6.2 - Metabolism and Pathways
1/172
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
173 Terms
Steroid synthesis (Mevalonic acid pathway)
Ketogenesis
Fates of HMG - CoA [2]
b. Steroid synthesis
Pathway for synthesis of sterols
a. Beta-oxidation
b. Steroid synthesis
c. Fatty acid synthesis
d. Ketogenesis
b. Mevalonic acid pathway
Steroid synthesis is also known as
a. Beta-oxidation pathway
b. Mevalonic acid pathway
c. Embden-Meyerhof pathway
d. HMP shunt
c. Cytosol (especially the liver)
[Fates of HMG - CoA - Steroid Synthesis / Mevalonic Acid Pathway]
Cellular site of steroid synthesis
a. Mitochondrial matrix
b. Nucleus
c. Cytosol (especially the liver)
d. Endoplasmic reticulum
a. True
[Fates of HMG - CoA - Steroid Synthesis / Mevalonic Acid Pathway]
Steroid synthesis timing is primarily genetic but also fed (stimulated by insulin)
a. True
b. False
b. HMG-CoA reductase
[Fates of HMG - CoA - Steroid Synthesis / Mevalonic Acid Pathway]
Rate-limiting enzyme of steroid synthesis
a. Acetyl-CoA carboxylase
b. HMG-CoA reductase
c. HMG-CoA synthase
d. Citrate synthase
Bile acids
Steroid hormones
[Fates of HMG - CoA - Steroid Synthesis / Mevalonic Acid Pathway]
In humans, cholesterol can be converted to ________ [2] whenever necessary
c. Ketogenesis
Production of ketone bodies
a. Steroid synthesis
b. Fatty acid synthesis
c. Ketogenesis
d. Beta-oxidation
Acetone
Acetoacetate
B-hydroxybutyrate
[Fates of HMG - CoA - Ketogenesis]
Ketone bodies [3]
c. Ketone bodies
[Fates of HMG - CoA - Ketogenesis]
Serve as emergency energy source
a. Glucose
b. Fatty acids
c. Ketone bodies
d. Amino acids
c. Mitochondrial matrix (especially the liver)
[Fates of HMG - CoA - Ketogenesis]
Cellular site of ketogenesis
a. Cytosol
b. Nucleus
c. Mitochondrial matrix (especially the liver)
d. Endoplasmic reticulum
d. Glucagon
[Fates of HMG - CoA - Ketogenesis]
Ketogenesis is stimulated by ________ , indicating it happens in the fasted state
a. Insulin
b. Cortisol
c. Epinephrine
d. Glucagon
c. HMG-CoA synthase
[Fates of HMG - CoA - Ketogenesis]
Rate-limiting enzyme of ketogenesis
a. HMG-CoA reductase
b. HMG-CoA lyase
c. HMG-CoA synthase
d. Acetyl-CoA carboxylase
c. HMG-CoA lyase
[Fates of HMG - CoA - Ketogenesis]
First enzyme to make the first ketone body
