VL Alkene und Alkine & E-Reaktionen
1/19
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
20 Terms
Beta Eliminierung
Wichtigste Nebenreaktion der nukleophilen Substitution von Halogenalkanen
Eliminierung führt zur Ausbildung der Doppelbindung
Reaktionsprodukten von HX Eliminierungen sind Alkene (Verbindungen mit CC Doppelbindungen)
Alkene und Alkine zählen zu ungesättigten Kohlenwasserstoffen (C-Atome binden nicht maximale Anzahl von Wasserstoffatomen)
Alkane (CnH2n+2) -2H -> Alkene (CnH2n) -2H -> Alkine (CnH2n-2)

C-Atome in Alkanen, Alkenen, Alkinen - Eigenschaften
C-Atome in Alkanen sp3 -> gesättigte Kohlenwasserstoffe, nur C-H und C-C Einfachbindungen, eine sigma Bindung, frei drehbare Bindungen, tetraedrisch, 153pm lang, 109,5° Winkel, 346 kj/mol
C-Atome in Alkenen sp2 -> ungesättigte Kohlenwasserstoffe, C-C Doppelbindungen dazu, trigonal planar, 134pm lang, 120° Winkel, 602 kj/mol, pi und sigma Bindung, Bindungen nicht frei drehbar
C-Atome in Alkinen sp -> C-C Dreifachbindung dazu, zweite pi Bindung dazu, linear, 121pm lang, 180° Winkel, 836 kj/mol
Nomenklatur
Wie Alkane nur anstatt Endung -an ist Endung -en (Alkene) und -in (Alkine)
Mehrere Doppel- und Dreifachbindungen durch di, tri etc. angedeutet (dien, triin)
1.Längste Kette
Stammname ergibt sich aus längster Kette die möglichst viele Mehrfachbindungen enthält
Nummerierung so, dass sich niedrigste Positionsziffern für Mehrfachbindungen ergeben
2.Substituenten und Mehrfachbindungen
Substituenten der Hauptkette immer mit Positionsziffer und alphabetisch geordnet dem Stammnamen voranstellen
Position der Mehrfachbindungen als einzelne Ziffer dem Stammnamen/Endung vorangestellt -> besagt, dass die Bindung zwischen dem C-Atom mit dieser Nummer und dem nächsten C-Atom der Hauptkette lokalisiert ist
3.Konfigurationsbezeichnungen
E/Z analog zu R/S dem Gesamtnamen vorangestellt
E/Z
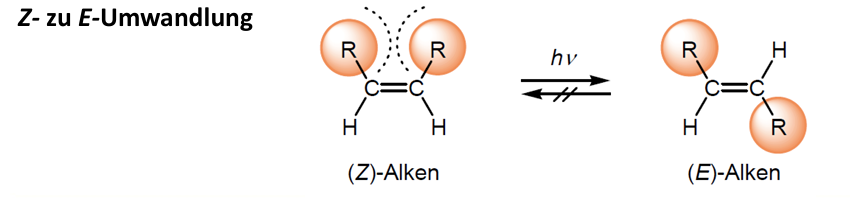
Durch C-C Doppelbindung keine freie Drehung -> unterschiedliche räumliche Anordnungen möglich
