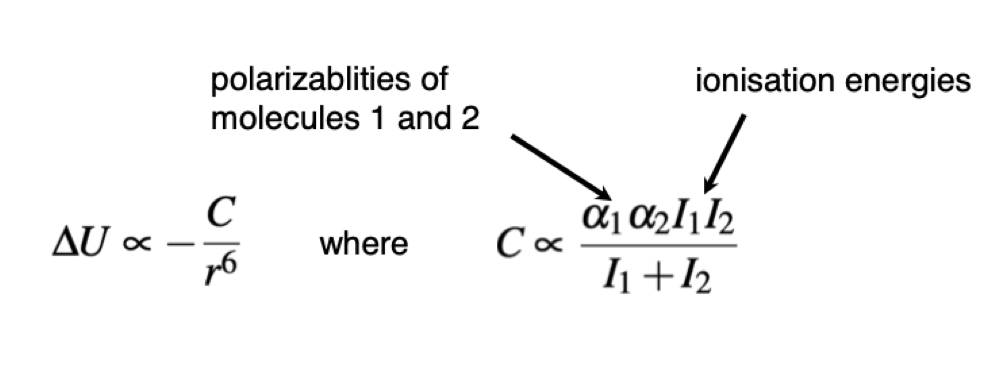
Describe London dispersion interactions and the equation associated with them
Molecules have no net charge
The instantaenous electron positions fluctuate, creating a small transient dipole
--> there is a fluctuation of different electron densities which induces a fluctuation in the moelcule next to it due to increase electron density or increased nucelus exposure, which causes attractions or repulsion of electrons
This induces another transient dipole in the second molecule
The magnitude and direction of this dipole remains correlated ith the fluctuating transient dipole in the first molecule
There is a net attraction between the two molecules
Present in all molecules
Why don’t molecules attract if there are attractive interactions?
There are a range of repulsive forces that act at a very short range
They arise from electrostatic repulsions between electron orbitals (classical effect - electron orbitals cant overlap) and the pauli exclusion principle (quantum effect)
--> there are limits where the atoms can o longer be close to each other
--> can no longer consider an atom as a simple point at short distances and no two electrons can have the same quantum numbers (cant occupy the same space)
Combined effect is modelled by ΔU∝ 1/r^n
--> n is usually 12
Describe the Lennard jones potential graph
For non polar molecules, the repulsive term (A/r^12) is balanced by the attractive london dispersion forces (B/r^6)
If n is taken to be 12, we get the Lennard Jones potential
The graph tends to infinity for small values of r, and tend to 0 at large vaues of r, there is a minimum where forces balance
--> there is an excluded volume at short values of r and the forces are short range
Repulsive force - positive energy, only in play at very close to the atom
Attractive force - negative energy, more negative the closer the atoms are
Combining them gives the lennard jones potential
When do forces balance at the lennord jones graph?
