Session 11: Prions & Prion Diseases
1/36
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
37 Terms
Prion disease
A disease caused by an abnormal protein particle that infects brain tissue.
Neurodegenerative disease.
Alternatively known as Transmissable Spongiform Encephalopathy (TSE).
Sheep prion disease
Scrapie
Cow prion disease
Bovine spongiform encephalopathy (BSE) or mad cow disease
Deer prion disease
Chronic wasting disease (CWD)
In humans, prion diseases include...
- Creutzfeldt-Jakob disease (CJD) and v-CJD
- Kuru
- Fatal familial insomnia (FFI)
- Gerstmann-Straussler-Scheinke syndrome (GSS)
Symptoms of prion disease
Anxiety/depression
Ataxia
Memory loss, loss of cognition
Dystonia
Incontinence (bowl & urinary)
Inevitably fatal (no cures, only symptomatic treatment)
Characteristics of prion disease
- Neurodegenerative
- Neuronal death leading to spongiform appearance of brain
- Proliferation of astrocytes & microglia
- Build up of amyloid plaques (protein aggregates)
- Evidence of oxidative stress
Prion diseases can occur via three mechanisms
1) Sporadic (spontaneous) = 85-90%
2) Genetic (familial) = 10-15%
3) Acquired (infectious/transmitted) = 2-5%
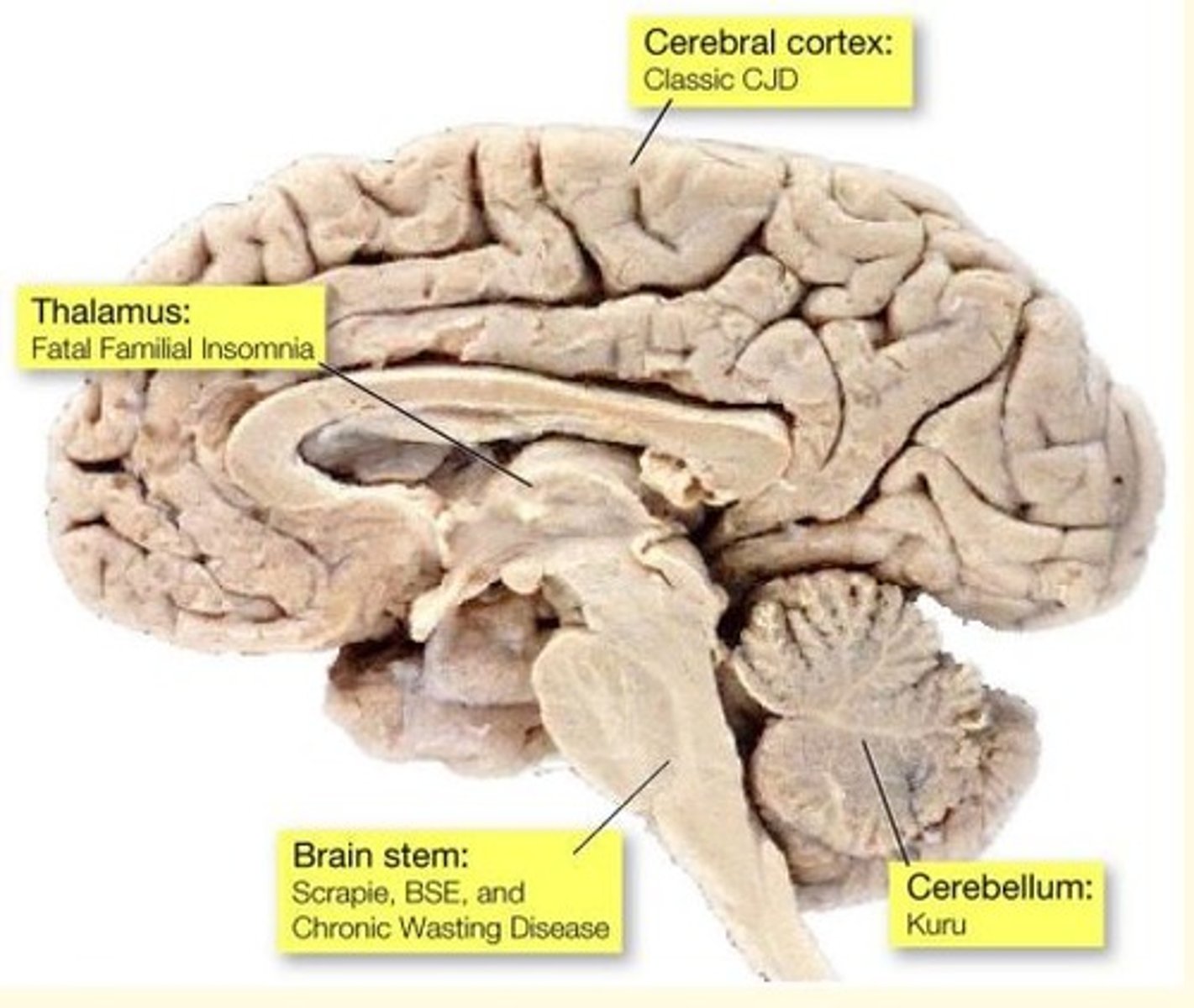
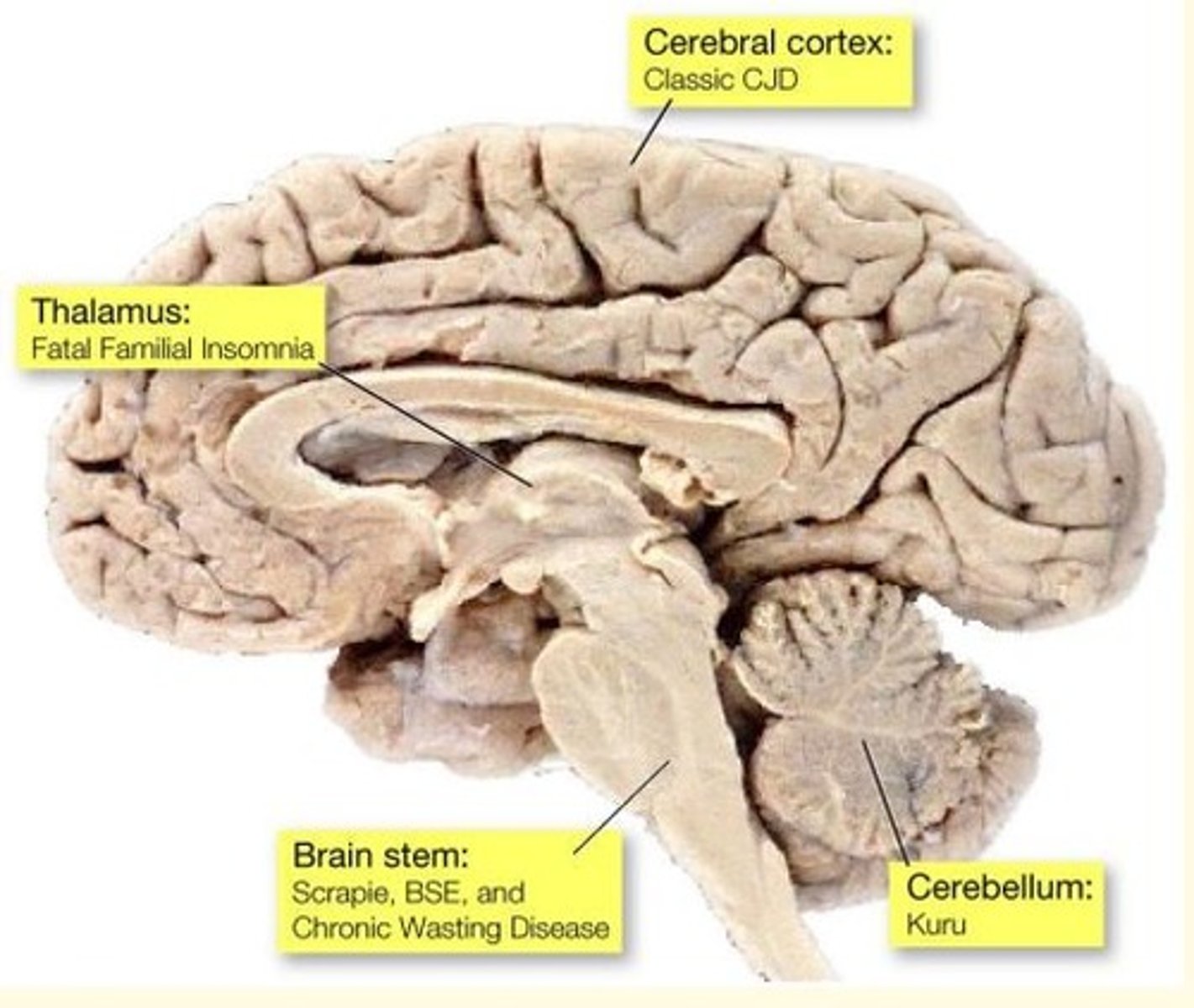
Different forms of prion disease affect different parts of the brain
Prion disease of the cerebral cortex causes what prion disease?