a. HMG-CoA reductase
b. HMG-CoA synthase
c. HMG-CoA lyase
d. Acetyl-CoA carboxylase
c. Brain
Organ that does not use fat as energy source
a. Liver
b. Muscle
c. Brain
d. Kidney
d. Ketosis
Uses ketone instead of sugar
a. Glycolysis
b. Gluconeogenesis
c. Ketoacidosis
d. Ketosis
b. Ketonemia
Presence of ketone in blood
a. Ketonuria
b. Ketonemia
c. Ketoacidosis
d. Ketosis
a. Ketonuria
Presence of ketone in urine
a. Ketonuria
b. Ketonemia
c. Glucosuria
d. Proteinuria
d. Ketoacidosis
Complication for people with uncontrolled DM
a. Ketonemia
b. Ketosis
c. Ketonuria
d. Ketoacidosis
b. Fruity breath due to ACETONE
What is the smell of ketone bodies?
a. Fishy breath due to AMMONIA
b. Fruity breath due to ACETONE
c. Garlic breath due to ARSENIC
d. Musty breath due to METHANOL
a. True
↑insulin, ↓ketone body
a. True
b. False
a. True
Carbohydrate and lipid metabolism are clearly influenced to a great extent by insulin and glucagon
a. True
b. False
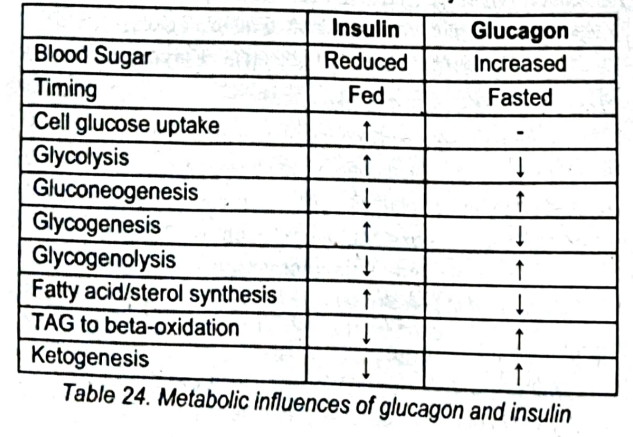
Table 24: Metabolic Influences of glucagon and insulin [diko na sasauluhin e2]
c. HMG-CoA Synthase
[Fates of HMG - CoA - Diagram]
3 acetyl-CoA → hydroxymethylglutaryl-CoA (HMG-CoA); enzyme involved
a. HMG-CoA Reductase
b. HMG-CoA Lyase
c. HMG-CoA Synthase
d. Acetyl-CoA Carboxylase
d. HMG-CoA Reductase
[Fates of HMG - CoA - Diagram]
Rate limiting enzyme of the mevalonate pathway
a. HMG-CoA Synthase
b. HMG-CoA Lyase
c. Acetyl-CoA Carboxylase
d. HMG-CoA Reductase
b. Statins
[Fates of HMG - CoA - Diagram]
Drug class that inhibits HMG-CoA Reductase
a. Fibrates
b. Statins
c. Niacin
d. Bile acid sequestrants
c. Mevalonate pathway
[Fates of HMG - CoA - Diagram]
HMG-CoA → Mevalonate → Squalene → Sterols; this pathway is called
a. Ketogenesis
b. Beta-oxidation
c. Mevalonate pathway
d. Glycolysis
c. HMG-CoA Lyase
[Fates of HMG - CoA - Diagram]
HMG-CoA → Acetoacetate; enzyme involved
a. HMG-CoA Reductase
b. HMG-CoA Synthase
c. HMG-CoA Lyase
d. Acetyl-CoA Carboxylase
Acetone
β-hydroxybutyrate
[Fates of HMG - CoA - Diagram]
Ketone bodies produced from Acetoacetate
a. Pyruvate and Lactate
b. Acetone and β-hydroxybutyrate
c. Mevalonate and Squalene
d. Acetyl-CoA and Oxaloacetate
d. Ketogenesis
[Fates of HMG - CoA - Diagram]
Pathway occurring in the matrix; produces ketone bodies from HMG-CoA