Minimum is the most stable point, but this is the firthest together they can go due to repulsions that overcome the attractions
Can be found bu differentiating
Describe the van Der Waals radio and compare it to the atomic radii
As the temperature is lowered, the atoms have less energy and the distance between them decreases until it reaches a minimum - d is reached
D= sum of the van der waals radii
Van der walls radii > atomic radii
The more electronegative the atom, the smaller the van der waals radius
How can the VDW raid be used to look at molecular structure in proteins
Van der waals radius is approximately length of covalent bond
Van der waals spheres are tightly packed against one another
VDW spheres allow us to have a more accurate representation of interactions between atoms in molecules
Don’t have much room to move atoms around
Describe torsion angles and their effects on protein strycture
Phi, Psi and Omega
Can rotate the atoms around the bonds, but this is determoined by van der waals radii
There will be specific orientations where there will be unfavourable clashes of the VDW radii
Phi - around Ca and N
Psi - around C and Ca
Omega - around Ca and N - this is around the peptide bond, we know this is very limites (basically static) so don’t really need to worry about it
If Phi is 0, (eclipsed) there will be clashesbetween atoms
In the stagered confromation for Phi, there will not be many clashes
Side chains will also have an effect on clashes
Some combination result in energy increasing due to the repusive nature of the van der waals potneital - such as CO and NH coming close to eachother
Describe how a ramachandran plot can be used to determine protein structure and the region glycine is in
Used a computer to study which phi and psi combination were possible
Take different phi and psi angles and look for clashes between atoms
We get very limited favourable regions, this allows you to look at where peptide bonds can be favourably
22% is allowed, and only 7% is favoured
We can identify the favoured regions with protein secondary structure
Glycine does not have a side chain, this gives us more freedom around the rotation of the phi and psi bonds due to less electrostatic repulsions from side chains
--> this means glycine can be used to study whether specific side chains are involved in the activity of a protein as it will disrupt the shape of the active site due to being very flexible if the substituted residue
--> this can also be done with alanine, there is still a side chain so the phi and psi angles are still limited, but it can determine whether a charged residue is involved in the catalytic activity of an enzyme
Describe polysaccharide torsion angles in cellulose and a-amylose and the effect on function
Analagous to proteins, for a 1-4 linked glycoside:
Cellulose
Cellulose is a polymer of β 1-4 linked D glucose
Forms chain of glycans, each flipped 180 relative to the preceding one
Parallel extended chains form sheets
Sheets stack to form fibres
Chains can pack in together, this is due to hydrogen bonding
The bonds are very strong, so cannot be used as energy as the atoms are not accessible
a-Amylose (starch)
A-Amylose is a polymer of ⍺ 1-4 linked D glucose
An isomer if cellulose
Acts as an energy store
Forms a left handed helix - this is good for enzymes as the atoms are accessible for bond breakage
Starch can be dissolved in solution and must be to retain its coiled structure, this could be an issue as it carries a lot of water
--> a simple conformational change has largely changed the properties of the materials
Describe the different torsions in nucelotdes and their effects on DNA and RNA structure
Bases adopt planar structure
Different conformation of the double helix result from: pucker of ribose sugar or rotation of N-glycosidic bond
Bases can adopt syn and anti positions (anti is more likely)
Sugars an be C2-endo or C3-endo
RNA tends to use C3, DNA tends to be C2
This is due to the hydroxyl group in ribose favouring the C3 and the lack in DNA favouring rthe C2 - DNA has more flexibility
RNA forms an A-DNA helix, quite skewed, whereas DNA adopts a B-DNA helix, which is less skewed. DNA is more stable
Describe what hydrogen bonding influences
Structure of water - hydrophobic effect
Protein secondary structure
Base pairing in DNA and RNA
Carbohydrate structure
Orientation of amino acid side chains
What is a hydrogen bond?
Hydrogen bond: When hydrogen is covalently bound to an electronegative donor, its electron is predominantly found near the donor. Its proton is almost unshielded due to electron attraction to the other nucleus when viewed along the axis of the covalent bond and can interact strongly with other electronegative atoms (high charge density)
Describe the observed vs expected hydrogen bonding distances
D and A approach closer than the sum of the VDW radii, so a hydrogen bond is stronger than a VDW interaction
H-A distance is shorter than expected, this is because the hydrogen is shared between D and A, so the hydrogen bond has some covalent character
Hydrogen bonds are only observed for a narrow range of orientation and distances, this shows the hydrogen bond is directional
Describe the effect on physical and chemical properties done by hydrogen bonding
Physical properties
Gases are non-ideal do not follow pV = nRT
Boiling points are raised - have to put energy in to break hydrogen bonds
The relative permittivity (dielectric constant) is affected
Chemical properties
HD is a Lewis acid, so a hydrogen bond is an intermediate in the transfer of a proton from an acid to a base
What is the radial distribution function of water?