Z (zusammen) -> die jeweils höher priorisierten Gruppen liegen auf derselben Seite der Doppelbindung
E (entgegen) -> die höher priorisierten Gruppen liegen auf gegenüberliegenden Seiten
Priorität bestimmt nach CIP (höhere OZ -> höhere Priorität)
Nicht bei Alkinen da Dreifachbindung linear -> alle Substituenten auf einer Linie
Im Z-Isomer liegen größere Gruppen auf selber Seite der Doppelbindung -> kommen sich näher und stoßen sich ab (sterische Verhinderung) -> hat größere Energie -> weniger stabil
Im E-Isomer liegen größere Gruppen weiter entfernt -> stoßen sich nicht ab -> geringere Energie -> stabiler (thermodynamisch)
Um Z zu E umzuwandeln müsste Doppelbindung rotieren (schwer durch pi-Bindung) -> viel Energie benötigt (270 kj/mol) -> wandeln sich bei Raumtemperatur nicht spontan ineinander um (Z-Isomere sind metastabil) -> können nur durch hohe Temperaturen (400-500°C) oder photochemische Isomerisierung umgewandelt werden
Anwendung: Licht-induzierte Isomerisierung einer Doppelbindung läuft am Retinal ab -> Sehen
cyclische Alkene/Alkine
Nomenklatur gleich -> Präfix cyclo wird Stammnamen vorangestellt
Doppelbindungen in kleinen Ringen (<7) fast immer cis (Z)
Trans (E) Doppelbindungen führen zu hoher Ringspannung -> erst ab 8-gliedrigen Ringen
Alkene
Ungesättigte Kohlenwasserstoffe
Früher Olefine
CnH2n
Sehr elektronenreiche Teilchen, relativ schwache pi-Bindung wird leicht durch Elektrophile angegriffen
Konfigurationsstabil
C2-4 Gase, C5-15 Flüssigkeiten, <C15 Feststoff
Unpolare (hydrophobe oder lipophile) Verbindungen nicht mischbar mit Wasser
Bilden mit Sauerstoff hochexplosives Gemisch
Elektrophile Addition: Elektrophil greift Doppelbindung an (Halogenaddition, Hydrierung, HX, Epoxidierung, Dihydroxylierung, Hydroborierung) - (+I, +M Substituenten erhöhen Reaktivität)
Radikalische Addition: Halogenaddition, HX-Addition
Konzertierte Addition: Cycloadditionen
Nukleophile Addition: Addition von Nukleophilen (-I, -M Substituenten erhöhen Reaktivität)