a. Mevalonate pathway
b. Beta-oxidation
c. Glycolysis
d. Ketogenesis
b. Mevalonate pathway
[Fates of HMG - CoA - Diagram]
Pathway occurring in the cytosol; produces sterols from HMG-CoA
a. Ketogenesis
b. Mevalonate pathway
c. Beta-oxidation
d. Gluconeogenesis
Synthesis
Breakdown
Nucleotide metabolism involves ______ [2]
De novo
Salvage
Two Main methods for Synthesis [2]
c. De novo
[Nucleotide Metabolism - Main methods for Synthesis]
Synthesis from scratch ("from the beginning")
a. Salvage
b. Catabolism
c. De novo
d. Beta-oxidatio
c. Inosine monophosphate
[Nucleotide Metabolism - Main methods for Synthesis - De Novo]
First purine
a. Orotidine monophosphate
b. Adenosine monophosphate
c. Inosine monophosphate
d. Guanosine monophosphate
d. Orotidine monophosphate
[Nucleotide Metabolism - Main methods for Synthesis - De Novo]
First pyrimidine
a. Inosine monophosphate
b. Cytidine monophosphate
c. Thymidine monophosphate
d. Orotidine monophosphate
b. Salvage
[Nucleotide Metabolism - Main methods for Synthesis]
Use of already-made purines and pyrimidines from the digestion of food (especially animal meat)
a. De novo
b. Salvage
c. Catabolism
d. Gluconeogenesis
c. Hypoxanthine-guanine phosphoribosyltransferase (HGPRT)
[Nucleotide Metabolism - Main methods for Synthesis]
Major enzyme in the salvage of purines
a. Adenosine deaminase
b. Xanthine oxidase
c. Hypoxanthine-guanine phosphoribosyltransferase (HGPRT)
d. Purine nucleoside phosphorylase
b. Lesch-Nyhan syndrome
[Nucleotide Metabolism - Main methods for Synthesis]
Deficiency in HGPRT
a. Severe combined immunodeficiency
b. Lesch-Nyhan syndrome
c. Gout
d. Hyperuricemia
b. Juvenile Gout
[Nucleotide Metabolism - Main methods for Synthesis]
Lesch-Nyhan syndrome is also known as
a. Juvenile Arthritis
b. Juvenile Gout
c. Juvenile Diabetes
d. Juvenile Hyperuricemia
a. True
[Nucleotide Metabolism - Synthesis]
Some enzymes in nucleotide synthesis are targets of rheumatologic and chemotherapeutic drugs
a. True
b. False
c. Severe combined immunodeficiency (SCID)
[Nucleotide Metabolism - Synthesis]
Genetic defect in immunity
Has many variations but some are related to purines (ex. adenosine deaminase, purine nucleoside phosphorylase)
a. Lesch-Nyhan syndrome
b. Hyperuricemia
