Fatty-Acid Oxidation Defects
1/45
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
46 Terms
Fatty Acids
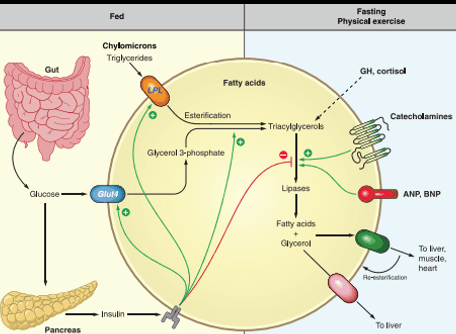
Under fasting conditions, fatty-acids are the stores used once 8-hour of carb derived glycogen is used up
Lipolysis: fatty acids get released into blood stream to be used by the body and converted into energy as Actyl-CoA and gluconeogenesis
Fatty acids can also produce ketone bodies via Bet-Oxidation to serve as energy for the BRAIN
Fatty acids:
3 acyl groups with attached glycerol group + carbon chain
Many fatty acids based upon the length of their carbon chain + the saturated double bonds
Most dietary fat stored as Palmaric acid (C16) or Stearic acid (C18)
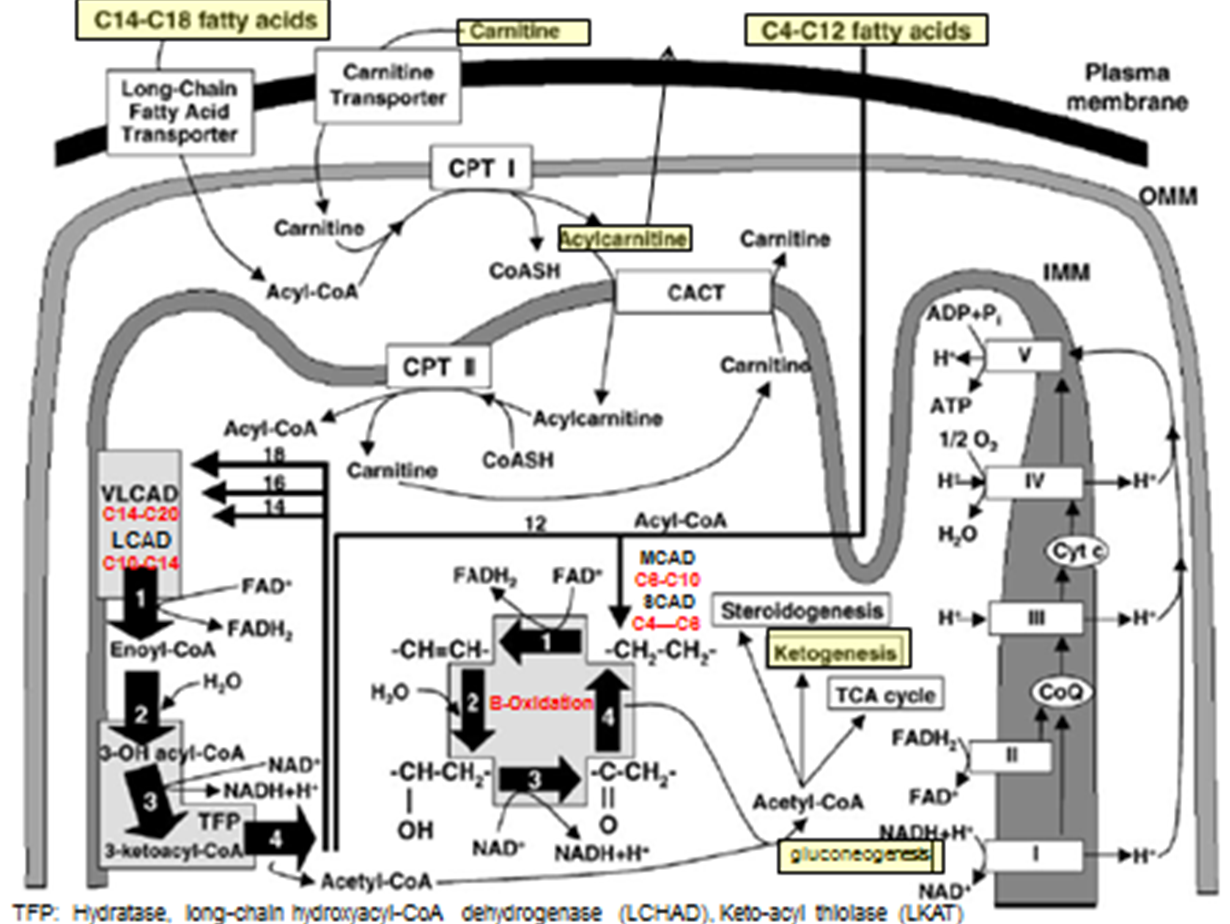
Fatty-Acid Metabolism Pathways: C14-C18 transport step
Fatty acids needs to be converted to Acetyl-CoA
C14-C18 Fatty acids → transport across plasma membrane via Long chain F.A transporter
Converted to Actyl-CoA (But cannot enter inner mitochondrial membrane)
Acyl-CoA + Carnitine → Acylcarnitine (of varying lengths) via CPT1 activity (TRANSPORT PREP STEP)
Acyl Carnatine moved into inner membrane in exchange free cranatine out via CACT activity (TRANSPORT STEP)
Acylycratine → Actyl CoA + Carnitine via CPTII activity (LIBERATION STEP)
Acyl-CoA can now enter into BETA OXIDATION
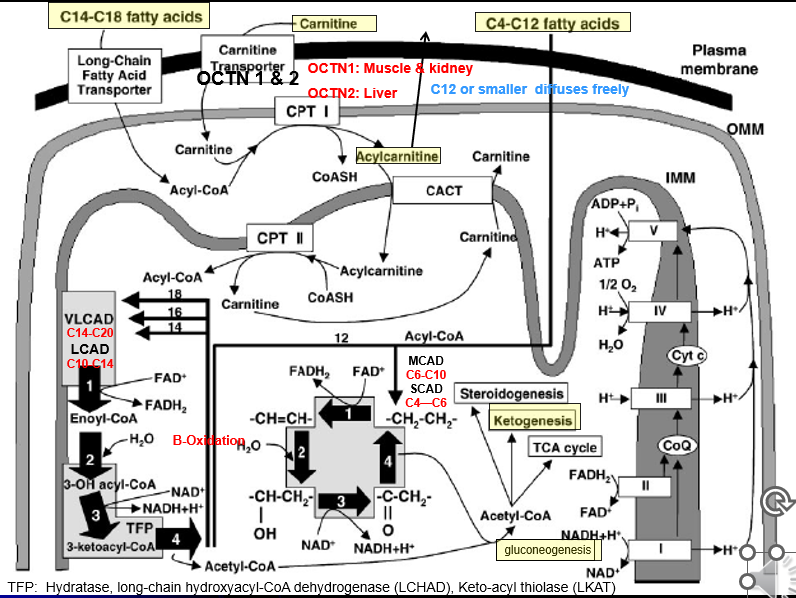
Fatty-Acid Metabolism Pathways: C14-C20 B-Ox
Special Cleavage Steps: Only done for fatty acidsC14 or longer