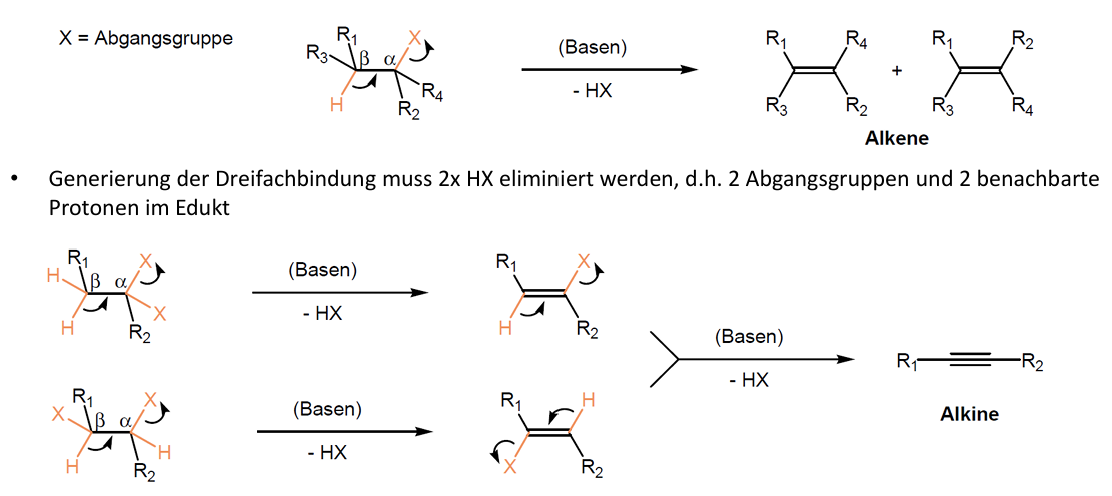
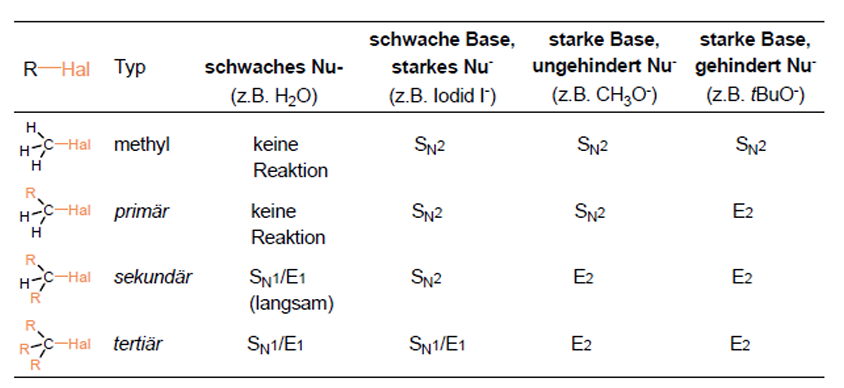
Synthese Alkene/Alkine - Eliminierung
Nebenreaktion der nukleophilen Substitution
Halogenalkane -> Alkene
Abgangsgruppe wird protoniert und entfernt -> Mehrfachbindung entsteht
Abgangsgruppe mit Proton am benachbarten beta-Kohlenstoff nötig
Nukleophil ist fast immer auch eine Base -> Nukleophile Substitution (Abgangsgruppe ersetzt) und Eliminierung (Base entfernt Proton) konkurrieren
Starke Basen, hohe Temperaturen und sterisch gehinderte Substrate -> Eliminierung bevorzugt
Für Dreifachbindung (Alkine) muss zweimal HX abgespalten werden
Beta-Eliminierung: das H Atom (Proton) stammt relativ zur Abgangsgruppe in Position 1 aus Position 2
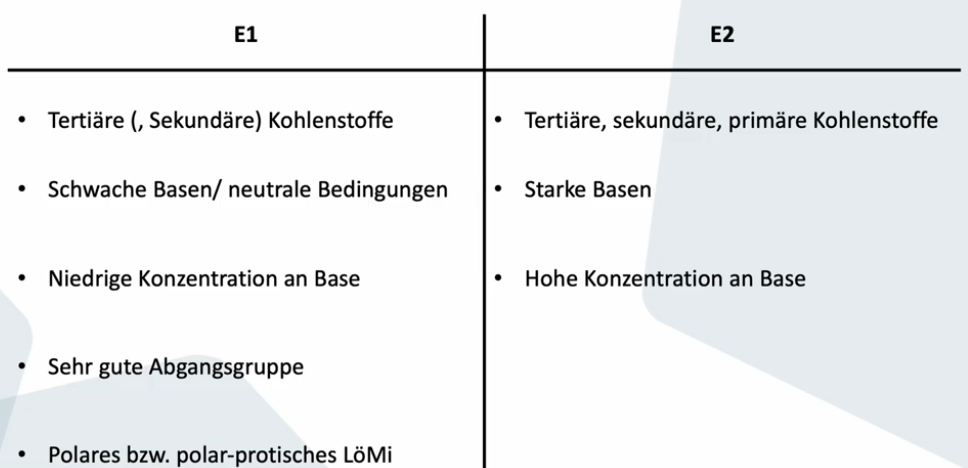
E1
Nur Substrat bestimmt Reaktionsgeschwindigkeit
Abgangsgruppe verlässt Molekül -> Carbeniumion entsteht, Base entfernt ein Proton -> Doppelbindung entsteht (Zwei Schritte nacheinander)
Carbeniumion als Zwischenstufe (nur drei Bindungen, sp2, planare Struktur) -> muss stabilisiert werden durch Hyperkonjugation oder +M oder polar protisches LM
Gute Abgangsgruppe nötig, wenn Abgangsgruppe an terminalem C -> keine E1
Räumliche Information geht verloren da kein Zusammenhang zwischen den beiden Schritten -> Gemische von E und Z entstehen -> stabileres überwiegt (meist E)
Höher substituiertes Produkt (Saytzew-Produkt) entsteht eher als Hofmann-Produkt
Reaktion kann auch im Neutralen oder bei schwacher Säurekatalyse stattfinden
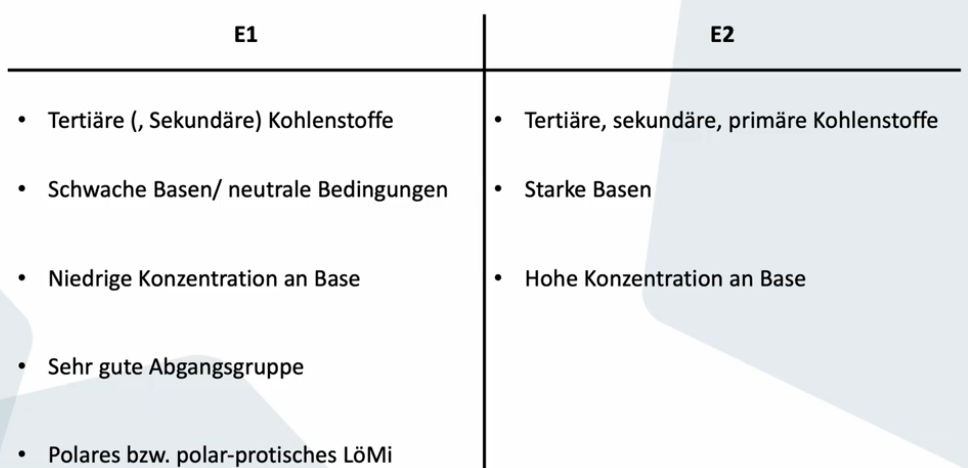
E2
Beide Reaktionspartner am geschwindigkeitsbestimmenden Schritt beteiligt
Base greift beta-H von gegenüberliegender Seite der Abgangsgruppe an, C-H Bindung bricht, Doppelbindung entsteht, Abgangsgruppe raus -> alles ein einziger Schritt
Nur ein Übergangszustand, keine Zwischenstufe
Auch 1,2-anti-Eliminierung (trans-Eliminierung) genannt -> bezeichnet NICHT Konfiguration des Produkts
Je mehr Base und je stärker desto schneller die Reaktion
