Trini Part 1
1/22
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
23 Terms
Lead compound
Compound showing desired, but not optimised, biological activity
Starting point for drug design and development
Analogue
A compound made by performing structural modifications on a lead compound
e.g. altering, removing, or masking specific functional groups
Prodrug
pharmacologically inactive when administered
must undergo biotransformation to exhibit pharmacological effects
Briefly discuss the use of natural products as sources of lead compounds
derived from: plants, animals, marine organisms, microorganisms, endogenous chemicals.
Challenges:
Low availability
Potential toxicity
Can be difficult to identify the specific active compound
e.g.s
Penicillin: through serendipity from a fungus (microorganisms)
Vinblastine: active compound isolated from Madagascar periwinkle - treats leukemia
In silico
CADD methods - measure binding affinity of drug to target virtually
In vitro
1st tests carried out on… Isolated target molecules (enzymes or receptors), cells, tissues, micro-organisms.
Measure binding affinity (e.g. IC50) and direct interaction with target.
IC50 increases/binding affinity drops after modification?
FUNCTIONAL GROUP WAS IMPORTANT !
In vivo
2nd: Tests carried out on live animals or humans
Ability to interact with and react target (movement through ADME)
side effects
effective prodrugs
Measures the Drugs potency, therapeutic index, LD50, ED50
Not effected after modification?
Functional group was not important !
Bioassay (probs won’t be asked)
Process for screening compounds for biological activity and quantifying that activity
What is IC50 + what does it tell you
concentration of a drug needed to inhibit target
Lower IC50 = better activity
Briefly outline the main approaches/strategies for the discovery of new lead compounds in medicinal chemistry (must include lead compound definition in answer)
Compound showing desired, but not optimised, biological activity - Starting point for drug design and development
Random Screening
- Testing large libraries of compounds for biological activity.
high throughput screening: automated system, tests large numbers of compounds in vitro and quickly using only small amounts of each substance.
- Can come from many different sources (natural/synthetic).
e.g. Teixobactin – discovered in a screen of uncultured soil bacteria (CADD library mining) - kills pathogens without detectable resistance.
Rational Drug Design
- structural info + natural ligands of drug targets (e.g. enzyme or receptor) to design effective drugs.
- methods: de novo design or CADD
e.g. Relenza – antiviral drug
Serendipity
- by chance
- e.g. Penicillin
Observation of Side Effects
- side effects of existing drugs – may reveal a new therapeutic application.
- e.g. Viagra (originally for its analgesic action/angina but repurposed for erectile dysfunction
Propose possible interactions question … write out all possibilities

Bioavailability
The fraction of a drug that reaches systematic circulation after a particular route of administration
Represents true amount of drug that is available at the site of action
IV = 100% bioavailability (no absorption needed)
Discuss the concept of “pH partition” (ion trapping)
The pH of different body compartments influences the distribution and accumulation of a drug across the membrane.
Uncharged molecules = lipid soluble - cross cell membranes
And visa versa
Therefore, a drug accumulates in the compartment where it becomes the most charged!

Discuss the most important physicochemical characteristics that need to be considered in the optimisation of the pharmacokinetic properties of a lead compound [13 marks]
Ionisation
The charge of a drug is determined by the drugs pKa and the pH of its environment.
Affects ADME…
Adsorption -solubility, ability to cross membranes (uncharged molecule = lipid soluble)
(E.g. stomach = low pH, acidic molecules will absorb here).
Lipophilicity
Describes the non–polar, “organic soluble” character of a molecule.
DRUG NEEDS BALANCE (between water solubility and lipid solubility)
Too lipophilic; won't dissolve in aqueous media (e.g. blood) or it could accumulate non-selectively in adipose tissue.
Too hydrophilic; won't cross membranes,(like BBB), and will be eliminated too quickly.
Ability to form H-Bond
Too many H-Bonds makes desolvation and formation of a neutral molecule unfavourable.
Preventing the drug from crossing membranes.
The polar surface area (PSA) - should be less than 140 A² (angstrom) for good oral bioavailability
Molecular size
Larger molecules face more difficulty in passive diffusion across membranes
Factors that affect molecular size:
Molecular weight (most important) - </= 500 Da is optimal !!!
Also; electron density, polar surface area (PSA), van der Waals surface.
Molecular flexibility
Determined by the no. of rotatable bonds
Reducing flexibility through rigidification can improve a drugs activity and selectivity.
Molecule must adopt a fixed active confirmation to bind to a target or cross a membrane.