Acyl 2 carbon is cleaved from it via the Mitochondrial Trifunctional Protein
the Acyl-2C’s liberated can then undergo Beta-Oxiadtion
Each turn liberated a 2C Acytl CoA group
Fatty-Acid Metabolism Pathways: C4-C12 B-Ox
Smaller fatty acids can diffuse freely across all membrane of the cell + mitochondria: no need for the transporter or Beta-oxidation steps
Each turn liberated a 2C Acytl CoA group
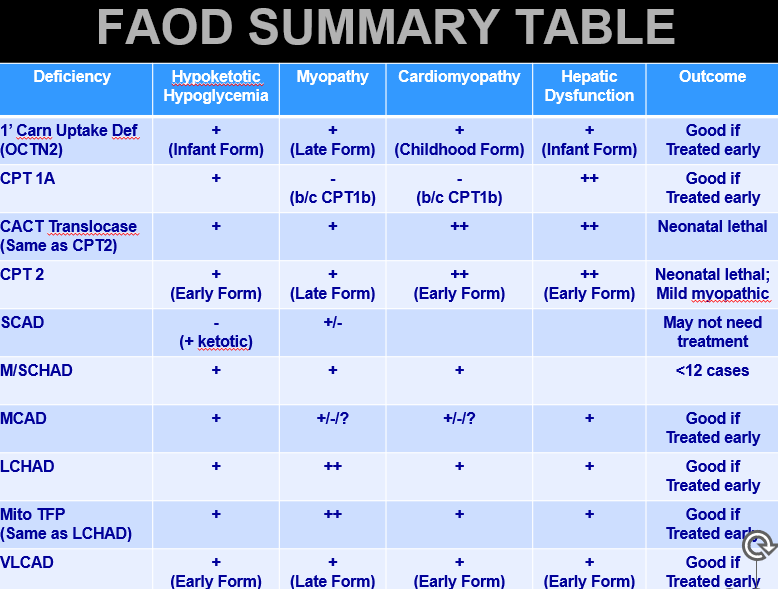
Fatty Acid Oxidation Defects: Pathophysiology
Defective utilization of Fatty Acids (Acyl groups) for energy
Rapid glycogen depletion→ hypoglycemia in fasting sate (can’t switch over to FA B-Ox to maintain blood glucose levels)
Deficiency of energy substrate for muscles and brain
Muscles: F.A → Acetyl-CoA for energy
Brain: F.A → Acetyl-CoA → Ketone bodies (ketogenesis step)
Accumulation of unmetabolized F.A in liver and muscle
Liver disease
Myopathy
Cardiomyopathy
Fatty Acid Oxidation Defects: Major Clinical phenotypes
Hypoketotic + hypoglycemic
Glucose levels drop quickly, Ketone bodies made at low levels
Cognitive and developmental insults to the brain
Body TRIES to breakdown F.A., but can’t convert smaller chunks to the 2-Acetyl-CoA via B Ox
F.A.s liberated into blood stream = buildup within muscle and liver → dysfunction
Myopathy + Cardiomyopathy
Hepatic failure/Liver dysfunction (Reye Syndrome)
Maternal liver disease → affected fetus can create issue in the mother w F.A build up