Look at the chances of a water molecule orientating itself with another water molecule
Shows that at specified dustances, there is a high chance of interacting with other water molecules
Enhanced probability of finding oxygen atoms at around 2.7Å due to hydrogen bonding between waters
Slightly enhanced probability of finding another oxygen atom at 4.2Å
Zero probability of finding another oxygen atom at <2.2Å due to the repulsive part of the van der waals potential
Describe how hydrogen bonds aid the catalytic triad of chymotrypsin
His is abstracting a proton from serine and therefore acting as a general base - this increases the nucleophicity due to an increased electron density (as the covalent pair are more closer to oxygen than hydrogen)
Histidine orients serine for nucleophilic attack
Describe the energy of a hydrogen bond

Describe the strengths of hydrogen bonds
If the Pka (acceptor) - Pka(donor) is large or the pKa(conjugate acid of base) - pKa(acid) is small, then the hydrogen bond is strong
Describe how hydrogen bonds form salt bridges in proteins
This is only a salt bridge if both residues are ionised
The pKa of these residues are measured in water
Hence at pH 7 in water both residues are ionised and a saltbridge forms
If these residues are buried within a protein they may no longer be ionised, this is because the permittivity will be different, meaning the pKas are different, so we cannot determine whether the residues will form free ions or not
Describe energy dispersal and how you can use it to get entropy
Macroscopic level: entropy measures how dispersed energy is between molecules
Molecular level: entropy measures how dispersed energy is within a molecule
--> energy dispersal in individual molecules - translational, rotational, vibrational, electron states
Can use the Boltzmann distribution to find the different ways and probabilities of energy dispersal
S = klnW
S = entropy
W = the number of ways the energy could be arranged
Describe degrees of freedom in molecules
Any molecule aslways has 3 translational degrees of freedom as It can move in x y and z
Molecules made up of more than one atom have vibrational degrees of freedom and rotational degrees of freedom
More degrees of freedom means more ways to disperse energy, so this means there is a higher entropy
Describe how we can use N-methyl acetamide to model backbone hydrogen bonding in CCl4 and water
CCl4
Aprotic, cannot form H bonds
N-methyl acetamide can form H bonds with itself, each molecule forms one hydrogen bond
Calculations show that it is entropically unfavoured to form hydrogen bonds as a dimer is more ordered than two monomers
It is favourable enthalpically
G is slightly negative, so somewhat favourable to form hydrogen bonds between the dimers
Water
Water can compete with a second molecule to form a hydrogen bond, the concentration of water is much more than that of the monomer
The hydrogen forms with the NMA is negligible as it equals those removed by that between water (as waters normally have hydrogen bonds between them)
--> so the enthalpic term is 0
The entropic term is negative due to the formation of dimers
So it is overall unfavourable
Describe hydrogen bonding in macromolecules and how it is favourable/unfavourable
If we are hydrogen bonding 2 separately chains, it is entropically unfavourable. But forming a second and third hydrogen bonds will be less unfavourable, they are entropically less unfavourable
Describe hydrogen bonding in a molecule and how it is favourable/unfavourable
The chain is already close to itself, so the entropic penalty is much less than expected
Only working for specific geometries
Describe the a-helix
Form when protein backbone groups hydrogen bonds
More than one helix can from
A-helix has 3.6 residues per turn and 13 backbone atoms
Hydrogen bonds between every 4 residues
Hydrogen bonds are forming throughout the chain
Describe the 3-10 helix
3 residues per turn and 10 atoms
Tighter helix, atoms are much closer together
Not favoured due to instability
Not very common
Smaller diameter than a-helix
Side chains experience greater steric clashes with main chain
In a mildly forbidden region of Ramchandran plot
Uncommon and often short
Describe the 4.4-16 helix
4.4 residues per turn
Larger in diameter and shorter than an a-helix
Large hole in the centre is too large for stabilising VDW, making it very unstable
Observed at the ends of a few a-helices/near functional sites
Describe B-sheets
Harder to form than an a-helix, as the geometry must be perfect anf they will not be adjacent in the primary sequence, they must be brought together to form hydrogen bonds
--> a-helices form much faster
IN the allowed region of the Ramachandran plot
In globular proteins need to change direction, this is done using B-turns, they are reverse turns of the peptide bonds, often contain prolines/glycine's
Describe B-turns
Connect elements of secondary structure
Used in proteins needing to turn their amino acids - globular
--> not in fibrous proteins as they form long chains
Describe the properties of water
Unusual properties stem from its geometry and ability to form hydrogen bonds
Radial distribution functions - how it organises itself in solution
High specific heat Cp
High surface tension
High dielectric constant
Small thermal expansion coefficient - when heated there is little expansion
What does the hydrophobic effect result from?
