Pharmacodynamics and IF Altering Drug Response
1/66
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
67 Terms
Age (Biological factor)
Biological factor influencing drug effects; extremes of age (neonates/infants and elderly) show heightened sensitivity due to organ immaturity/degeneration and polypharmacy.
Neonates and Infants (Age factor)
Altered pharmacokinetics: absorption, distribution (low albumin, immature BBB), hepatic metabolism, and renal excretion are reduced or variable.
Elderly (Age factor)
Increased susceptibility to adverse effects due to organ degeneration, polypharmacy, and reduced hepatic/renal function.
Body Weight/Composition
Dose calculations based on body size; adults around 70 kg; children require smaller doses using specialized formulas.
Psychological factors/Placebo
Non-pharmacologic influences that can alter response; placebos can elicit real responses in some individuals.
Genetic Factors
I.e. Inherited difference in coagulation factors enhance DVT risk in women using oral contraceptives
Idiosyncrasy
Rare, unpredictable drug responses not explained by known factors; type B adverse reactions.
ie malignant hypergthermia or neuroleptic malignant syndrome
Metabolic and Pathologic disturbances
Low acidity = low iron and aspirin absorption
Liver disease = more bioavailability of drugs w/ high first pass metabolism
Renal disease = low excretion of drugs
Diarrhea/vomiting = orally given drugs ineffective
Pharmacogenomics
Study of how genetic differences affect drug response and tailored therapy.
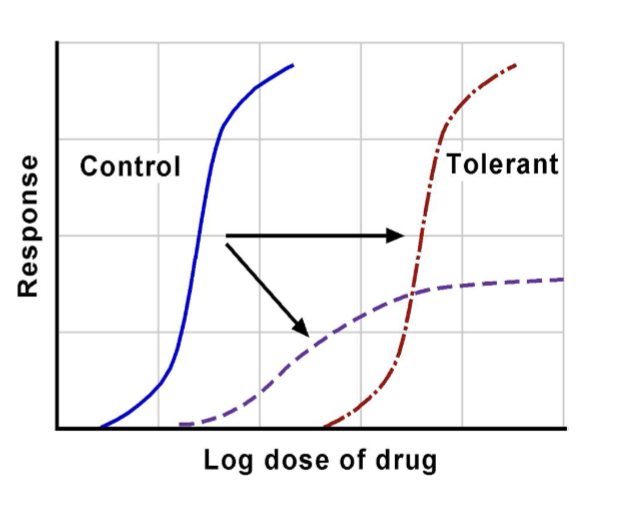
Pharmacokinetic tolerance
Acquired tolerance due to changes in drug disposition (absorption, metabolism, excretion) reducing effect.
Pharmacodynamic tolerance
Acquired tolerance due to cellular adaptive changes (receptor changes) reducing response.
Innate/Natural tolerance
Genetically determined first-time non-sensitivity to a drug.
-ie Chinese tolerant to purgative action of castor oil
Acquired tolerance (overview)
Tolerance that develops after repeated drug exposure; may involve pharmacokinetic, pharmacodynamic, or acute tolerance/tachyphylaxis mechanisms.

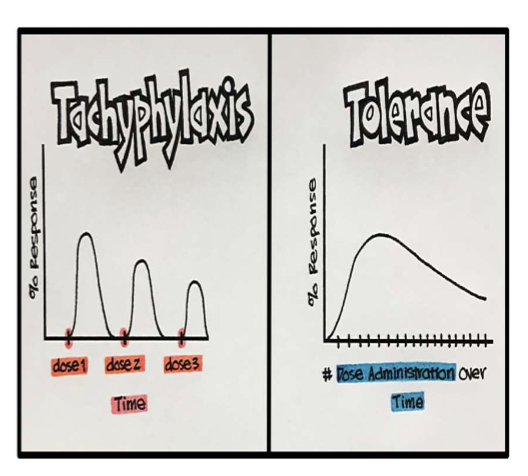
Acute Acquired Tolerance (Tachyphylaxis)
Rapidly developing decrease in responsiveness when a drug is repeatedly administered over a short period of time
Results from an intermediate needed for response being depleted, receptors getting internalized, or signal transduction becoming uncoupled
Max response usually reduced
Cross tolerance
Tolerance among the drugs belonging to the same class
PPL tolerant to morphine are also tolerant to heroin and other opioids
Desensitization
Gradual, usually reversible decrease in receptor response despite ongoing presence of agonist.