Fatty Acid Oxidation Defects: Etiologies
Disease of the Carnitine pathway:
primary: defects in the protein pathways
secondary: nutrition deficiencies
Disease of Fatty Acid B-Oxidization
L-Carnitine Amino Acid
Binds to Acyl (organic +inorganic) residues (F.A) in the blood and enables transport into cells + their elimination → (why we supplement with Carnitine in Organic Acidemias: for their elimination w carnitine)
Long chain (>C12) needs it to get their long “acyl” chains into the inner mitochondria membranes where “Digestion” step + B-Ox can occur
Sources: we can make a-little, but not enough on its own
Diet: Milk + meat
Reabsorption: Kidneys via Caratine trnasporter protien
Carnitine Testing
Plasma Carnatine levles: Free, unbound and Acyl-Cartines (boudn to Fas and organic acids)
Plasma Acyl Carnatine prolei: quantity the diffrent types of bound-caratine resdiues
Fatty Acid Oxidation Defects: Key Concepts
All Autosomal Recessive
Spectrum of Severity
Phenotypes vary but always have
Hypoketotic + Hypoglycemia
Myopathy and/or Cardiomyopathy
Liver failure (Reye Syndrome)
SCIDs can ocure fruently: overnight fast casues sudden hypoglycemi even that kills them
Fatty Acid Oxidation Defects: Therapy
Acute Illness/Fastine
IV Dextrose (immediate)
Carnitine (some FOADs): overdrive membrane import of FA and remove excess FAs
Monitor for
hypoglycemia
liver failure
Muscle breakdown
Chronic
Avoid prolonged fasting
frequent feedings: CORNSTARCH (McArdles?)