Extensive and cohesive hydrogen bonding networks in water
Non-polar molecules having stronger interactions with themselves than with water
How can the partition coefficient be used?
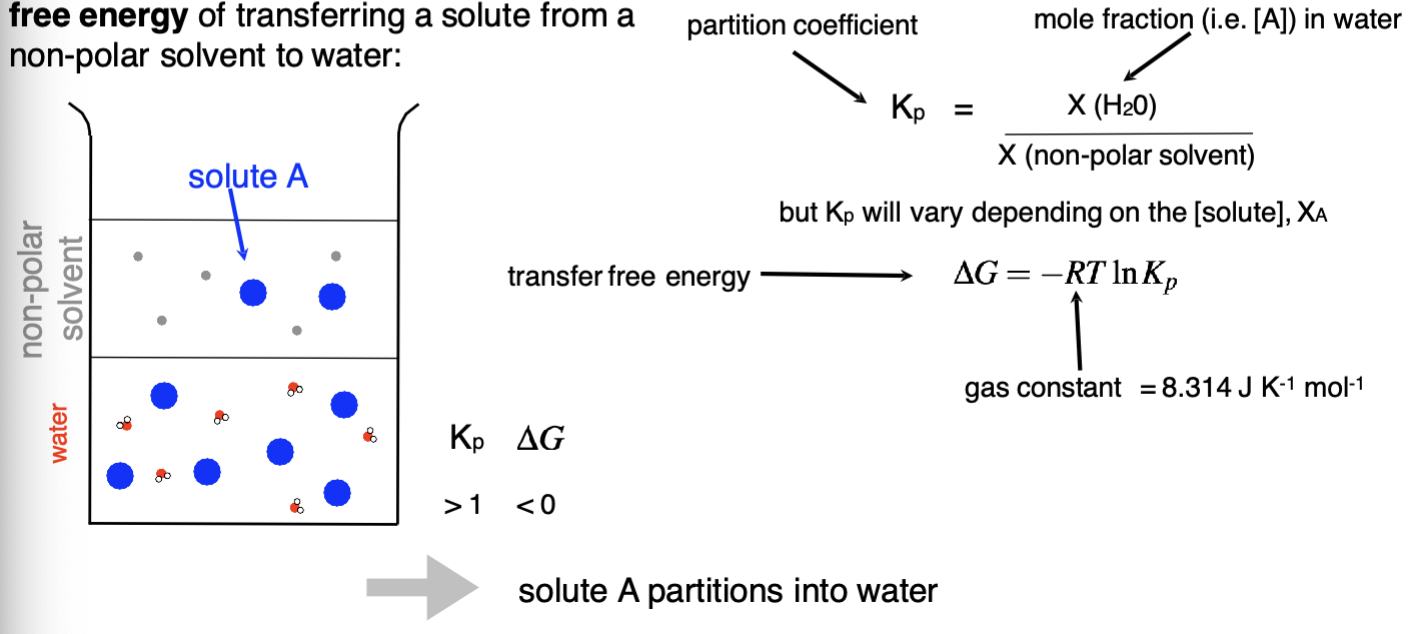
Describe the molecular contribiting factors for free energy of transfer
ΔH determined by
Interactions between solute and non polar solvent
Interactions between solute and water
Resulting changes in the number of water hydrogen bonds
Need to determine which is more favourable
TΔS
TS changes in the local order of water due to the solute presence
This therefore changes the number of energy states of the water, changing the entropic term
For some hydrophobic molecules, the water forms an organised clathrate around the molecule, this is highly ordered and will therefore reduce the entropic term
Describe the effect of temperature on the hydrophobic effect
At higher temps, hydration shell outside of the hydrophobic solute breaks down and interactions over come
Water H bonds between each other reduced
At physiological temperatures the hydrophobic molecules associate with one another (increases entropy) so that water can from the maximum number of hydrogen bonds
--> hydrophobic molecules are reorganising to present the minimum surface to water
Describe the hydropathy of amino acid residues
Whether an amino acid is hydrophilic or hydrophobic
Data used such as the transfer free energy from a nonpolar solvent to water can be used to construct a hydropathy scale
How often an amino acid is found buried in the interior of a protein compared to how often it is found on the surface of a protein correlates well with its hydropathy
Describe the consequences of folding of proteins
Hydrophobic effect is the primary driver of protein folding
Non polar residues are more likely to be found in the interior of proteins
Polar residues and caged residues are much more likely to be found on the surface of proteins (membrane proteins are an exception)
Hydrogen bonds form in the interior of the proteins in clusters, in a cooperative way
The interior of proteins is densely packed environment (side chains are interdigitated)
Describe how protein folding is a 2 step process
It is first order and dependent on the concentration of the unfolded state
Intermediate states exist
Describe the first step of protein folding
Hydrophobic collapse occurs