self defense mechanism by nature to avoid over stimulation of cells
Down-regulation
Prolonged exposure to agonist decreases receptor numbers on cell surface.
results due to endocytosis or internalization of receptors from cell surface
ie diminished response to albuterol over time
Up-regulation & Supersensitivity
Prolonged antagonist exposure increases receptor numbers and/or sensitivity.
results due to externalization of receptors on cell surface
ie Thyrotoxicosis up regulation causing tachycardia and palpitations
Rebound hypertension after abrupt discontinuation of antihypertensive drugs clonidine and beta blockers
Supersensitivity
Increased receptor response due to up-regulation or receptor changes; heightened responsiveness.
Concurrent administration effects
Multiple drugs can interact to produce summation, potentiation, synergism, or antagonism.
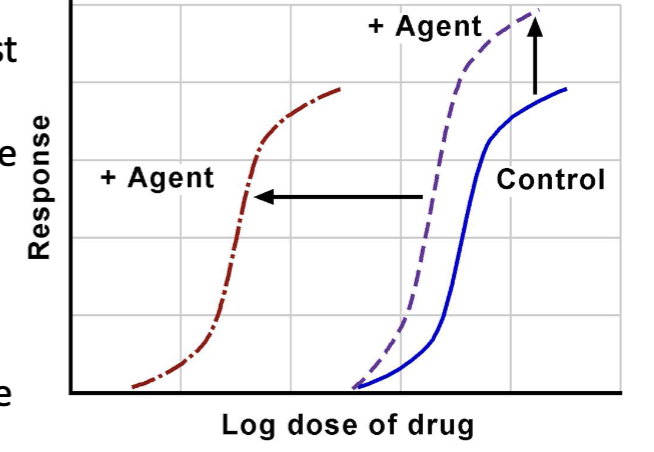
Potentiation
Potentiating agent increases effect of another drug without producing its own effect.
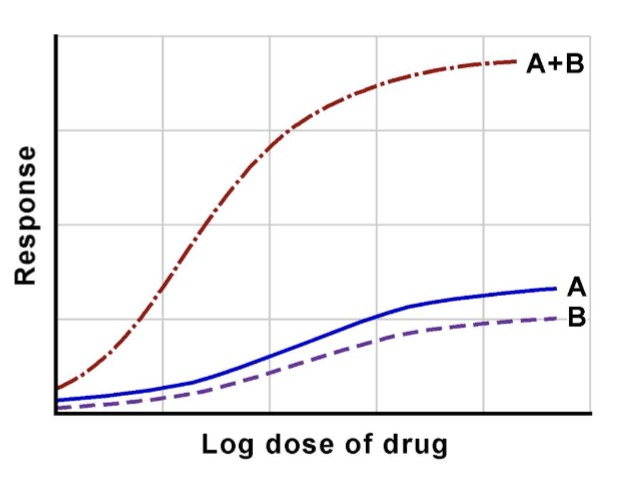
Synergism
Combined effect greater than the sum of individual effects.
- levodopa + carbidopa in Parkinsons
Antagonism
One drug reduces or blocks the effect of another.
Route of administration differences
Different routes yield different pharmacokinetic/pharmacodynamic profiles and effects.
Precision Medicine
Tailoring treatment based on individual genetics, liver/renal function, and other factors to optimize response.
Affinity
The ability of a drug to bind to its receptor to form the drug–receptor (DR) complex; governs receptor occupancy and is quantified by the dissociation constant (Kd); smaller Kd means higher affinity.