Supplment with Medium-Chain-Triglycrides (“MCT”) oil <C10 for some (not MCAD/SCAD) → doesn’t need transporter or Digestion steps before B-Ox
Supplment with L-Cartine for some (not LCHAD)
Avoid liver toxic or carantine lowerin medication (valproic acid, salicylates, some anethetics)
Overview of FAOD Clinical Issues
OCTN2 Deficiencey: Metabolism
OCTN1 and OCTN2 transport Carnitine across plasma membrane
OCTN1: Primarily in liver
OCTN2: Primarily in Muscle + Kidney
No OCTN2 = No carnitine = Long F.A.s don’t undergo B-Oxidation in the Muscle
Also: Kidney→ not enough Carnitine is re-absorbed and therefore too much excreted = very low levels of carnitine
OCTN2 Deficiency
Also called Primary Carnitine Uptake Defiencey
Etiology
SLC22A5 gene mutation→ reduced Carn. uptake by Kidney +muscle→ low blood and muscle Carn+ Acyl-Carn
Can also just be caused by LOW-Diet CARTNATINE
OCTN2 Deficiency: presentation
Forms: Severe Infantile, Mild Child/late Onset
Infancy:
Hypoketotic hypoglycemia
Liver failure
Child:
Myopathy + Cardio myopathy
Liver affects are less: when Carn is severe enough, B-Ox affected through whole body → but Child type, Carn is enough to avoid defieciny inthe liver but not the rest of the body
Late
mild myopathy or asymptomatic
OCTN2 Deficiency: Diagnosis
NBS: Low C0 and Acyl-Carnitine levels (Low level of ALL the species) → NEED TO TEST MATERNAL LEVELS AS WELL
Confirmation
Carnitine and Acyl-Carnitine levels
Enzyme activity: Fibroblasts
Molecular: finds 70%
OCTN2 Deficiency: Treatment and outcome
Treatment
high-dose Carn
Avoid fasting
IV Dextrose
Outcome
Good if treated before severe decompensation
CPT 1+2 Deficiency: Metabolism
CPT 1 and CPT2 present at outer and inner mitochondrial membrane
CPT1: Outer Mito membrane → Acyl-Coa + Carnitine = Acylcarnitine
High Free-Carn levels
Low Acylcarnitine levels
CPT 2: Inner Mito membrane
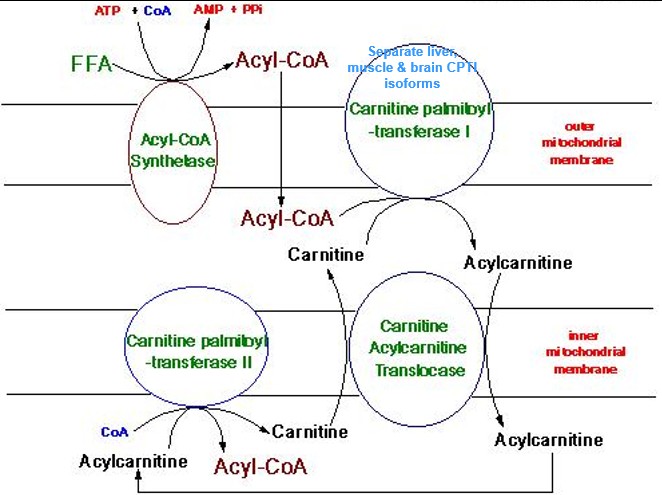
CPT1A Deficiency
CPT1A → Liver
CPT1B → Muscle
CPT1C→ Brain
CPT1A gene mutation→ Low hepatic longer chain acyl carntine
CPT1A Deficiency: presenation