Classical CJD
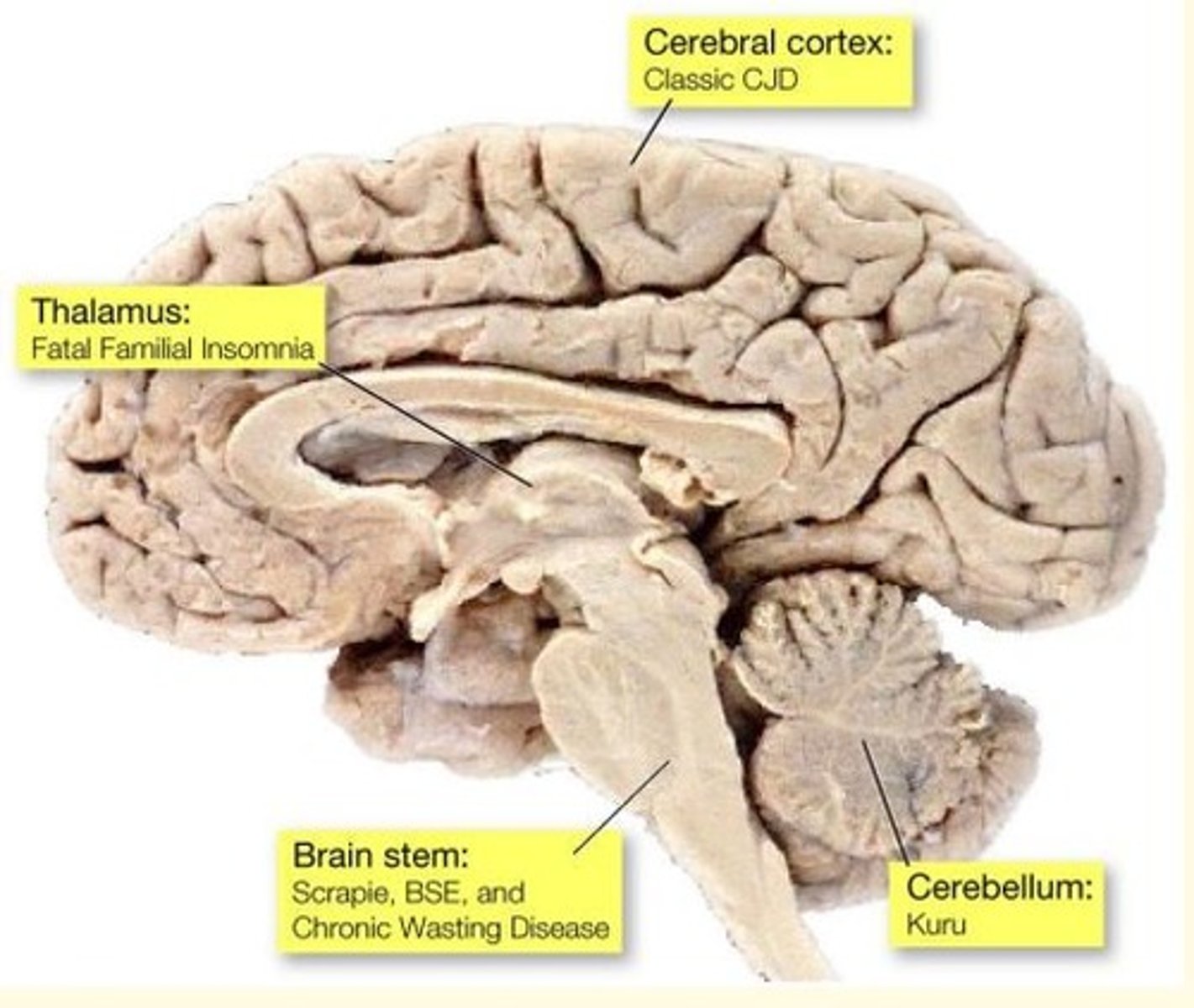
Different forms of prion disease affect different parts of the brain
Prion disease of the thalamus causes what prion disease?
Fatal Familial Insomnia
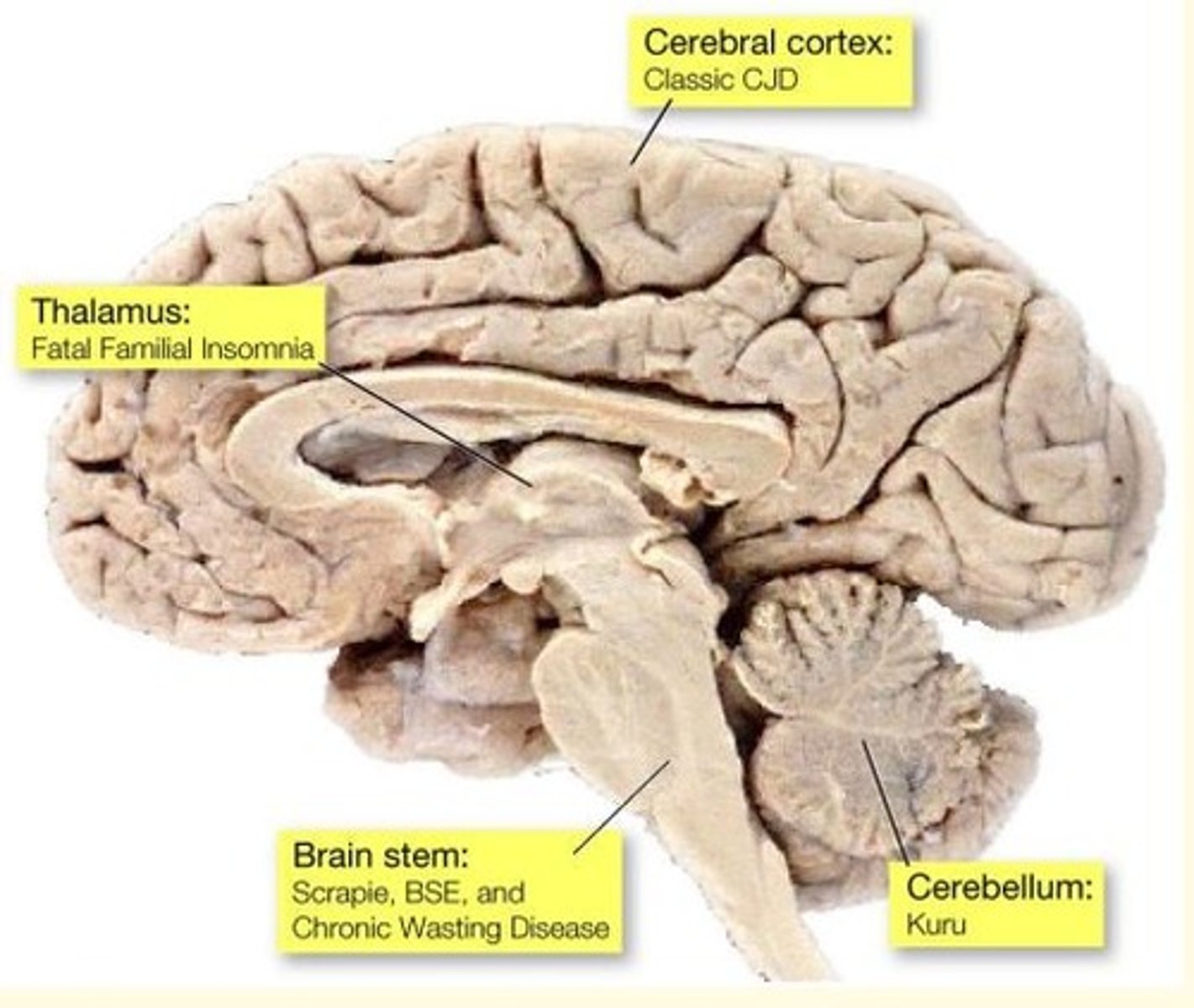
Different forms of prion disease affect different parts of the brain
Prion disease of the brainstem causes what prion disease?