c. Severe combined immunodeficiency (SCID)
d. Xanthinuria
c. Both a and b
Adenosine deaminase
Purine nucleoside phosphorylase
[Nucleotide Metabolism - Synthesis]
Severe combined immunodeficiency (SCID) is a genetic defect in immunity ; has many variations but some are related to purines. Example ______ [2]
a. Adenosine deaminase
b. Purine nucleoside phosphorylase
c. Both a and b
b. Xanthine
[Nucleotide Metabolism - Breakdown]
All purine nucleotides in the cytosol converge along the way to become_______
a. Uric acid
b. Xanthine
c. Hypoxanthine
d. Inosine
d. Xanthine oxidase
[Nucleotide Metabolism - Breakdown]
Last enzyme in purine catabolism for humans
a. HGPRT
b. Adenosine deaminase
c. Purine nucleoside phosphorylase
d. Xanthine oxidase
d. Xanthine oxidase
[Nucleotide Metabolism - Breakdown]
Enzyme that produces uric acid
a. HGPRT
b. Adenosine deaminase
c. Purine nucleoside phosphorylase
d. Xanthine oxidase
c. Gout
[Nucleotide Metabolism - Breakdown]
Excess xanthine oxidase = hyperuricemia → ________
a. Lesch-Nyhan syndrome
b. SCID
c. Gout
d. Xanthinuria
c. Xanthine
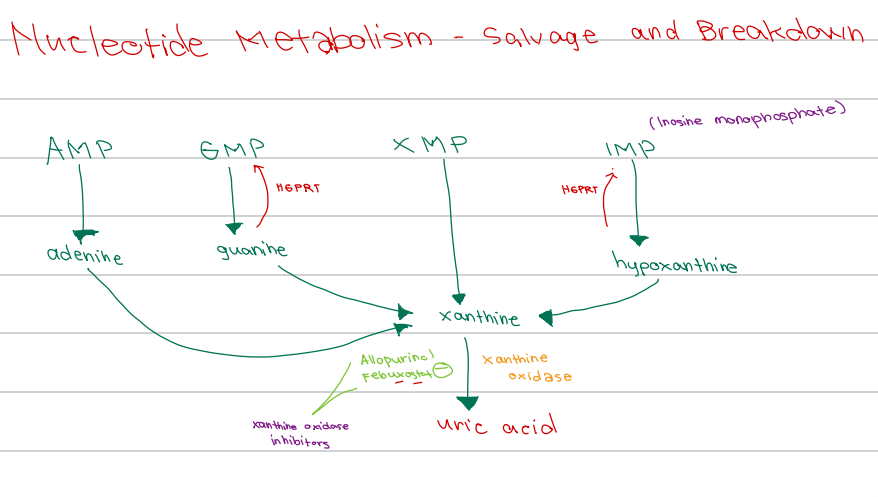
[Nucleotide Metabolism - Diagram]
AMP → adenine
GMP → guanine
IMP → hypoxanthine
All converge to form
a. Uric acid
b. Hypoxanthine
c. Xanthine
d. Inosine
b. Inosine monophosphate
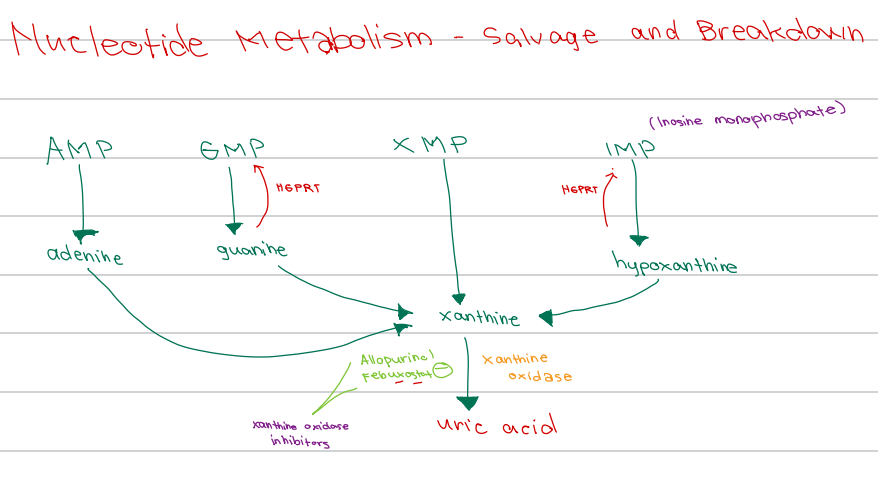
[Nucleotide Metabolism - Diagram]
IMP is also known as ______
a. Guanosine monophosphate
b. Inosine monophosphate
c. Inosine methylphosphate
d. Inositol monophosphate
d. HGPRT
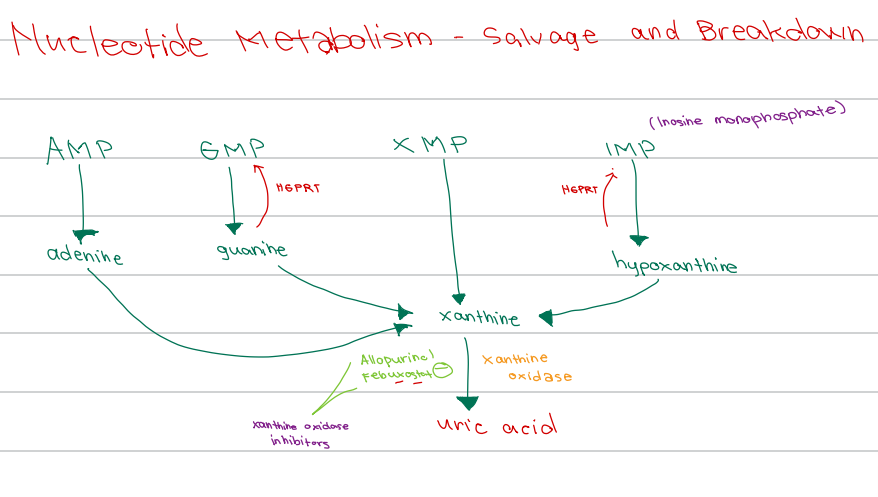
[Nucleotide Metabolism - Diagram]
Enzyme responsible for salvage of:
Guanine → GMP
Hypoxanthine → IMP
a. Adenosine deaminase
b. Xanthine oxidase
c. HMG-CoA reductase
d. HGPRT
c. Xanthine oxidase
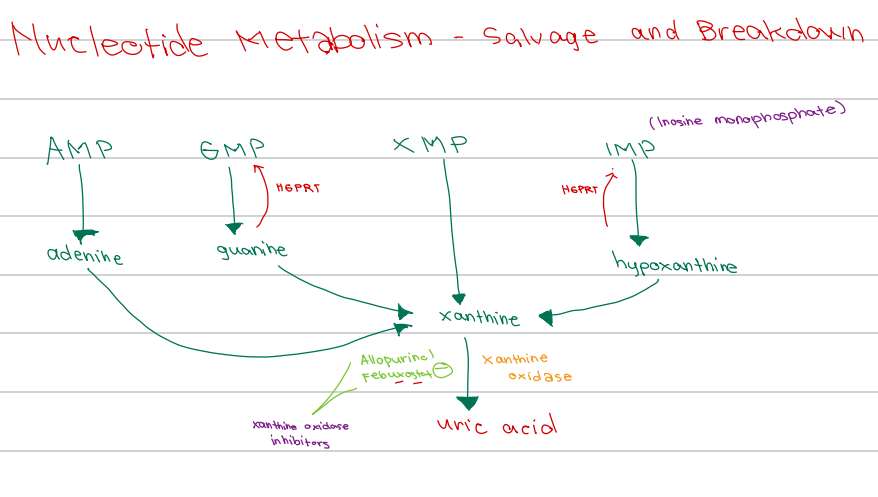
[Nucleotide Metabolism - Diagram]
Enzyme involved:
Xanthine → uric acid
a. HGPRT
b. Adenosine deaminase
c. Xanthine oxidase
d. Purine nucleoside phosphorylase
b. Xanthine oxidase inhibitors
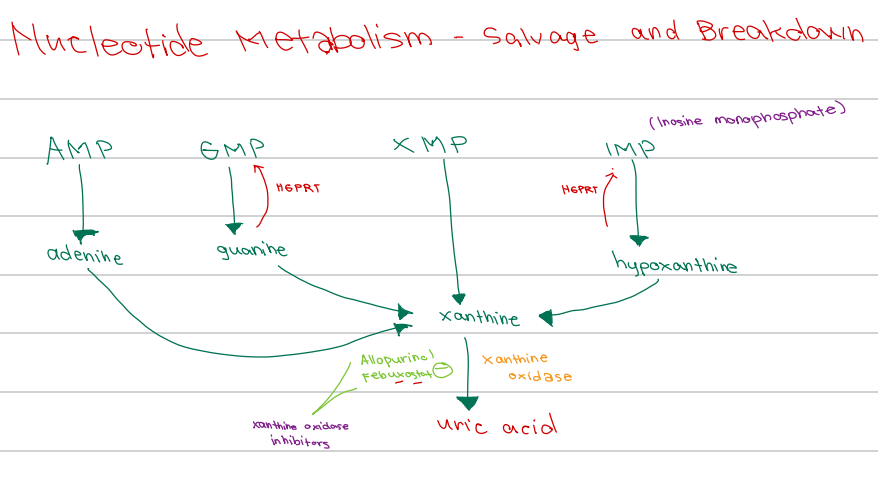
[Nucleotide Metabolism - Diagram]
Drug class of Allopurinol and Febuxostat
a. Uricosuric agents
b. Xanthine oxidase inhibitors
c. HGPRT inhibitors
d. Adenosine deaminase inhibitors
c. Xanthine
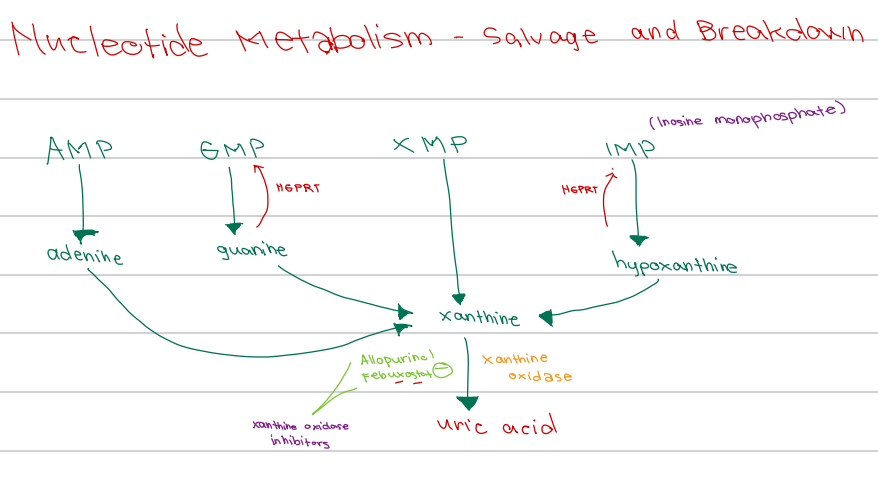
[Nucleotide Metabolism - Diagram]