Hydrophobic residues cluster together, this excludes water from the interior
This causes a large increase in the entropy of water as it is no longer ordered around the protein, meaning that the free energy is negative and favourbale
What is hydrophobic collapse driven by?
the increase in entropy of water
Compare a extended unfolded state and the compact molten globule
In the unfolded, the entire chain is accessible to water
In the folded, water cannot access the hydrophobic core that gas formed
Describe the formation of the native state from the compact molten globule
For the solvent, there is little difference between the molten globule and the folded native state, so the entropic and enthalpy terms are 0
The protein is becoming more ordered (less conformations can be accessed) so the entropic term is negative and unfavoured
Therfore for the folding to occur, the enthalpy of forming additional non-covalent interactions must overcome the reduction of entropy
Descruve how non-cooperative interactions must form and why
At first, the formation is unfavoured due to the increasing reduction in entropy
It must be cooperative, stabilising the native state in comparison to the molten globule
Descrie adding more hydrogen bonds to drive a molten globule to a native state
When adding more hydrogen bonds, the reduction in entropy is less than the one before as the one before has already partially contained the protein
The enthalpy is favoured as less H2O can compete
The G is decreasing, and eventually becomes negative, we can calculate when the formation of n hydrogen bonds becomes favoured
What is the equation for the formation of cooperative intra-molecular hydrogen bonding?
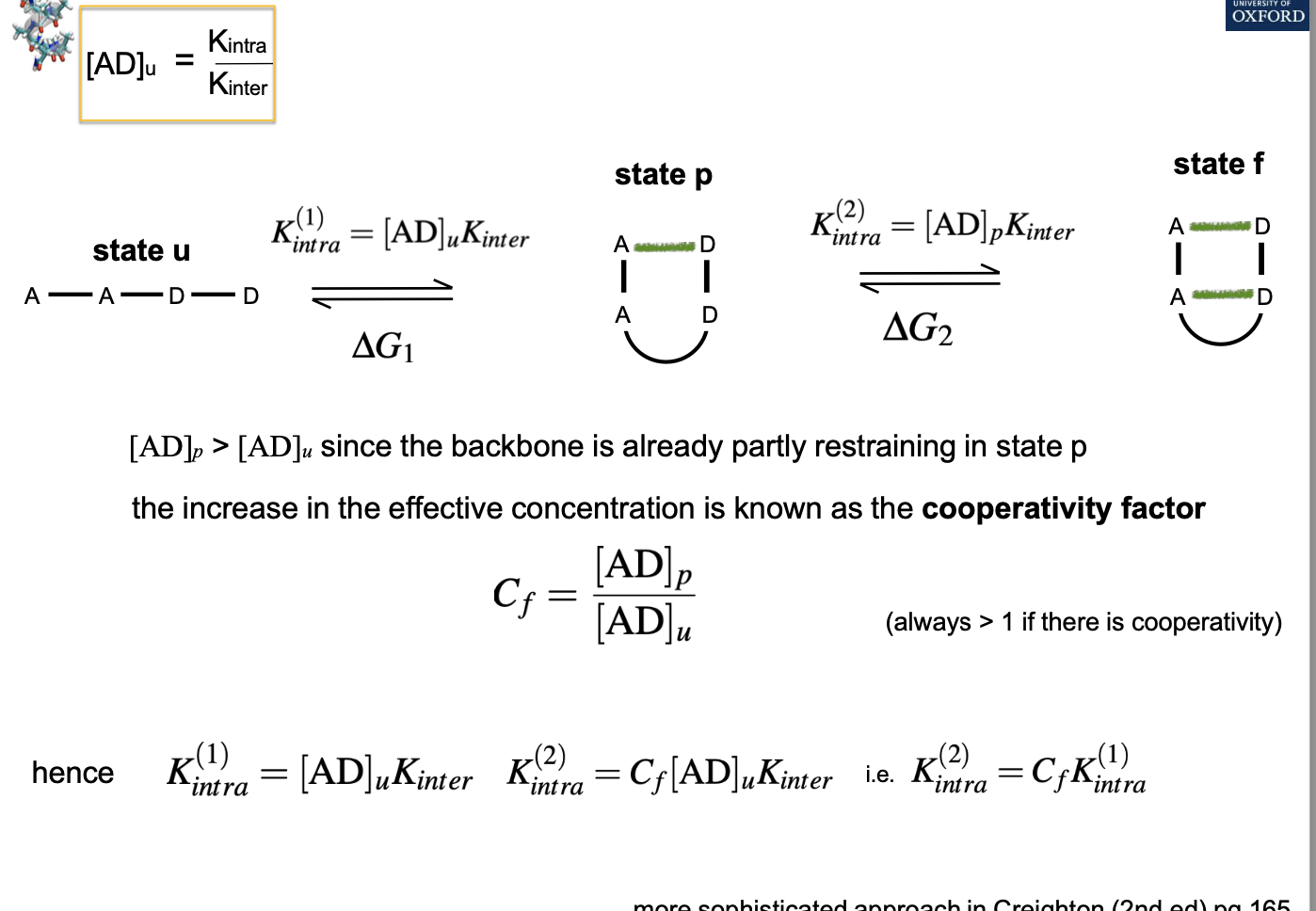
How does the hydrophobic effect drive protein folding?
Causes the extended chain to collapse and form a compact dynamic molten globule
Clusters of non-covalent interactions form cooperatively, forming secondary structures and the dense packing of a hydrophobic interior
Describe how unfolding is cooperative
Breaking one hydrogen bond decreases the favourability of the free energy, so it becomes easier to break another hydrogen bond
Describe how temperature affects stability of proteins
Affects the strength of the hydrophobic effect (as it is equilibrium dependent)
Affrcts the configurational entropy of the unfolded state