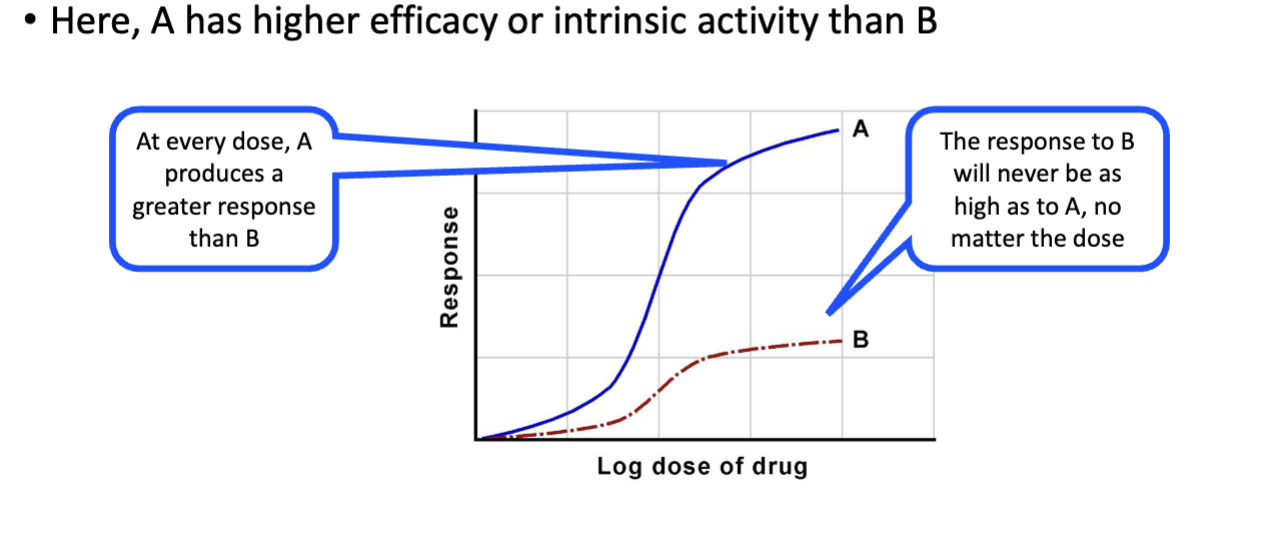
Intrinsic Activity (Efficacy)
The ability of a bound drug to activate the receptor and produce a pharmacologic response; governs receptor activation.
Agonist
A drug/ligand with high affinity and high intrinsic activity that can activate the receptor to produce a maximal response.
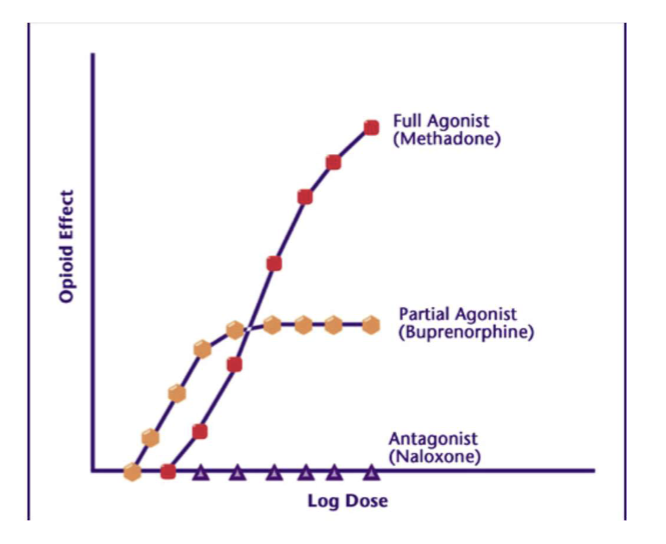
Partial Agonist
A drug/ligand with high affinity but submaximal intrinsic activity, producing a submaximal response even at full receptor occupancy.
Inverse Agonist
A drug with high affinity that has negative intrinsic activity, producing effects opposite to an agonist in the absence of the agonist.