XMP converges to form
a. Hypoxanthine
b. Uric acid
c. Xanthine
d. Guanine
a. True
[Amino Acid Metabolism]
In order to make the 20 proteinogenic amino acids, there exist several independent pathways
a. True
b. False
c. Food
[Amino Acid Metabolism]
The only way humans can get essential amino acids is to absorb them from ________
a. Sunlight
b. Biosynthesis
c. Food
d. Salvage pathway
a. True
[Amino Acid Metabolism]
There exist genetic defects in transporter proteins, causing malabsorption issues (ex. Hartnup disease for tryptophan)
a. True
b. False
c. Hartnup disease
[Amino Acid Metabolism]
Genetic defect in transporter proteins causing malabsorption of tryptophan
a. Lesch-Nyhan syndrome
b. Phenylketonuria
c. Hartnup disease
d. Maple syrup urine disease
a. True
[Amino Acid Metabolism]
In addition to the amino acids themselves, these are also highly valuable
a. True
b. False
b. Transamination
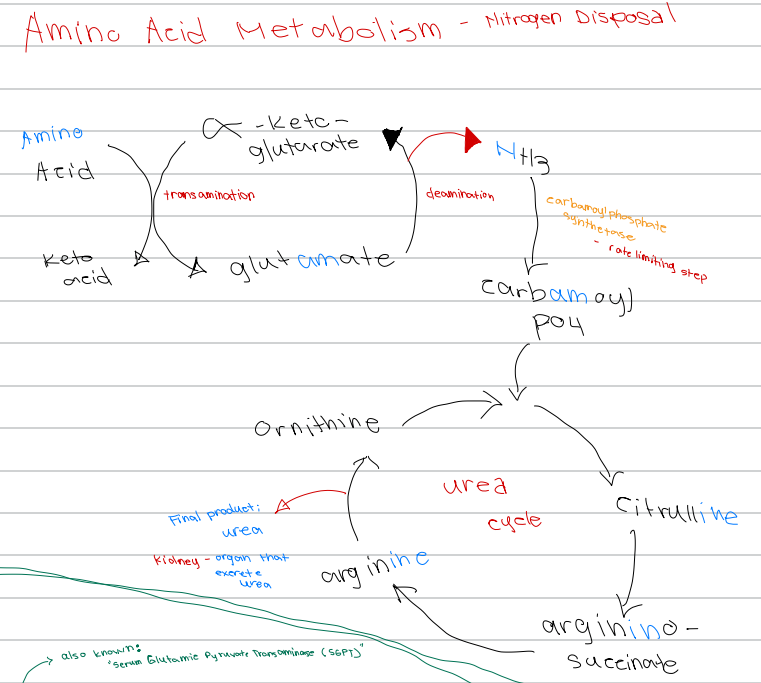
[Amino Acid Metabolism - Nitrogen Disposal - Diagram]
Amino acid + α-ketoglutarate → keto acid + glutamate; process involved
a. Deamination
b. Transamination
c. Urea cycle
d. Oxidative phosphorylation
b. Nitrogen balance
[Amino acid Metabolism - Nitrogen Disposal]
Either positive (intake > output) or negative (outtake > input)
a. Nitrogen waste
b. Nitrogen balance
c. Nitrogen disposal
d. Nitrogen excretion
a. True
[Amino acid Metabolism - Nitrogen Disposal]
Positive nitrogen balance = sufficient protein
Negative nitrogen balance = insufficient protein
a. True
b. False
c. Ammonia
[Amino acid Metabolism - Nitrogen Disposal]
_______ is the initial nitrogen waste , but is toxic
a. Urea
b. Creatinine
c. Ammonia
d. Uric acid
c. Encephalopathy
[Amino acid Metabolism - Nitrogen Disposal]
When ammonia accumulates in the brain, it can result to _________
a. Hepatic failure
b. Renal failure
c. Encephalopathy
d. Neuropathy
b. Excreted
[Amino acid Metabolism - Nitrogen Disposal]
Ammonia is converted to urea and eventually _____
a. Stored in the liver
b. Excreted
c. Recycled
d. Converted to glucose
c. Liver
[Amino acid Metabolism - Nitrogen Disposal]
The process starts once amino acids are in the
a. Kidney
b. Pancreas
c. Liver
d. Intestine
Transamination (cytosol)
Oxidative deamination (matrix)