Scrapie
BSE
Chronic wasting disease (CWD)
Different forms of prion disease affect different parts of the brain
Prion disease of the cerebellum causes what prion disease?
Kuru
Histology of prion diseases
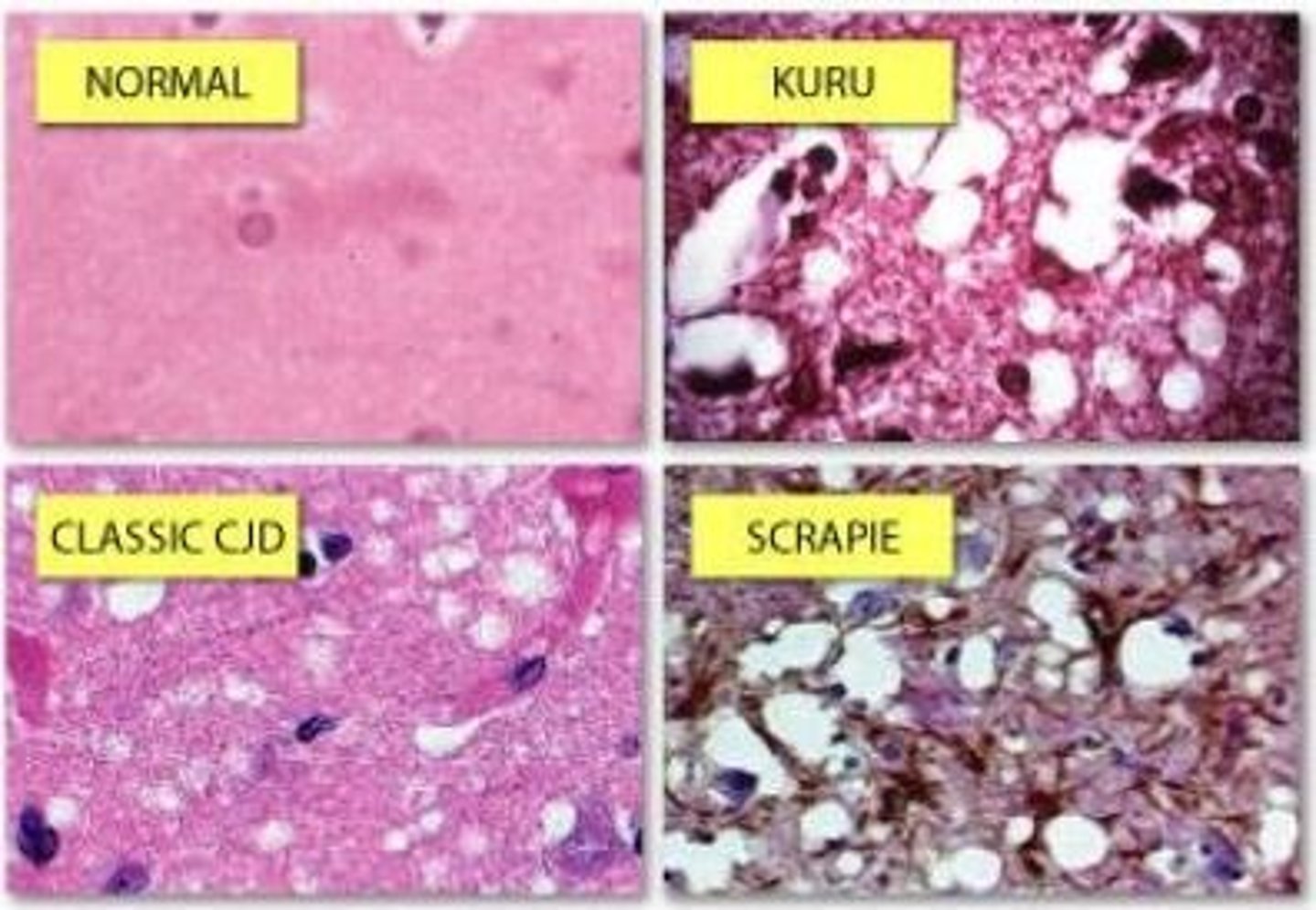
Characteristic prion pathology
- Spongiform changes
- Microglia
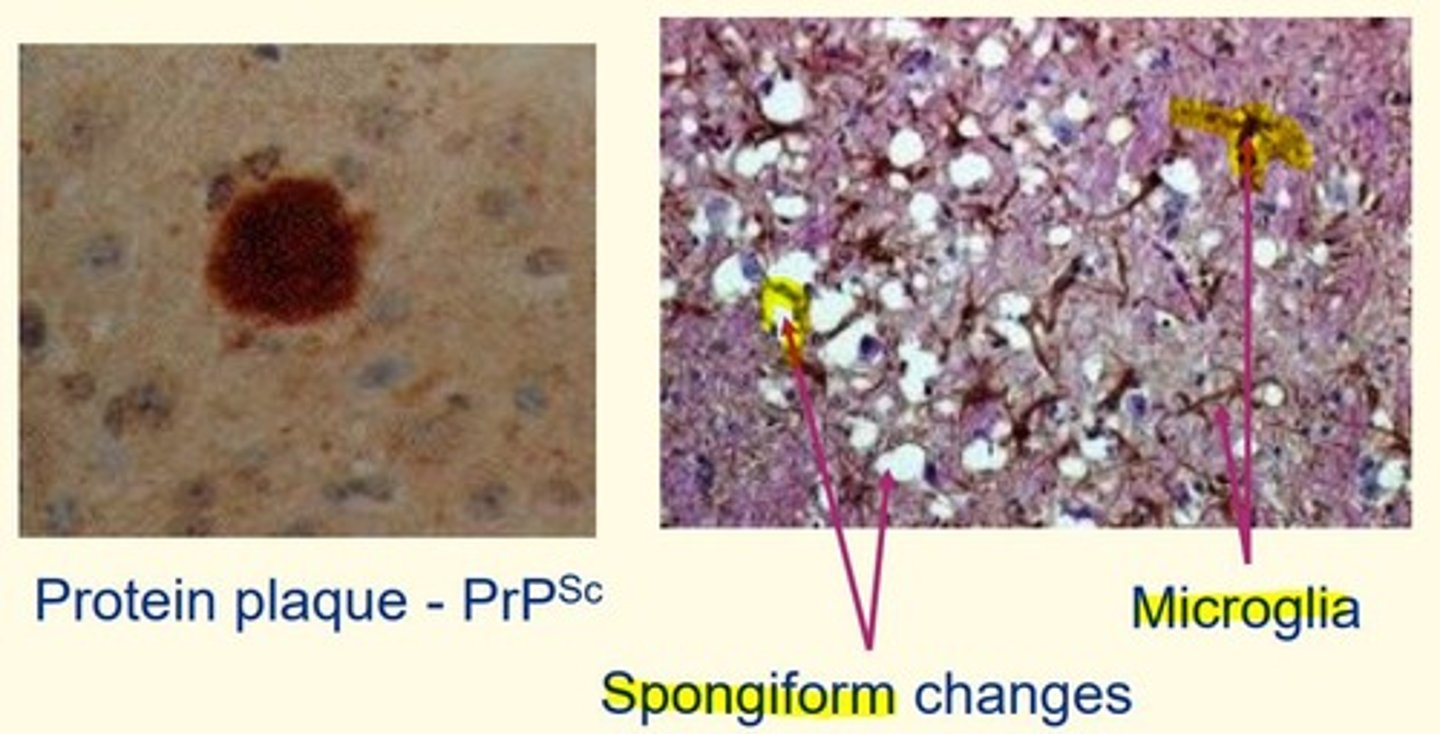
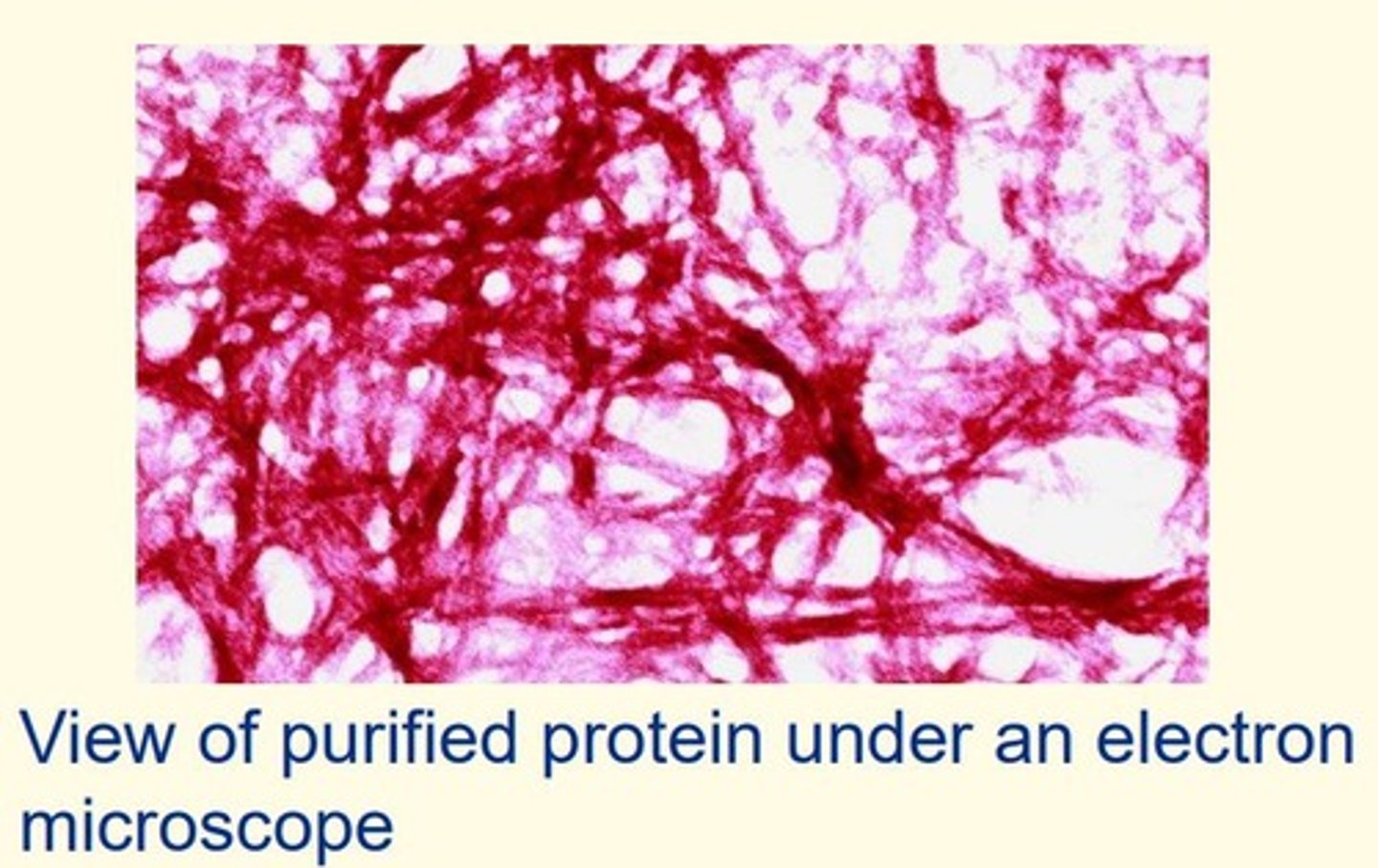
Amyloid plaques
Comprised of protein
B-pleated sheet rich
Proteins stack up to make fibrils (fibres)
Clump together to make a plaque
Prion protein (PrP)
Prion fibrils
Protein found in prion diseases (TSEs)
Prion proteins
Protein found in Alzheimer's disease
Amyloid-beta protein
Protein found in Parkinson's disease