Abgangsgruppe und H was deprotoniert wird müssen anti-periplanar zueinander sein -> Stereochemie des Edukts spiegelt sich in Stereochemie des Produkts wieder (stereospezifisch)
Konkurrenzreaktionen abschätzen
Je stärker die Basizität eines Nucleophils, umso eher Eliminierung
Je höher das C-Atom substituiert ist, das die Abgangsgruppe trägt, umso eher Eliminierung
Je größer und sterisch anspruchsvoller ein basisches Nucleophil ist, desto eher Eliminierung
In Gegenwart von starken Basen -> E2
In Anwesenheit von starken Basen oder guten Nucleophilen -> E1
Regioselektivität - Eliminierung
welches Alken entsteht
Mehrere beta-H die entfernt werden können -> mehrere mögliche Alkane die gebildet werden können
Bei E1 Eliminierung ist das höher substituierte Alken, das Saytzev Produkt, das Hauptprodukt
Bei Verwendung sterisch sehr anspruchsvoller Basen kann der Angriff an der höher substituierten Position soweit sterisch gehindert sein, dass das thermodynamisch weniger stabile Hofmann Produkt zum Hauptprodukt wird
Stereoselektivität - Eliminierung
welche räumliche Form des Alkens entsteht
E vor Z da stabiler
E2 -> anti-periplanare Anordnung wichtig (H und Abgangsgruppe müssen gegenüberliegen)
E1 -> oft Gemisch aus E und Z wegen Zwischenprodukt -> meist überwiegt E da stabiler
Additionsreaktion
Gegenreaktion zur Eliminierung
Pi-Bindung der C-C-Doppelbindung aufgebrochen und durch 2 neure Sigma-Bindungen ersetzt
Passiert weil Pi-Bindung deutlich schwächer ist als Sigma-Bindung -> Energiegewinn
Alkene/Alkine (Nucleophil) reagieren mit Elektrophil
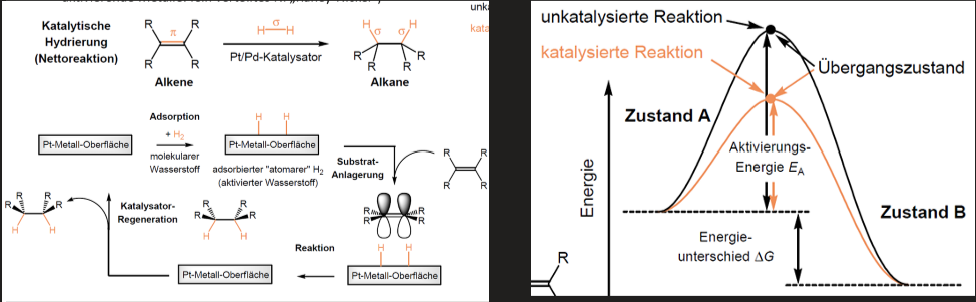
Katalytische Hydrierung
Alkene -> Alkane
Wasserstoff reagiert kaum mit Alkenen da hohe Aktivierungsenergie
Katalysator nötig (Edelmetalle: Pd, Pt, auch aktivierende Metalle: fein verteiltes Ni)
Reaktionsbeschleunigung durch Absenkung der Aktivierungsbarriere
Katalysator wirkt nicht thermodynamisch sondern kinetisch und wird in Reaktionen nicht verbraucht (Regeneration)
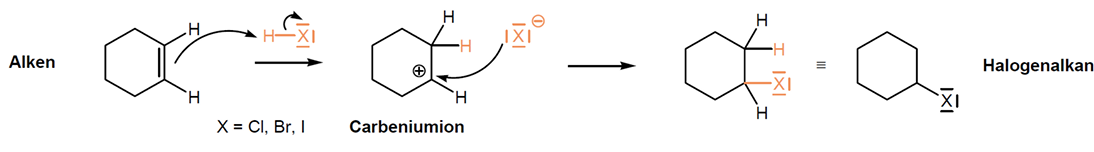