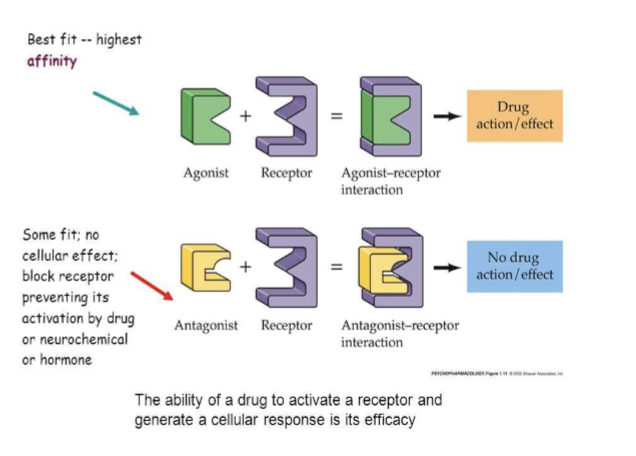
Antagonist
A drug with high affinity and zero intrinsic activity that occupies a receptor and prevents endogenous agonist binding or activation; no receptor activation.
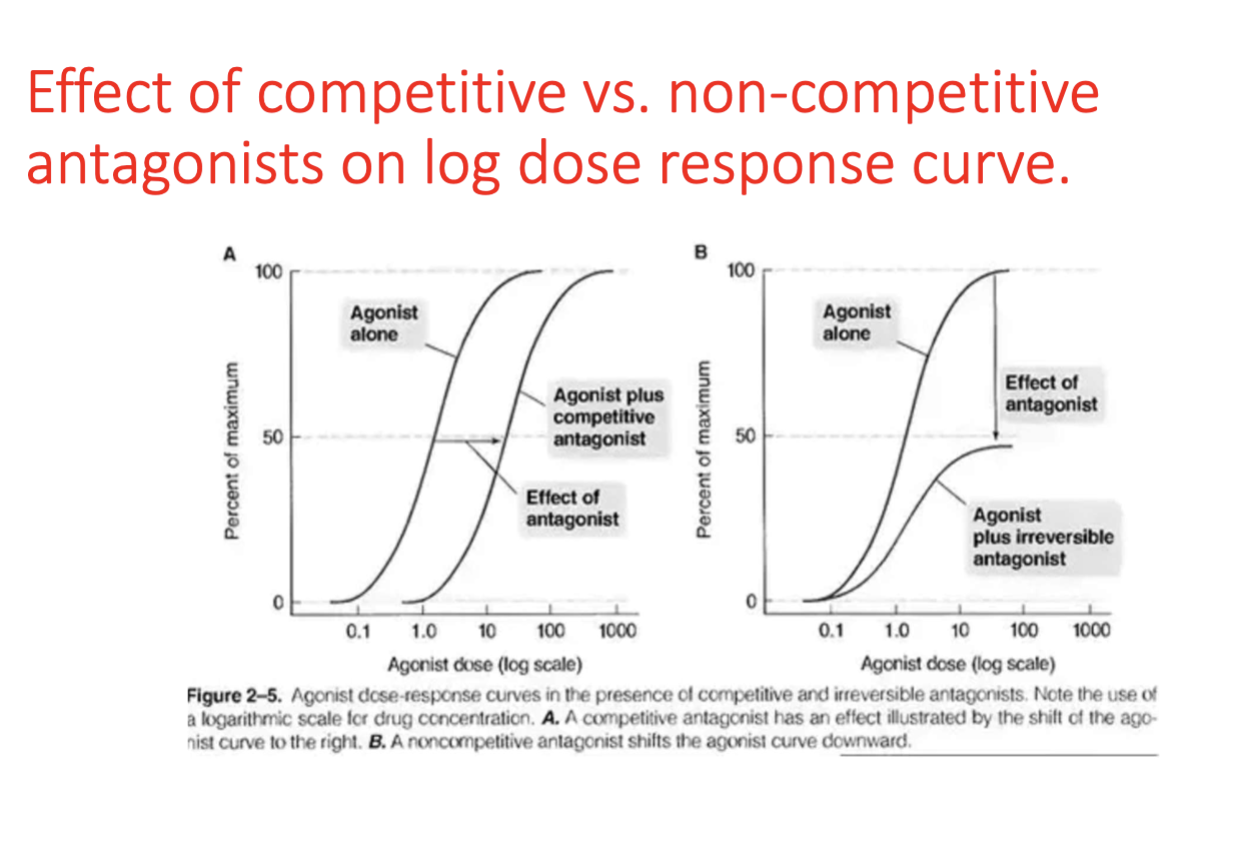
Competitive (Reversible) Antagonism
Antagonist binds the same receptor as the agonist; effects can be overcome by increasing agonist concentration; curve shifts to the right without changing maximal response.