Alpha-synuclein
Protein found in Huntington's disease
Huntingtin
Protein found in Wilson's disease
Defects in copper metabolism
Protein found in ALS
Defects in superoxide dismutase
Which prion diseases are transmissible?
- Kuru
- BSE
- v-CJD
Evidence that prion diseases are transmissible
- Injections of diseased brain tissue into another healthy animal of same species transmits the disease
- Suggests an infectious agent such as a virus BUT no viral evidence has been found in brain extracts
- Treating the brain extracts with agents (e.g., ultraviolet light/nucleases) that destroy nucleic acids does NOT reduce their infectiousness
- This evidence indicates that the infectious agent in the TSEs is a protein
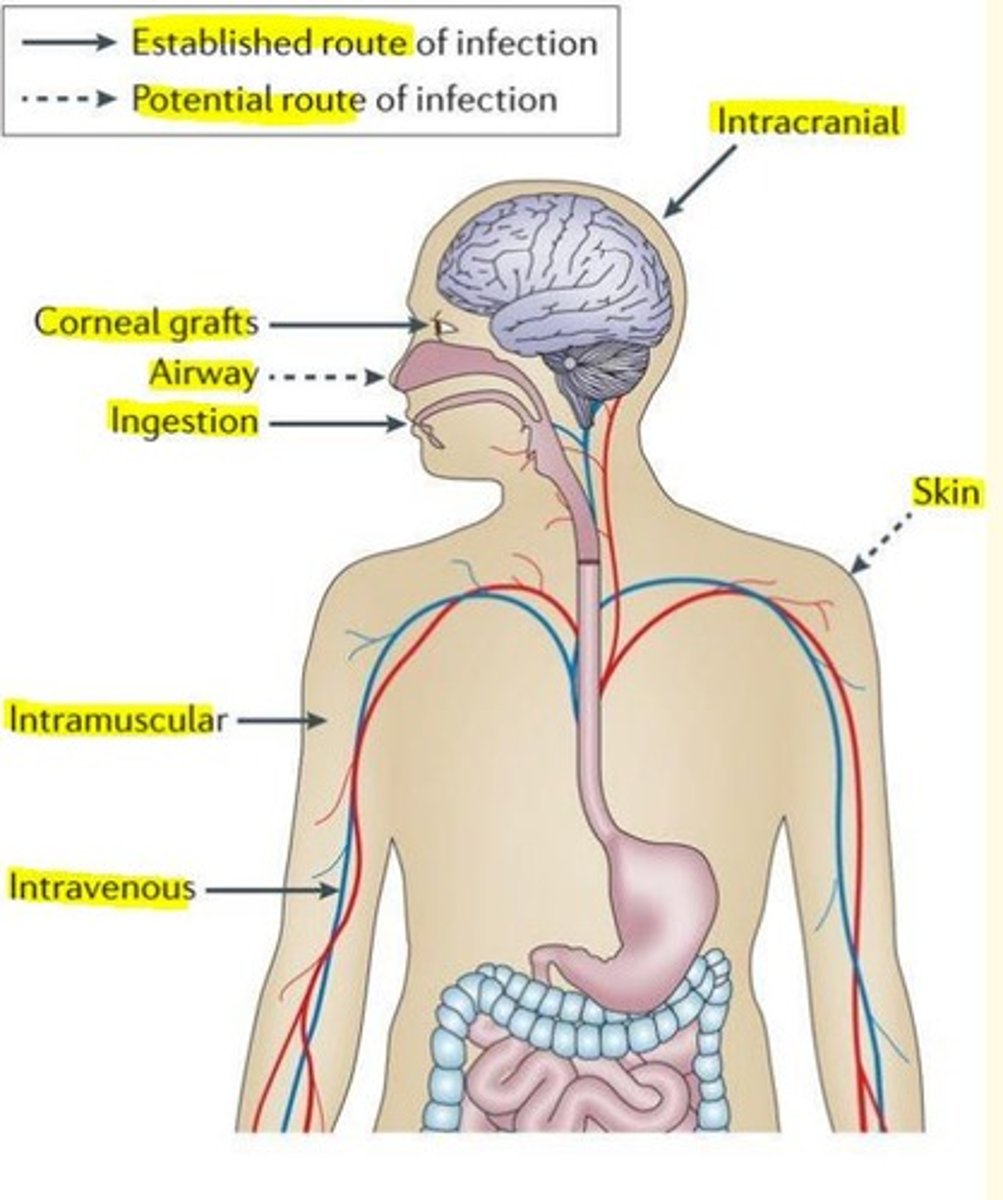
Routes of infection of prion disease
Ingestion (kuru, v-CJD)
Iatrogenic
Corneal grafts
Dura mater grafts
Human derived growth injection hGH
Experimental transmission in animal models
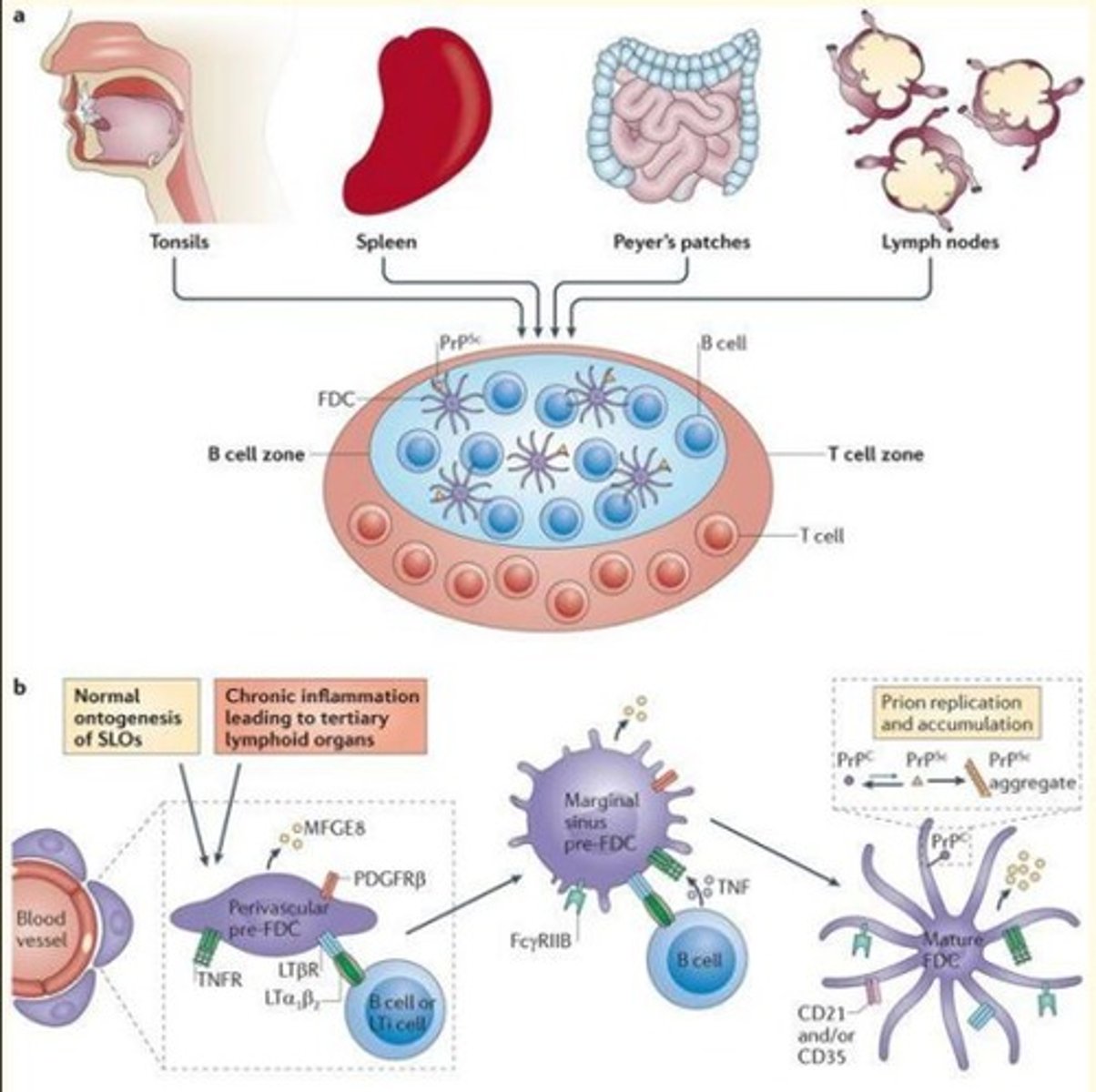
Describe pathogenesis of prion disease
1) Prions detected by immune system (resistant to proteolysis)
2) Prions replicate inside lymphoid tissue (especially follicular dendritic cells) e.g., tonsils, spleen, peyer's patches, lymph nodes
3) Prions enter the CNS via nerves of the autonomic nervous system (ANS)
Key event in prion disease pathogenesis
Seroconversion of PrPc (normal cellular form of prion protein) to PrPSc