Elektrophile Addition - Halogenwasserstoffsäuren
Iodwasserstoff HI, Bromwasserstoffsäure HBr, Salzsäure HCL
Elektrophiler Angriff eines Protons an Doppelbindung des Alkens -> Carbeniumion entsteht (tertiäres ist am stabilsten -> Markovnikoff-Regel)
Halogenid greift Carbeniumion nucleophil an -> zweite Sigma-Bindung entsteht -> Halogenalkan
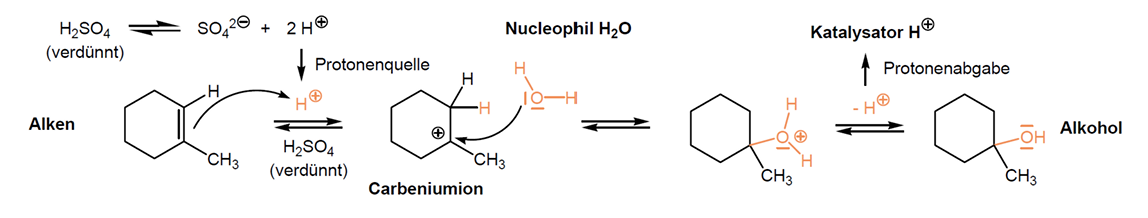
Elektrophile Addition - Hydratisierung
Addition von Wasser an C-C Doppelbindung läuft nicht spontan da Alkene nicht mit Wasser mischbar und Wasser ist kein Elektrophil sondern Nucleophil
Findet Säure-katalysiert statt -> analog zur Addition von Halogenwasserstoffsäuren (Protonierung der Doppelbindung, Carbeniumion, Angriff Wasser, Deprotonierung)
Cis-Addition
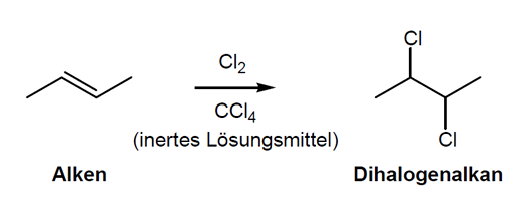
Elektrophile Addition - Halogenierung
Passiert mit Cl2 und Br2 aber nt mit F2 (zur reaktiv) oder I2 (zu unreaktiv) -> Dihalogenalkane
Trans-Addition
1.Halogen greift senkrecht zur pi-Bindung des Alkens an
2.Halogen-Halogen Bindung wird polarisiert (Vorkomplexierung in pi-Komplex)
3.Bruch der Halogen-Halogen Bindung -> cyclisches 3-Ring Halogeniumion gebildet
4.verbleibendes Halogenid macht Rückseitenangriff auf ein C-Atom des Halogeniumions in Sn2-artiger Reaktion -> trans-Additionsprodukt gebildet
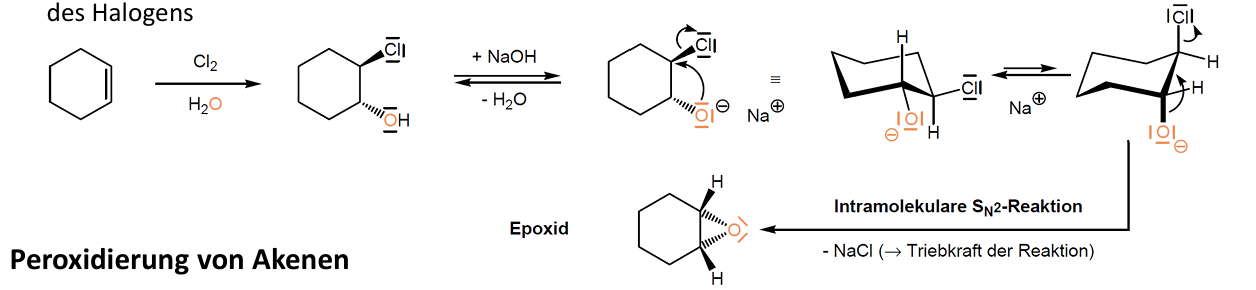
Elektrophile Addition - Epoxidierung
Epoxide sind dreigliedrige Sauerstoffringe
Entstehen durch Chlor- und Bromhydrine -> basenkatalysierte intramolekulare Sn2 Reaktion unter Substitution des Halogens -> Ring schließt sich
Alternative: direkte Oxidation der Alkene mit Persäuren
Stereoselektive Synthese von Diolen
Diole haben zwei Hydroxygruppen, wenn diese an benachbarten Kohlenstoffen sitzen -> 1,2-Diol
Cis-1,2-Diol -> syn-Addition ODER Trans-1,2-Diol -> anti-Addition
1.Aus Alken wird Epoxid gebildet durch Reagenz mCPBA
2.Epoxid wird hydrolysiert (Ringöffnung durch Sn2-artigen Rückseitenangriff)
Oxidative Spaltung von C-C Doppelbindungen
C=C -> C=O + O=C