Describe how denaturants affects stability of proteins
Increase the solubility of non polar residues - chaotrophic agents
Increase the stability of the unfolded state and reduce the strength of the hydrophobic effect
Experiments: an increase in viscosity and NH D exchange due to lack of secondary structure
Describe how protein modification affects the stability of proteins
Affect the configurational entropy of unfolded states
May change electrostatics that hold specific parts together
Describe how mutations affect the stability of a protein
Effect the strength and number of cooperative non covalent interaction as they change the primary sequence
Describe how pH affect the stability of a protein
Effect the strength and number of cooperative non covalent interaction
Buried groups can become ionised, this can lead to electrostatic repulstions
Saltbridges may increase the stability of a specific state
Describe the reversible folding experiment
Denature and reduce disulphide bonds in a protein
Allow refolding in different ways
Removing urea and then mercaptoethanol allows H bonds to reform and then cysteines to oxidise, this forms a 100% active protein as the cysteines are in the correct positions to form the correct disulphides in the correct orientation
Reducing mercaptoethanol and then urea allows cysteines to oxidise and then H bonds to reform. The protein formed is 1% active, this is because the disulphides form randomly, many different conformations can form
Descruve the protein folding funnel
Cooperative formation of non-covalent interactions drive the protein towards the folded state, the hdyrophobic effect drives the unfolded state towards the compact molten globule
Some proteins may aid folding
All conformations can enter the funnel, but it excludes a lot of unstable states
Proteins may get stuck in specific shapes, this can encourage unfolding and aggregate other proteins, leading to intermolecular bonds forming
Describe chaperones to aid protein folding
Can provide a specific cavity where proteins can fold, preventing interactions with the rest of the cell
Can bind to hydrophobic residues to prevent premature folding
Describe the ion dissociation of water
Water undergoes ionic dissociation:
This equilibrium can be affected by both physical and chemical factors
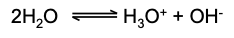
What is an Arrhenius acid and base?