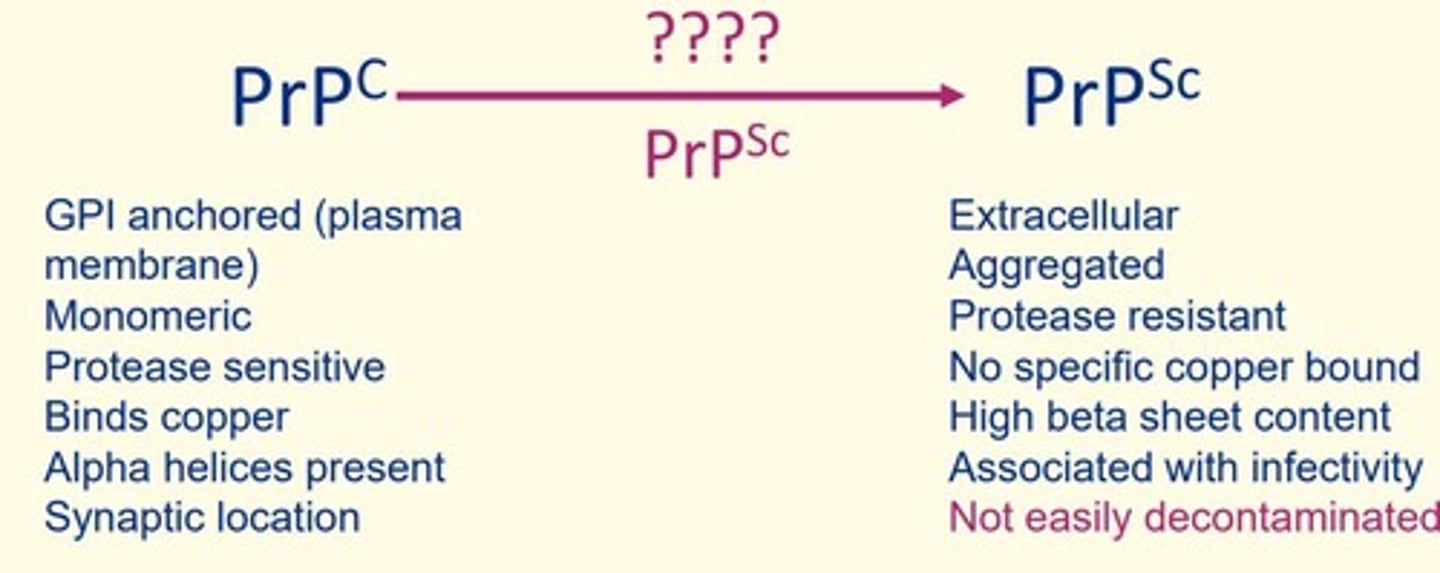
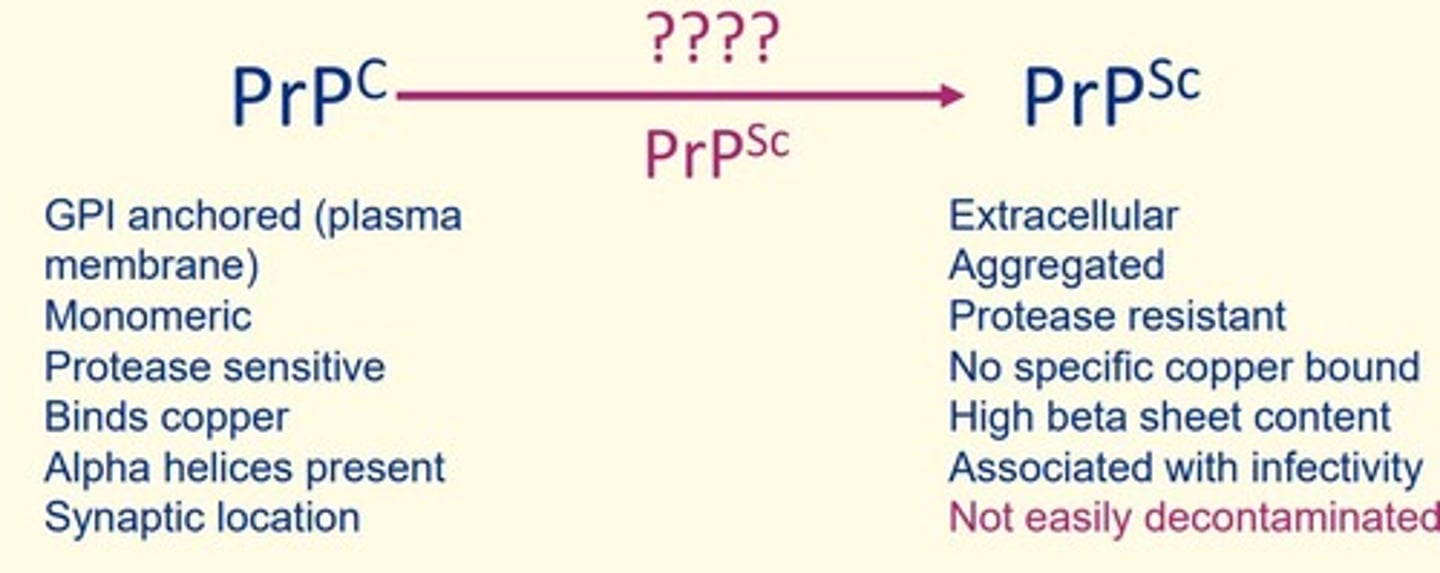
Some features of normal cellular form of prion protein (PrPc)
GPI anchored (plasma membrane)
Monomeric
Protease sensitive
Binds copper
Alpha helices present
Synaptic location
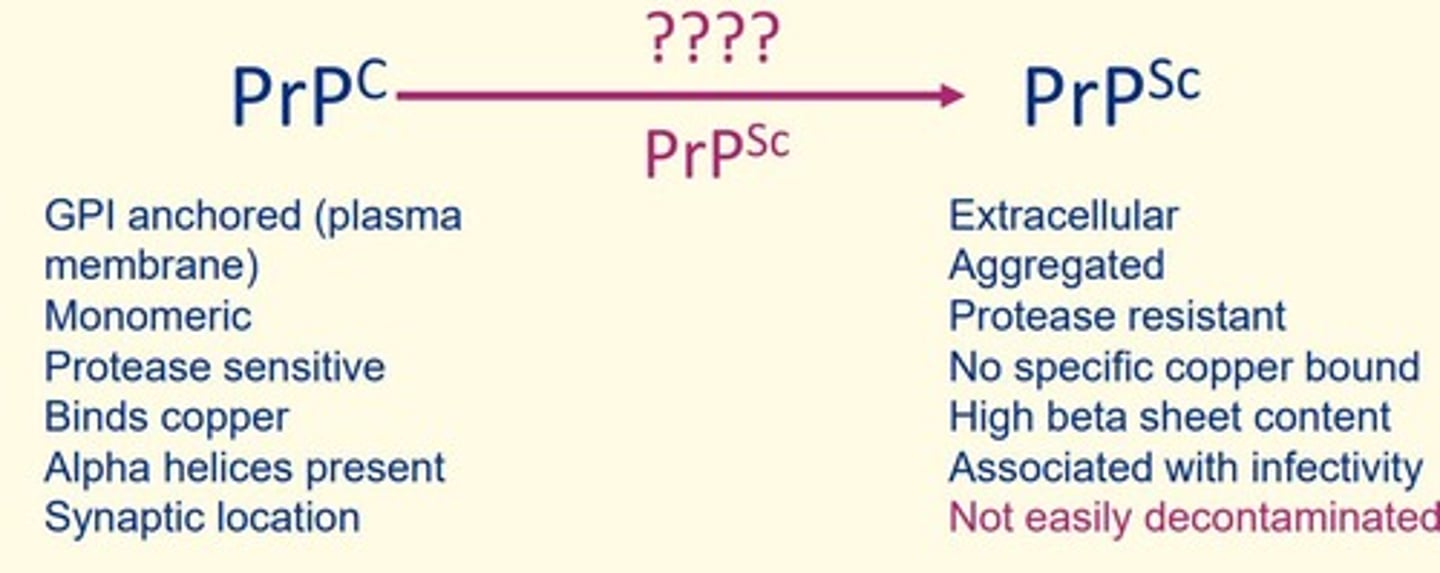
Some features of diseased form of prion protein (PrPSc)
Extracellular
Aggregated
Protease resistant
No specific copper bound
High beta sheet content
Associated with infectivity
Not easily decontaminated
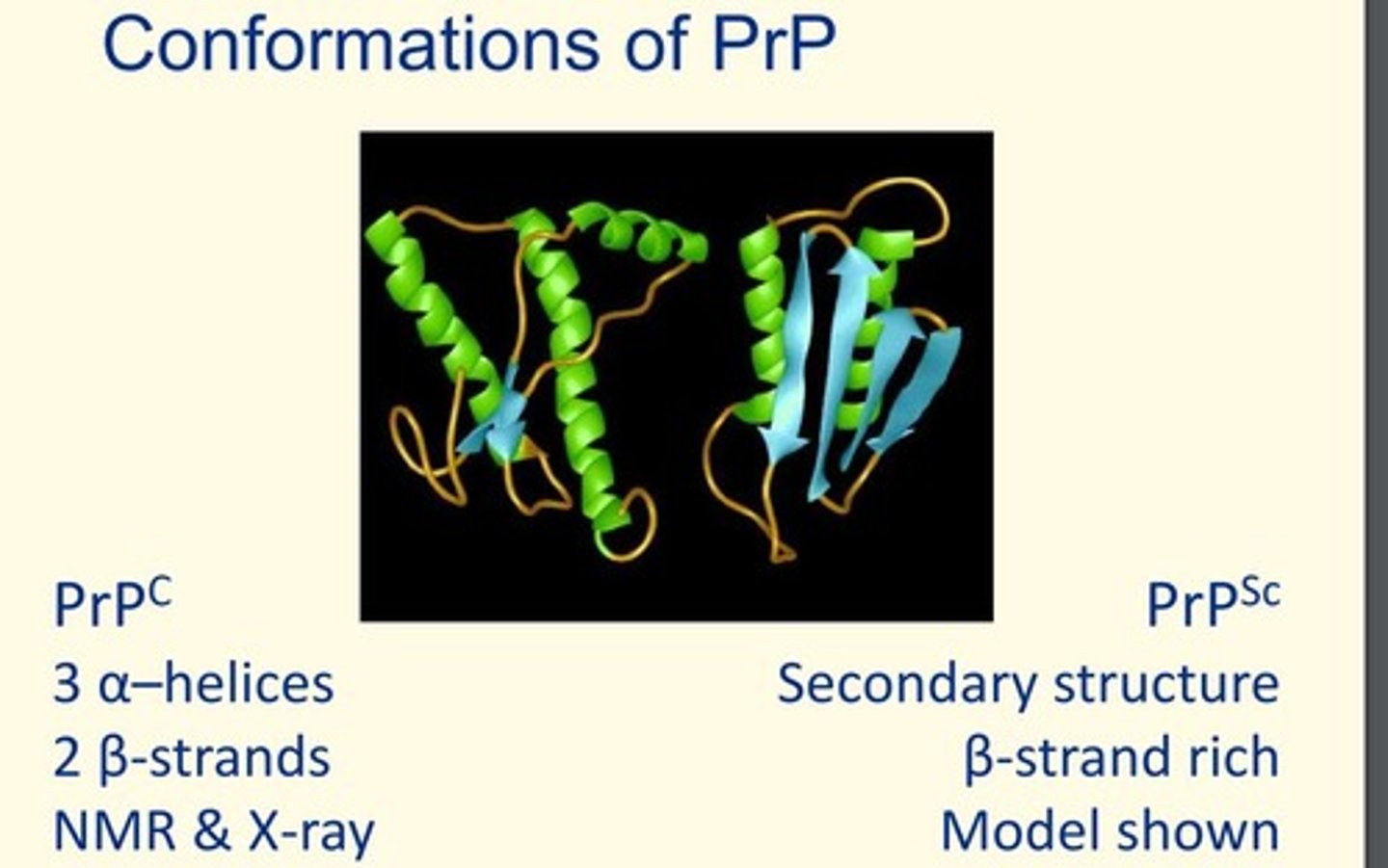
Conformations of PrP
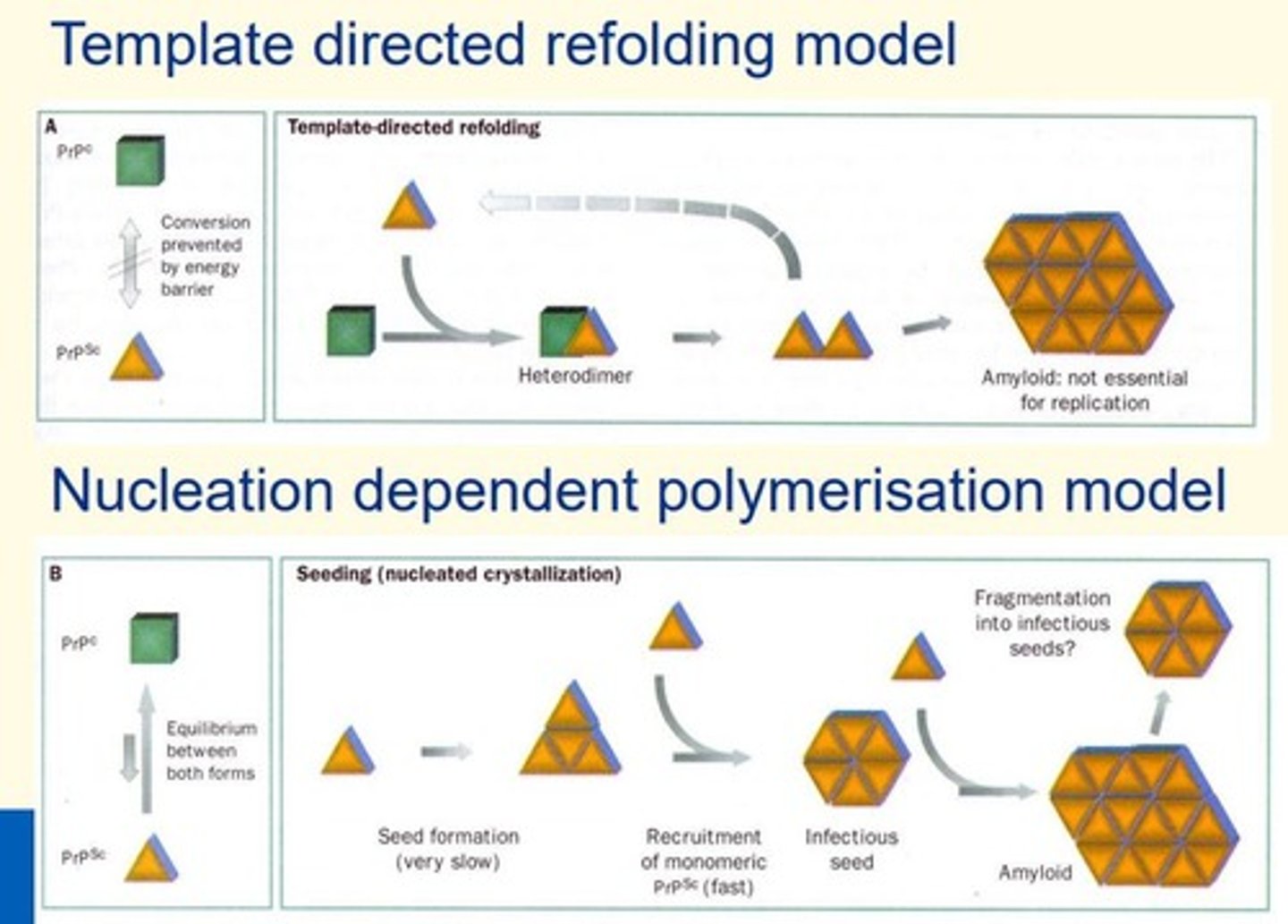
Proposed mechanisms of PrPSc catalysed conversion