Early onset
Hypoketotic + hypoglycemic
Liver fialure with Fasting/illnes
CPT1A Deficiency: Diagnosis
NB: Elevated free Carn (C0) + Low long chain F.A.s (C0:C16 ratio increased
Confirmation
Carnatine and Acylcarnitine levels
Enzyme: Skin + CVS/Amnio
Molecular: >90%
CPT1A Deficiency: Treatment+ Outcome
Treatment
Avoid fasting→ frequent feedings +
IV dextrose when Ill
Cornstarch
MCT Oil
High Carb/low-fat diet
Outcome
Good if severe decompensations prevented
Maternal liver disease reported with CPT1A offspring
CPT2 and CACT Deifiecnies
Bothe present very similarly
Once Acyl-Coa + Carnitine = Acylcarnitine, it must be brought through the inner mitochondrial membrane via Carnitine Acylcartine Translocase (CACT)
CACT: exchanges Acycl carnitine in, free carnatine out of the membrane
Acylcarnatine needs to be reconverted to Acyl-Coa via Carnitine palmibly-transferase II
Acyl-CoA needs to be liberated before it can enter into B-Oxidation
When CPT2 or CACT is deficient, Acyl carnitine generated via CPT1 cannot be used and BUILDS UP
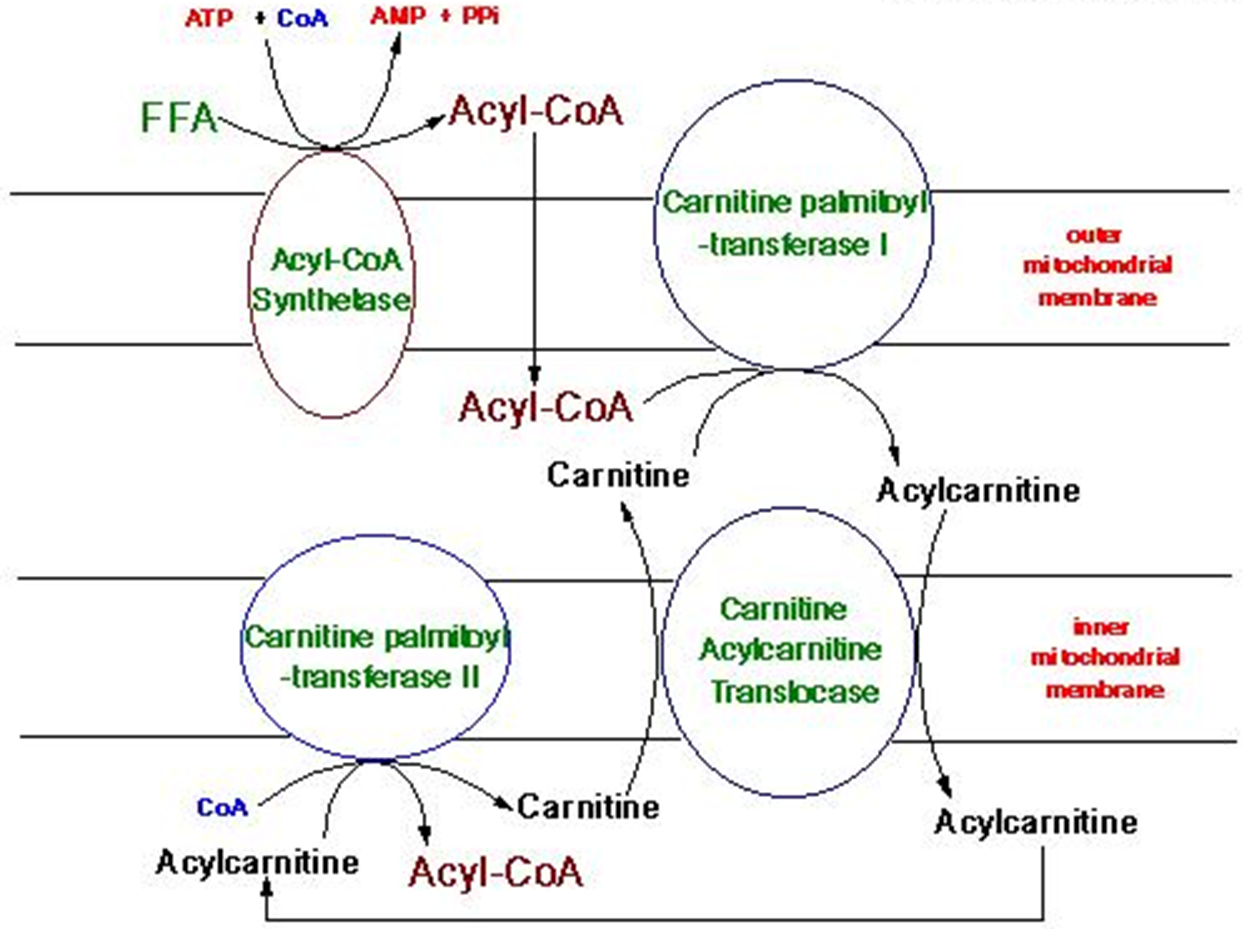
CPT2 and CACT Pathophysilogy
CPT2 or CACT gene mutation→ increased levels of long chain acyl-carnitine
CPT2 and CACT Def: Presentation
Very rare condition where symptoms begin IN UTERO: some energy deficit that can create dysmoprhic features at birth
Neonatal:
Hypoketotic
Liver fialure
Cardiomyopathy
Dymorphic feautres /renal cyts
Later onset form:
EXCERISE INDUCED MUOPATHY (build up of Creatine Phospho Kinase)
CPT2 and CACT Def: Diagnosis
NBS: Elevated C16 and or C18:1 ratio (long to short F.A. chain ratio)
Confirmation: Only way to differienate the two
Carnitine and
Enzyme activity: Skin, Muscle, CVS/Amnio
Molecular: >95%
CPT2 and CACT Def: Treatment + Outcome
Treatment:
Avoid fasting with frequent feeding and IV Dextrose when Ill
Cornstarch,
MCT oil
Carnitine,
High-carb/low-fat diet
Outcomes
Neonatal: Lethal
Infantile: Variable
Late: Myopathy is mild if treated
Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD) Deficeny