(High flexibility allows a molecule to adopt many conformations, but this results in a loss of entropy - we want rigidity !).
List lipinskis rule of 5 - may need to incorporate in physicochemical question.
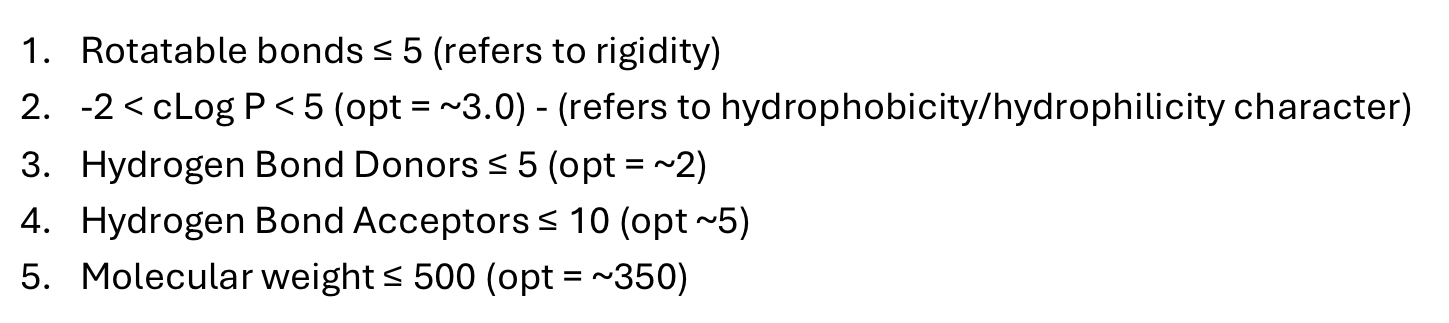
What is the difference between LogP and LogD? Why are these parameters important in drug development?
LogP: Partition coefficient
Measures the drugs relative affinity for the lipid and aqueous phases in its non-ionised state.
LogD: Distribution coefficient
Measures the effective lipophilicity at a specific pH, accounting for both ionised and non-ionised forms (depends on pKa).
important because….
determine the hydrophilic/lipophilic balance which affects ADME
Explain how you would expect the LogD of ibuprofen to vary at a pH range between 1 and 10. pKa of ibuprofen = 4.9
LogD: Distribution coefficient
Measures the effective lipophilicity at a specific pH, accounting for both ionised and non-ionised forms (depends on pKa).
Ibuprofen = weak acid (presence of COOH)
pH = 1; Stays ionised - highly lipophilic - highest LogD values
As pH increases larger than pKa…
COOH ionises, becomes neg charged - molecules now hydrophilic - lower LogD values.
LOGD OF IBUPROFEN DECREASES AS PH INCREASES
List the steps for chemical modification of a lead compound (basic)
Remove parts of the molecules to find the pharmacophore
Modify different functional groups (SAR)
Functional group transformations
Bioisosteric replacements
Rigidification: introduce confirmational restriction
Pharmacophore (2D + 3D)
Minimum structure of drug needed to exert a certain activity (considering a target).
2D pharmacophore: essential binding groups (basic molecular framework)
3D pharmacophore: Based on active confirmation of drug with target.
Specifies positions of essential binding groups in 3D space (distances and angles)
Active confirmation
Exact shape a drug adopts when it binds to its biological target.
Must be a fixed and specific shape to bind effectively + cross membranes.
What is a bioisosteric replacement?
Swapping a functional groups in a lead compound with a different compound that maintains similar biological activity
(e.g. spatial arrangement, electronic properties, critical functional groups etc)
but increases the pharmacokinetic profile of the drug (e.g. improving solubility, stability or adsorption)
What is rigidification? + methods
Endogenous compounds have many rotatable bonds (flexible) + can adopt many confirmations.
Conformational restriction: Limits no. of possible confirmations to increase…
Activity (higher chance of adopting active confirmation (needed to identify 3D pharmacophore)).
Selectivity (less chance of binding to undesired targets)
A rigid molecule has a lower loss of entropy when binding to target
Methods:
Introduce…
Rings
Rigid functional groups
Steric blockers
Briefly explain what a SAR study is and discuss its objectives in the context of medicinal chemistry [7 marks]
Structural modifications made to a lead compound…
To identify which functional groups are essential for its biological activity (pharmacophore)
By altering or removing specific groups + testing the resulting analogues (In vitro/In vivo) - we can see how these changes affect the compounds ability to bind to the target.
INFO CAN BE USED FOR OPTIMISATION OF DRUG