1) Template directed refolding model
2) Nucleation dependent polymerisation model (seeding)
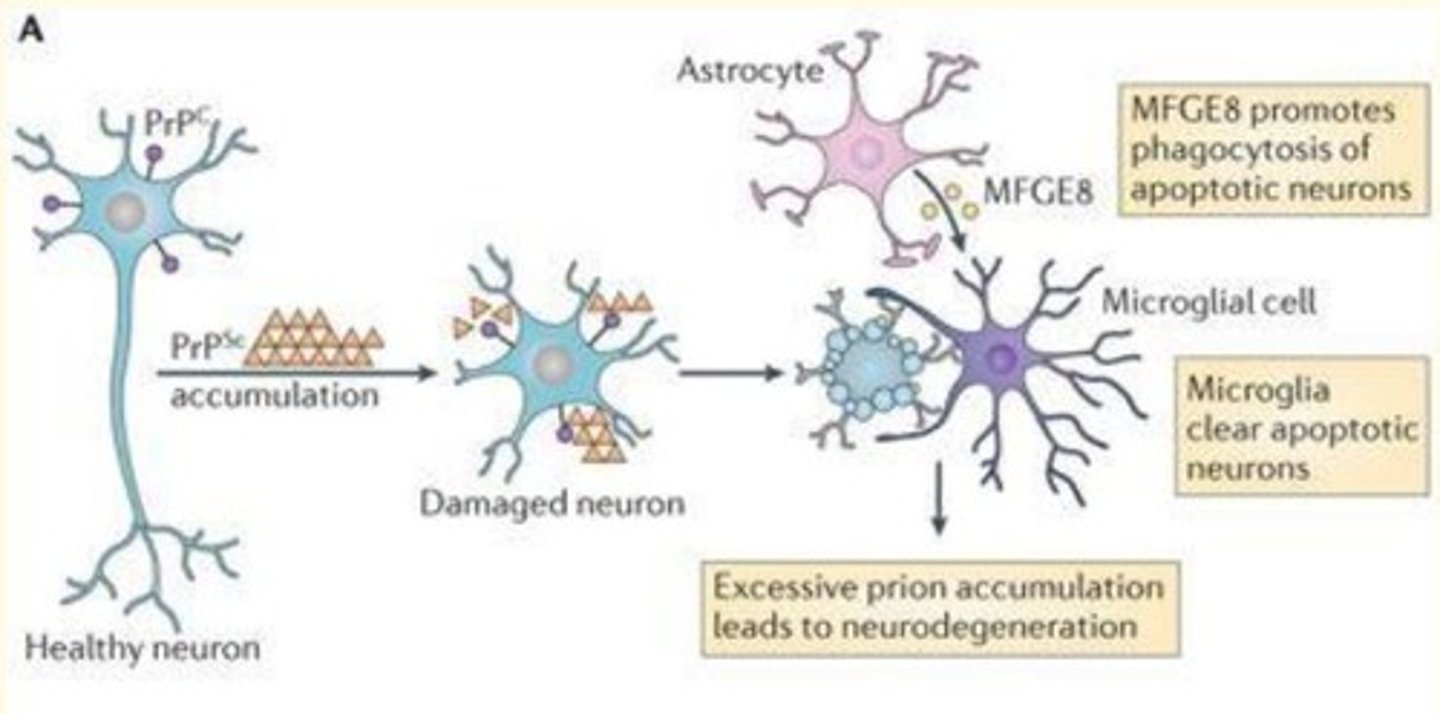
Mechanism of neurodegeneration in prion disease
1) PrPSc is cytotoxic - it accumulates in cells causing damage to neurons
2) Excess prion accumulation leads to neurodegeneration
3) MFGE8 secreted by astrocytes - promotes the phagocytosis of apoptotic neurons
4) Microglia clear apoptotic neurons
How do inherited prion diseases come about?
Mutations destabilise PrPc and make PrPSc more energetically favourable
CJD can occur with no family history of the disease and no known exposure to infectious prions & late-onset.
How do sporadic prion diseases come about?