Urea cycle (matrix and cytosol)
Nitrogen Disposal Processes [3]
c. Transamination
[Amino acid Metabolism - Nitrogen Disposal]
Nitrogen disposal process that occurs in the cytosol
a. Oxidative deamination
b. Urea cycle
c. Transamination
d. Beta-oxidation
d. Oxidative deamination
[Amino acid Metabolism - Nitrogen Disposal]
Nitrogen disposal process that occurs in:
Matrix → ammonia
a. Transamination
b. Urea cycle
c. Carbamoylation
d. Oxidative deamination
c. Urea cycle
[Amino acid Metabolism - Nitrogen Disposal]
Nitrogen disposal process that occurs in:
Matrix and cytosol → urea
a. Transamination
b. Oxidative deamination
c. Urea cycle
d. Beta-oxidation
c. Carbamoyl phosphate synthetase I (CPS I)
[Amino acid Metabolism - Nitrogen Disposal]
Rate-limiting enzyme of the urea cycle
a. Ornithine transcarbamylase
b. Argininosuccinate lyase
c. Carbamoyl phosphate synthetase I (CPS I)
d. Glutamate dehydrogenase
c. Liver
[Amino acid Metabolism - Nitrogen Disposal]
All three nitrogen disposal processes happen in the
a. Kidney
b. Pancreas
c. Liver
d. Intestine
d. Glutamate
[Amino acid Metabolism - Nitrogen Disposal]
When ammonia is made in other organs before getting to the liver. In those cases , ________ works differently but eventually gets the nitrogen to the liver
a. Aspartate
b. Alanine
c. Glutamine
d. Glutamate
RECYCLED for energy (ex. enter the Kreb’s)
Converted to SPECIAL WASTE products
[Amino acid Metabolism - Carbon Skeleton]
After the nitrogen is gone, the remaining skeleton is either ________ [2]
a. True
[Amino acid Metabolism - Carbon Skeleton]
If the carbon skeleton is fated to be special waste products , special enzymes and pathways are used
a. True
b. False
c. Aminoacidopathies
[Amino acid Metabolism - Carbon Skeleton]
Some people are born with inborn errors of metabolism and lack those enzymes, these are called ________
a. Glycogen storage diseases
b. Sphingolipidoses
c. Aminoacidopathies
d. Lipodystrophies
b. Black urine disease
[Popular Aminoacidopathies]
Alkaptonuria is also known as
a. White urine disease
b. Black urine disease
c. Red urine disease
d. Brown urine disease
c. Tyrosine
[Popular Aminoacidopathies]
Amino acid affected in Alkaptonuria
a. Methionine
b. Phenylalanine
c. Tyrosine
d. Branched-chain amino acids
d. Homogentisate oxidase