VLCAD Drives the initial steps of fatty acid beta oxidation for the long chain fatty acids (>C14)
VLCAD Deficiency: Pathophysiology
ACADVL mutation → Deficient C14-20 B-oxidation
Generally more mild, later onset, exercise
VLCAD Def: Presentation
Infant:
Hypoketoitic hypoglycemia
Liver failure
myopathy
cardiomyopathy
Child: Cardio myopathy
Late: *******EXCERCISE MYOPATHY: WAY MORE COMMON FORM *****
VLCAD Def: Diagnosis
NBS: Elevated C14:1 ratio and longer chain Acyl-carnitine
Confirmation:
Carnatine and AC
Enzyme activity: Skin, white blood cell, aminotic fluid
Moelcualr: 85-93%
VLCAD Def: Treatment + Outcome
Treatment:
Avoid fasting with frequent feeding and IV dextrose when ill
Cornstarch
MCT Oil
+/- Carnitine
High-carb/low-fat diet
Outcome
good if treated before severe decompensation
Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency
Most common FAOD and the first to be added to NBS
the meatbolism of C8-C10 length acyl-CoA Fatty Acid groups
These can move freely acros the inner mitocondrial memebrane WITH OUT then eed for Carnatine complex (so no Acylcarantine needed
Once inside,C8-C10 Acyl-CoAs → *****BETA OXIDATION via MCAD****→ C4-C6 AcylCoAs
MCAD Deficiency = C8-C10 Buildup in blood and tissue + C8 Octanoyl Acycl Carainatase (Toxic)
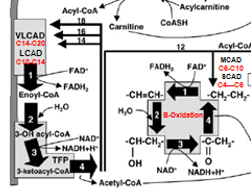
MCAD Def: Pathophysiolo
ACADM gene mutation → lowered C6-C10 Fatty acid B-Oxidation = build up of C8-C10 in tissues
MCAD Def: Presentation:
Typical presentation at 3-24months old with Fasting/Illness
Hypoketotic hypoglycemic
Liver disease
Often Sudden Infant Death syndrome→ usually the first personation you will see (sleeping longer through the night, greater chance for fasting affects to kick in)
MCAD Def: Diagnosis
NBS: Elevated C8, lesser C6 and C10 : (C8 >C6/C10)
Confirmation
Carnitine
AC
uAG
Enzyme activity: Skin, WBC, amino/CVS
Molecualr: 90%
MCAD Def: Treatment + Outcome
Treatment
Fasting with Frequent feeding and IV dextrose when ill
Cornstarch
+/- Carnitine
High carb/low-fat diet
****NO MCT OIL!!!!****
Outcome