An acid increases the concentration of H3O+ ions in solutions
A base increased the concentration of OH- ions in solution
Only refers to acids and bases in solution
What is a bronzed-lowry acid and base?
An acid is a H+ donor
A base is a H+ acceptor
This definition includes Arrhenius acids and bases, and also some water insoluble substances and gas phase reactions
What is a lewis acid and base?
A Lewis acid is an electron pair acceptor
A Lewis base is an electron pair donor
What is pH?
pH = -log[H3O+]
How can the pH of pure water be calculated?
H2O is mostly in its standard state, so [H2O]/[H2O]° = 1
So Kw = [H3O+][OH-]
At 298K, Kw = 10^-14
In pure water, [H3O+]=[OH-], so Kw = [H3O+]^2 = 10^-14
So [H3O+] = 10^-7
So pH = 7, this is neutral
If temperature changes, the pH of water will also change as pH is temperature dependent as Kw is temperature dependent
![<p></p><p></p><ul><li><p><span>H2O is mostly in its standard state, so [H2O]/[H2O]° = 1</span></p></li><li><p><span>So Kw = [H3O+][OH-]</span></p></li><li><p><span>At 298K, Kw = 10^-14</span></p></li><li><p><span>In pure water, [H3O+]=[OH-], so Kw = [H3O+]^2 = 10^-14</span></p></li><li><p><span>So [H3O+] = 10^-7</span></p></li><li><p><span>So pH = 7, this is neutral</span></p></li><li><p><span>If temperature changes, the pH of water will also change as pH is temperature dependent as Kw is temperature dependent</span></p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/0fabce77-b3bf-4dd9-88a7-2d503515d09b.png)
Describe an acid and its conjugate base
Consider an acid HA, this can donate a proton to water
HA + H2O <==> A- + H3O+
A- can then accept a proton from water and act as a base, this is the conjugate base
Any acid has a conjugate base and any base has a conjugate acid
Describe the strengths of acids
The position of equilibrium is a measure of the strength of an acid, and how easily it dissociates a proton
So the equilibrium constant is called the acid dissociation constant (Ka)
Ka = [H3O+][A-]/[HA]
In the same way we define pH, we can define pKa
pKa = -logKa, this can be used to compare different acids
The strength of acids and bases is measured by their degree of dissociation in water, so by their pKa
A strong acid is one that is almost fully dissociated in water, has a very low pKa
What is the base dissociation constant and what os the issue with it?
BOH <==> B+ + OH-
Kb = [B+][OH-]/[BOH]
pKb = -logKb
So we have separate scales for the strengths of acids and bases, this is difficult to relate acids and bases, so want them on the same scale, use the strength of the conjugate acid
How can we put the strengths of bases and acids on the same scale?
We can put acids and bases on the same scale using the strength of the conjugate acid as a measure of the strength od the base
This gives the ionic product of water, so pKa + pKb = 14
The strength of a base is usually given as a pKa value, so this is the strength of its conjugate acid
What os the isoelectric point?