[Popular Aminoacidopathies]
Enzyme deficient in Alkaptonuria
a. Tyrosinase
b. Phenylalanine hydroxylase
c. Cystathionine synthase
d. Homogentisate oxidase
c. Crippling arthritis
[Popular Aminoacidopathies]
Notable effect of Alkaptonuria
a. Retardation
b. Light complexion
c. Crippling arthritis
d. Ectopia lentis
d. Tyrosine
[Popular Aminoacidopathies]
Amino acid affected in Albinism
a. Methionine
b. Phenylalanine
c. Branched-chain amino acids
d. Tyrosine
b. Tyrosinase
[Popular Aminoacidopathies]
Enzyme deficient in Albinism
a. Homogentisate oxidase
b. Tyrosinase
c. Cystathionine synthase
d. Phenylalanine hydroxylase
Light complexion
Photophobia
[Popular Aminoacidopathies]
Notable effects of Albinism
a. Crippling arthritis and osteoporosis
b. Light complexion and photophobia
c. Retardation and ketonuria
d. Ectopia lentis and osteoporosis
a. Albinism
[Popular Aminoacidopathies]
The term originates from the Latin word albus, meaning "white.
a. Albinism
b. Alkaptonuria
c. Hemocystinuria
d. Phenylketonuria (PKA)
c. Methionine
[Popular Aminoacidopathies]
Amino acid affected in Homocystinuria
a. Tyrosine
b. Phenylalanine
c. Methionine
d. Branched-chain amino acids
d. Cystathionine synthase
[Popular Aminoacidopathies]
Enzyme deficient in Homocystinuria
a. Tyrosinase
b. Homogentisate oxidase
c. Phenylalanine hydroxylase
d. Cystathionine synthase
Ectopia lentis
Osteoporosis
[Popular Aminoacidopathies]
Notable effects of Homocystinuria
a. Light complexion and photophobia
b. Crippling arthritis
c. Ectopia lentis and osteoporosis
d. Retardation and ketonuria
d. Phenylketonuria (PKA)
[Popular Aminoacidopathies]
Too much phenylalanine
a. Albinism
b. Alkaptonuria
c. Hemocystinuria
d. Phenylketonuria (PKA)
c. Phenylalanine
[Popular Aminoacidopathies]
Amino acid affected in Phenylketonuria (PKU)
a. Tyrosine
b. Methionine
c. Phenylalanine
d. Branched-chain amino acids
d. Phenylalanine hydroxylase
[Popular Aminoacidopathies]
Enzyme deficient in Phenylketonuria (PKU)
a. Tyrosinase
b. Cystathionine synthase
c. Homogentisate oxidase
d. Phenylalanine hydroxylase
c. Retardation
[Popular Aminoacidopathies]
Notable effect of Phenylketonuria (PKU)
a. Crippling arthritis
b. Ectopia lentis
c. Retardation
d. Light complexion
c. Aspartame
[Popular Aminoacidopathies]