Good if treated before severe decompensation
Maternal liver disease reported (AFLP/HELLP)
Short Chain Acyl-CoA (SCAD) Deficiency
C4-C6 Acyl-CoAs are B-Oxidized by SCAD activity
Much more MILD production of Acetyl-Coa (C1)
SCAD Def: Pathophysiology
ACADS gene mutation→ deficient C4 +C6 fatty acid B-Oxidation
SCAD Def: presentation
Incidence is not well known: Most SCAD inididuals are ASYMPTOMATIC
Possible NEONATAL form
Ketotic hypoglycemia: lood gluclose can be low, but can still produce KETONE BODIES for energy→ B-Ox can still be done on longer chains FAs to produce Acytl CoAs (stopping at C4-C6)
Myopathy
Lethargy
Sizures
SCAD Def: Diagnosis
NBS: Elevated C4: BUT other IEM (like Organic Acidemias) also high C4 → Urine Organic Acid profiles can distinguish
Confirmation
Carnitine and AC
Urine Organic Acid Profile
Enzyme activity: Skin
Molecular: 100%
SCAD Def: Treatment + Outcome
Treatment
Avoid fasting with frequent high-carb feeding and IV dextrose when ill
Outcome
Good if treated before severe decompensation
Maternal liver disease reported (AFLP/HELLP)
Long-Chain HydroxyAcy-CoA l Dehydrogenase (LCHAD) or Tri-Function Protein Deficiency
TFP is involved in the B-Oxidation of a number of long-chain FAs
when Deficient: inability to metabolize long chain Acyl-Coa molecules
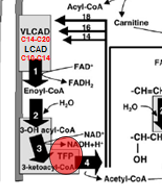
LCHAD/TFP Def: Pathophysiology
LCHAD gene mutation:
CHAD/TFP Def: Presentation
Typical presentation in Infancy
Hypoketotic hypoglycemia
Myopathy
Cardiomyopathy
Liver deiase
c16-oh and c18 elevated = pigmented retinopathy + neuropathy, MANY SCIDS cases
CHAD/TFP Def: Diagnosis
NBS: Elvated C16-OH w
Confrimation
Carnatine and AC
Enzyme acitivty: Skin, WBC
Molecular: HADHA gene
CHAD/TFP Def: Treatment + Outcome
Treatment
Avoid fasting with frequent feedings and IV dextrose when ill
Cornstarch
MCT oil
High-carb/low-fat diet
******NO CARNITINE ******
Outcome:
Believed to be good if treated before severe decompensation
MOST PREVELANT FOAD found in Mom’s who devlope liver dysfunction with fetus’s thatare affected with FOAD