- A spontaneous somatic mutation may have occurred in one of the prnp genes in a cell
- Normal PrPC protein may have spontaneously converted into the PrPSc form
- Tends to be a susceptibility polymorphism in their prnp genes
Guidance when treating patients with prion disease
- Perform the intervention in an operating theatre
- Schedule at the end of the list, to allow cleaning
- Involve the minimum number of personnel required
- Protective clothing should be worn, i.e., liquid repellent operating gown, over a plastic apron, gloves, mask and goggles, or full-face visor; for symptomatic patients – this protective clothing should be single-use
- Single-use disposable surgical instruments and equipment used wherever possible – subsequently destroy with incineration or sent to instrument store
- Effective tracking of reusable instruments should be in place, so that instruments can be related to use on a particular patient
Blood & blood product guidance (safety)
Safety measures to reduce any possible risk of spreading v-CJD through blood…
- Withdrawal & recall of any blood components donated by anyone who develops v-CJD & importing plasma from USA
- Removing white blood cells from all blood used for transfusions (leucodepletion)
- Not accepting donations from people who may have received blood transfusion since 1980
- Promote appropriate use of blood, tissues and alternative through NHS
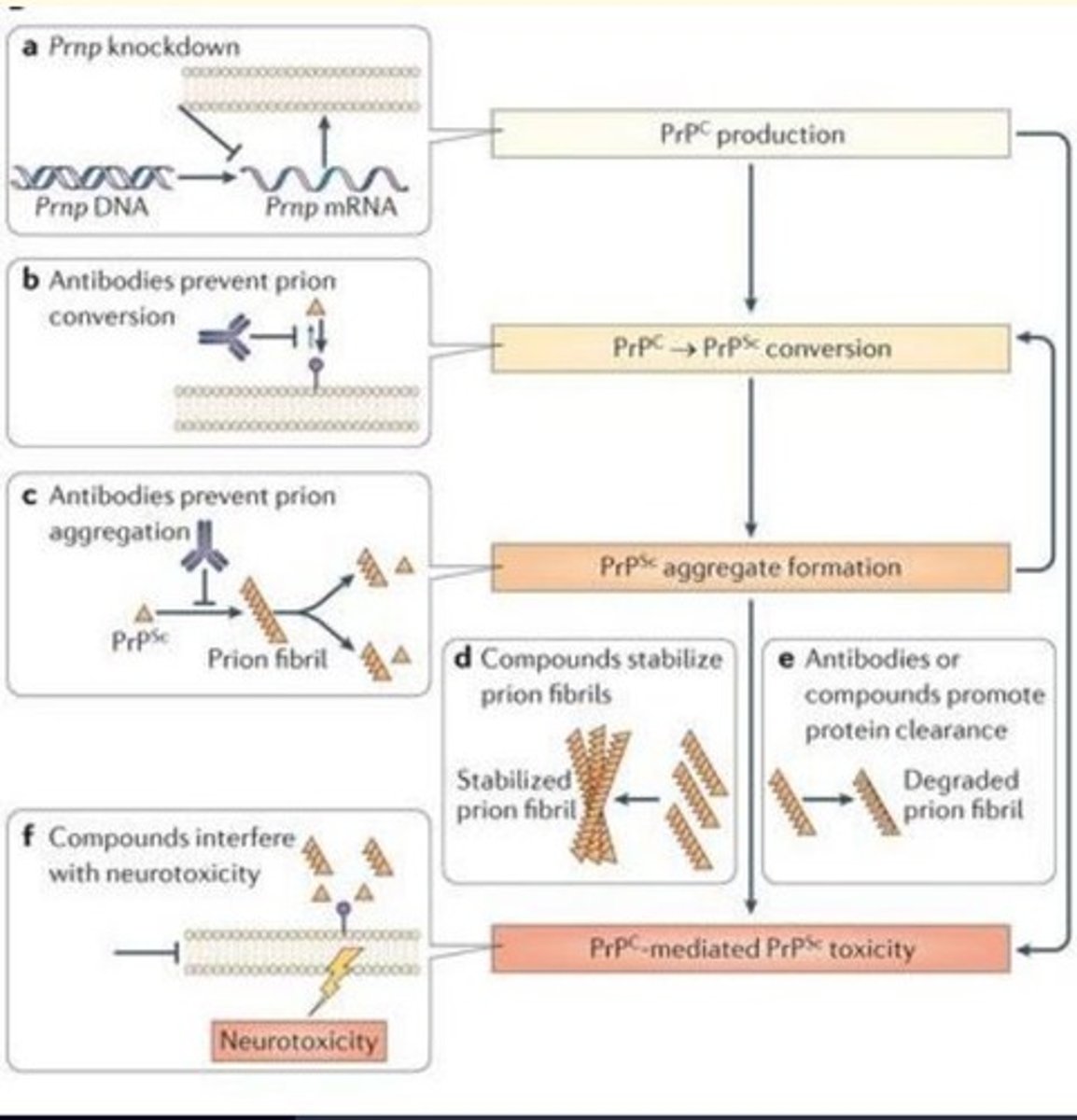
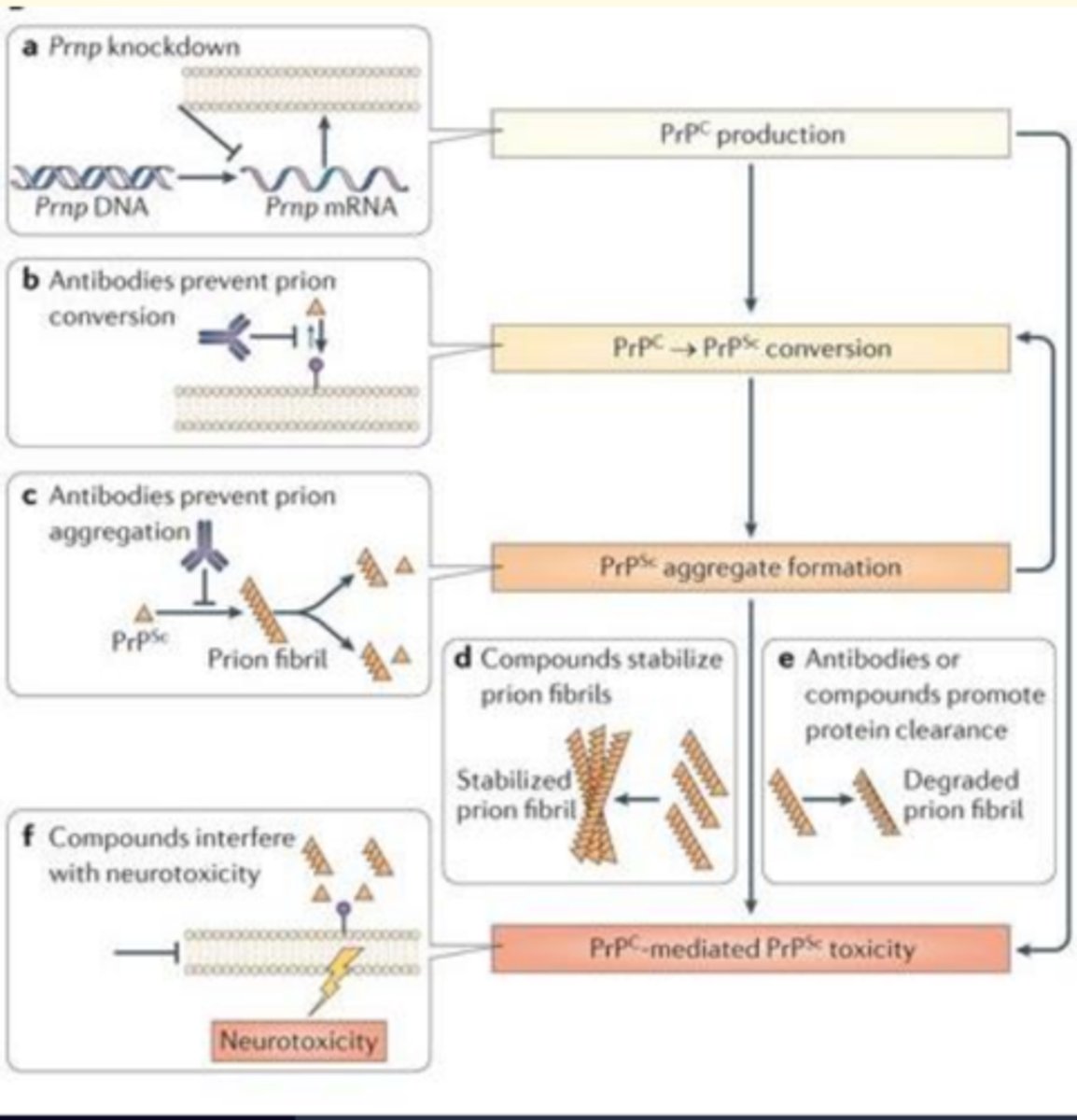
Potential pharmaceutical interventions for prion disease
- Prnp knockdown
- Antibodies prevent prion conversion
- Antibodies prevent prion aggregation
- Compounds interfere with neurotoxicity