Artificial sweetener that contains phenylalanine, dangerous for PKU patients
a. Saccharin
b. Sucralose
c. Aspartame
d. Stevia
d. Branched-chain amino acids
[Popular Aminoacidopathies]
Amino acids affected in Maple Syrup Urine Disease (MSUD)
a. Tyrosine
b. Methionine
c. Phenylalanine
d. Branched-chain amino acids
c. Branched-chain keto acid dehydrogenase
[Popular Aminoacidopathies]
Enzyme deficient in Maple Syrup Urine Disease (MSUD)
a. Cystathionine synthase
b. Tyrosinase
c. Branched-chain keto acid dehydrogenase
d. Phenylalanine hydroxylase
b. Ketonuria and diet restrictions
[Popular Aminoacidopathies]
Notable effects of Maple Syrup Urine Disease (MSUD)
a. Crippling arthritis and photophobia
b. Ketonuria and diet restrictions
c. Ectopia lentis and osteoporosis
d. Light complexion and retardation
d. Deamination
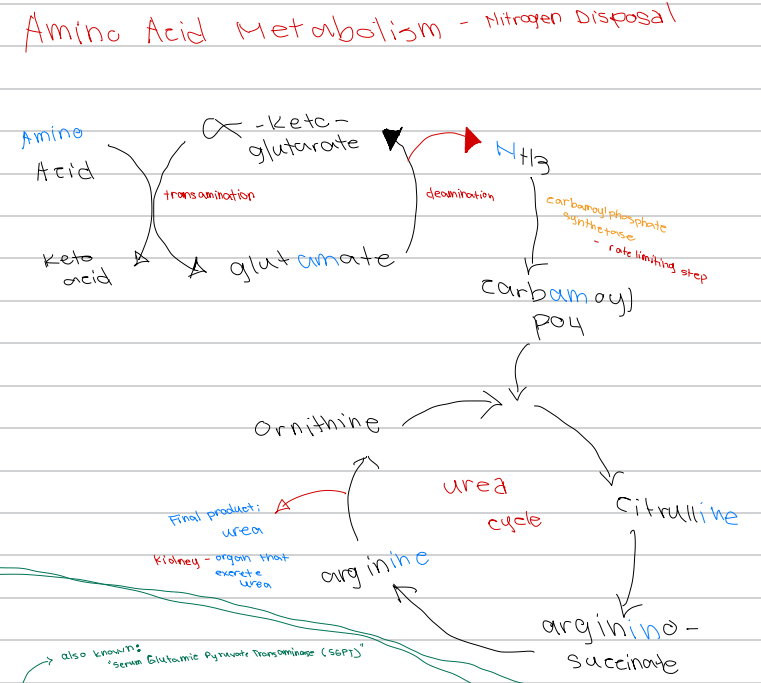
[Amino Acid Metabolism - Nitrogen Disposal - Diagram]
Glutamate → α-ketoglutarate + NH3; process involved
a. Transamination
b. Carbamoylation
c. Urea cycle
d. Deamination
d. Carbamoyl phosphate synthetase
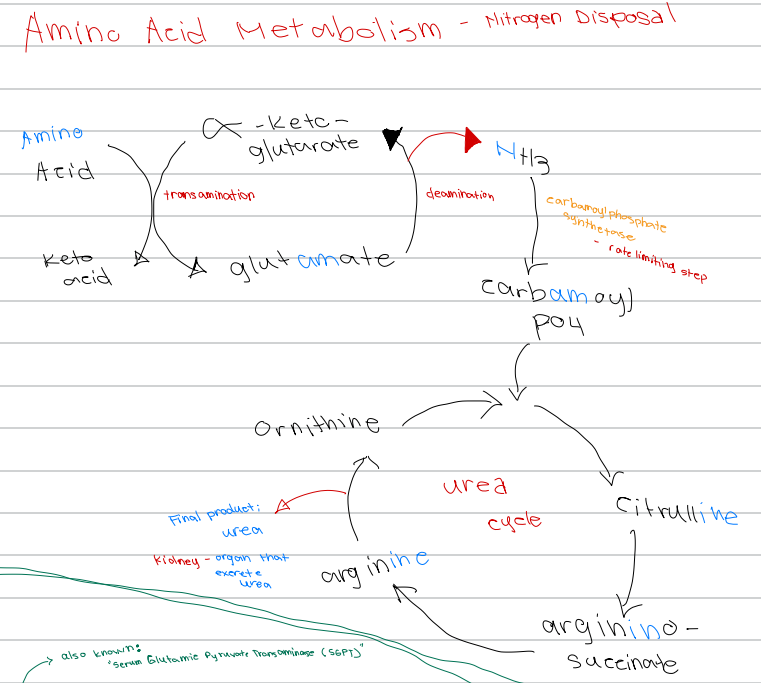
[Amino Acid Metabolism - Nitrogen Disposal - Diagram]
NH3 → carbamoyl phosphate; enzyme involved
a. Glutamate dehydrogenase
b. Ornithine transcarbamylase
c. Argininosuccinate lyase
d. Carbamoyl phosphate synthetase
c. Carbamoyl phosphate synthetase
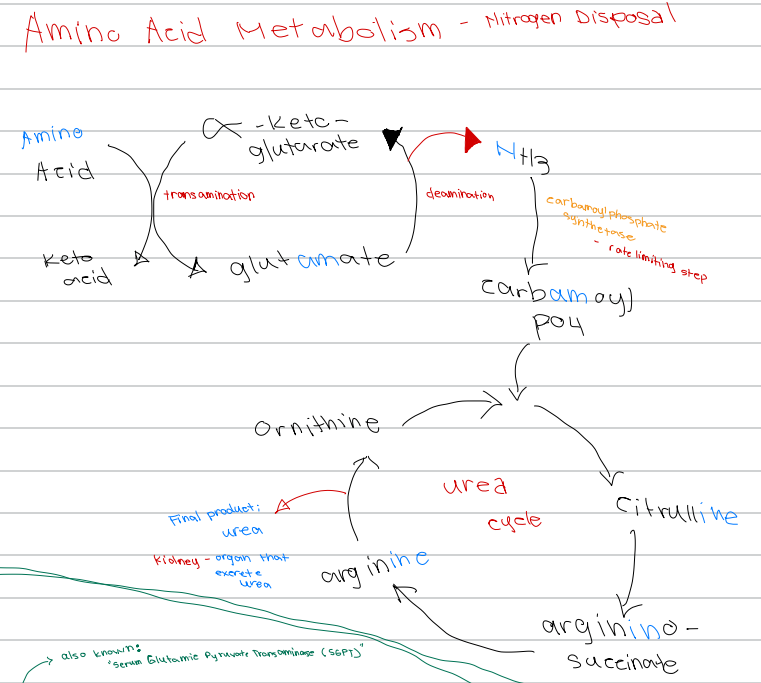
[Amino Acid Metabolism - Nitrogen Disposal - Diagram]
Rate limiting step of nitrogen disposal
a. Transamination
b. Deamination
c. Carbamoyl phosphate synthetase
d. Ornithine transcarbamylase
c. Ornithine
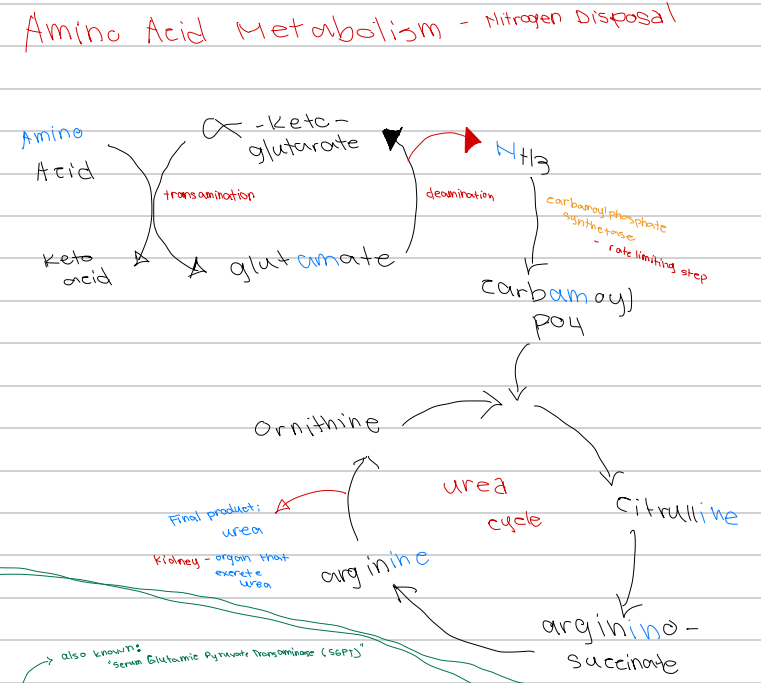
[Amino Acid Metabolism - Nitrogen Disposal - Diagram]
First intermediate of the urea cycle
a. Arginine
b. Citrulline
c. Ornithine
d. Argininosuccinate
c. Citrulline
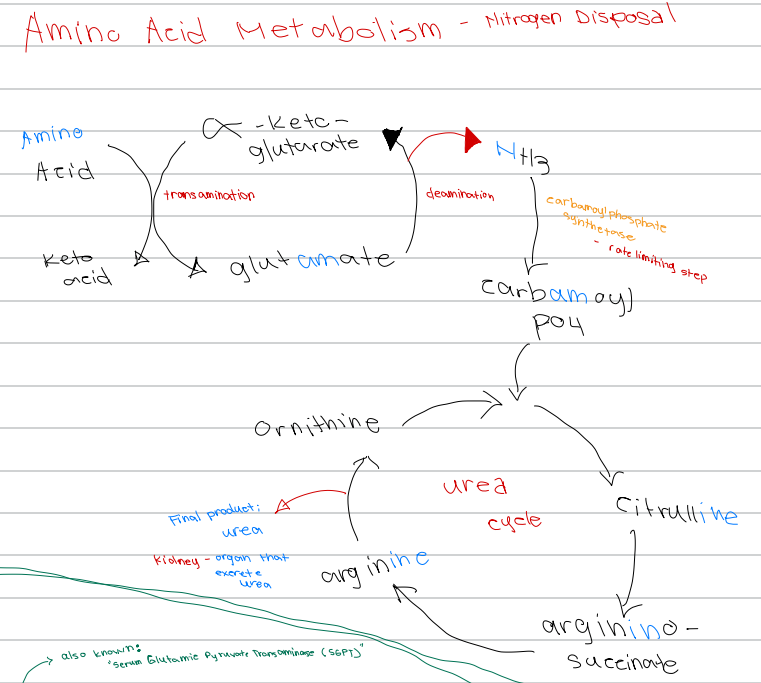
[Amino Acid Metabolism - Nitrogen Disposal - Diagram]
Ornithine + carbamoyl phosphate → ______ intermediate of the urea cycle
a. Arginine
b. Argininosuccinate
c. Citrulline
d. Glutamate
d. Argininosuccinate
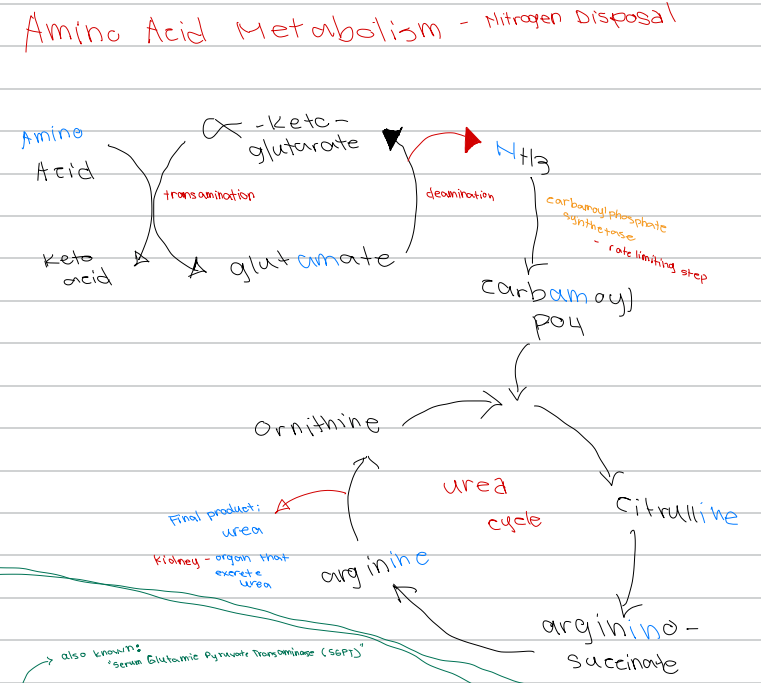
[Amino Acid Metabolism - Nitrogen Disposal - Diagram]
Citrulline → _______ intermediate of the urea cycle
a. Ornithine
b. Urea
c. Citrulline
d. Argininosuccinate