The pH at which the net charge on a molecule with more than one ionizable group is 0 (individual groups will be charged but will cancel each other out)
If you know the isoelectric point, can change the pH either side to have different groups charged, this will vary the protein charges, can be used for purification methods - one protein is the only one with a positive charge so can use negative chromatography to remove it from a solution
This can be extended to include all the charged groups in a protein, isoelectric focussing and ion exchange chromatography use separation on the basis of pI value
Describe the neutralisation of different acids or bases
For strong acids, there is no buffering or equilibrium, once NaOH is added it dissociates and all H+ is used for neutralisation
For weak acids like Ch3COOH, there is an equilibrium acheived at first and then a large pH increase
Neutralisation occurs when all acid has been dissociated and all the H+ liberated has been used up
Describe polyprotic acids
If the compound contains more than one titratable group, such as H3PO4, then the titration cirve depends on the difference of the pKas of the different groups. If they are significantly diferent, then separate dissociataions can be seen on a plot.
We are moving from one pKa to another
At the equivalence points, the pH swaps from being determined by one equilibroum to being determined by the next
Describe buffers and how they work
Buffers are solutions that resist changes in pH, they therefore increase the amount of acid or base that must be added to change the pH of a solution
--> do not completely top pH changes, instead they slow the change in pH, meaning it remains approximately constant
The buffer consists of an equilibrium mixture of an acid and its conjugate base - or base and conjugate acid
HA + H2O <==> A- + H3O+
Describe buffering capacity equation and what it means
ß = amount of acid or base added / change in pH
The higher the buffering capacity, the better the solution is a buffer
The buffering capacity of a solution depends strongly on the solution concentration of the buffering acid or base and on the pH of the solution
As pH moves away from pKa, the buffering capacity decreases
How can pH be measured?
Electrochemically
Indicators, use of pH dependent properties
--> properties include colour, fluorescence, nuclear magnetic resonance
Describe how pH indicators using colour can be used?
Different indicators turn a different colour at a specific pH
How can NMR be used to determine side chain peas?
Can look at individual side chains as they will give different peaks
Look at the chemical shift of the equilibrium and find the pKa of a specific residue
What are the different scenarios for ligand binding to a protein/receptor?
Binding of one ligand to one site on a protein
Multiple types of ligands competing for a single binding site (for example some cases of competitive inhibition of enzymes)
Multiple types of ligands binding to multiple sites on a protein, binding is independent
Multiple types of ligand binding to multiple sites on a protein, binding events effect subsequent ligand binding
One type of ligand binding to multiple identical sites on a protein and each binding event is independent
One type of ligand binding to multiple identical sites on a protein, and once binding event affects subsequence ligand binding
One type of ligand binding to multiple non-identical sites on a protein, can be independent or cooperative
One multivalent ligand binding to multiple sites on a protein
Describe the thermodynamic approach for one ligand and one binding site
One ligand binding to one site on a protein follow this equilibrium:
P + L <==> PL
K = [PL]/[P][L]
Kd = 1/K, units are M
Kd can be used to calculate ΔG for binding
When calculating ΔG', it is normally for the forward reaction, so need to do 1/Kd instead of Kd (as Kd is the dissociation constant, not the association constant)
It is also useful to know the average number of ligands bound per molecule of protein, ṉ (but the bar will be on top)
Ṉ = concentration of L bound to P / total concentration of P
Combining the equation for Kd and Ṉ gives:
Ṉ = [L]/ (Kd + [L])
For a single ligand binding to a single site, n ≤ 1, and is the fractional saturation of the protein, the fraction of ligand binding sites that have a ligand bound
Fractional saturation, θ = Ṉ/n, n is the total number of binding sites for the ligand, and θ has a value between 0 and 1
--> completely unbound = 0, completely bound = 1
How can Kd be determined using the Scatchard plot?
A plot of θ/[L] vs θ gives a straight line with a slope of -1/Kd
[L] is the concentration of free, unbound ligand. If the ligand is in large excess, [L] = [L]tot - [PL] , which is approximately [L]total