Noncompetitive (Irreversible) Antagonism
Antagonist binds irreversibly or very tightly to the receptor; higher agonist doses cannot restore maximal response; lowers the maximal effect.
Chemical Antagonism
Antagonism that occurs via a chemical reaction between drugs, not via receptor interactions (e.g., neutralization of gastric acid by antacids).
Physiologic (Functional) Antagonism
Two drugs produce opposite effects on the same physiological system through different receptors or pathways.
insulin and glucocorticoids on blood sugar
Acetylcholine and epinephrine on heart rate
Pharmacokinetic Antagonism
One drug alters the absorption, metabolism, or excretion of another, reducing its concentration at the site of action.
urinary alkilizers
Pharmacodynamic/Neutral/ receptor Antagonism
Antagonism where the antagonist blocks the action of an agonist by occupying the same receptor site.
Competitive (equilibrium/reversible) antagonism
Reversible due to weak Hydrogen bonds
competes w/ agonist for same receptor
Noncompetitive (nonequilibrium/irreversible) antagonism
Some bind to receptor w strong covalent bonds
Agonist unable to reduce their receptor occupancy even at high dose = irreversible
Log dose response curve shortens in height = diminished receptor activation, intrinsic activity and efficacy of the agonist
Allosteric Modulator
A compound that binds to a site distinct from the orthosteric (active) site and modulates receptor activity, either increasing (PAM) or decreasing (NAM) activity.
Negative Allosteric Modulator (NAM) - Allosteric Inhibitor
An allosteric inverse modulator that reduces receptor activation by binding an allosteric site.
receptor unable to bind w agonish or inhibits post ligand binding signaling
reversible effects
Positive Allosteric Modulator (PAM)
An allosteric activator that increases receptor activity or efficacy when bound.
Allosteric Site
A binding site on a receptor separate from the orthosteric site where the endogenous ligand binds.
Orthosteric Site
The primary active site where the endogenous ligand or agonist binds to elicit a response.
Receptor
High affinity binding site for endogenous substance - causes change in cellular funciton
most are regulatory proteins
Cell surface receptors - for water soluble & Intracellular - for fat soluble ligands
Ligand-Gated Ion Channel
Receptors that open or close ion channels in response to ligand binding (e.g., acetylcholine, serotonin, GABA, glutamate).
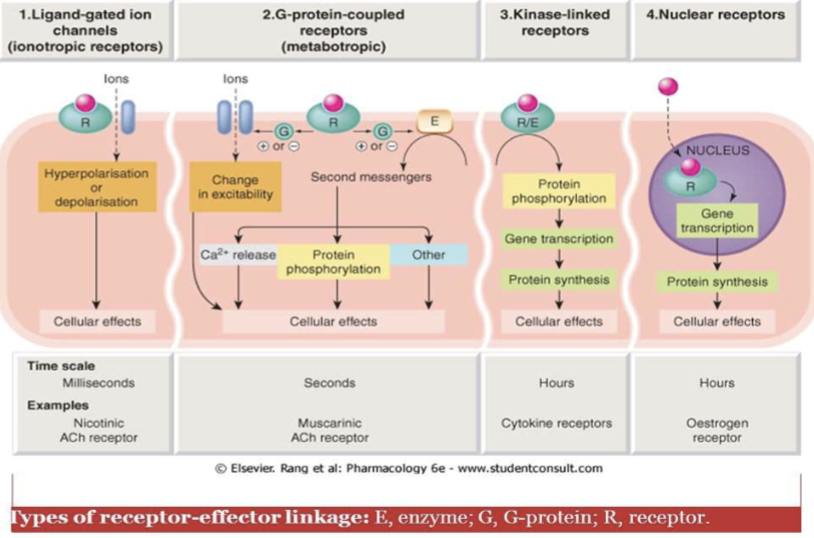
G Protein-Coupled Receptor (GPCR)
A receptor with seven transmembrane domains that activates intracellular G proteins (Gs, Gi, Gq) and downstream signaling such as AC or PLC.
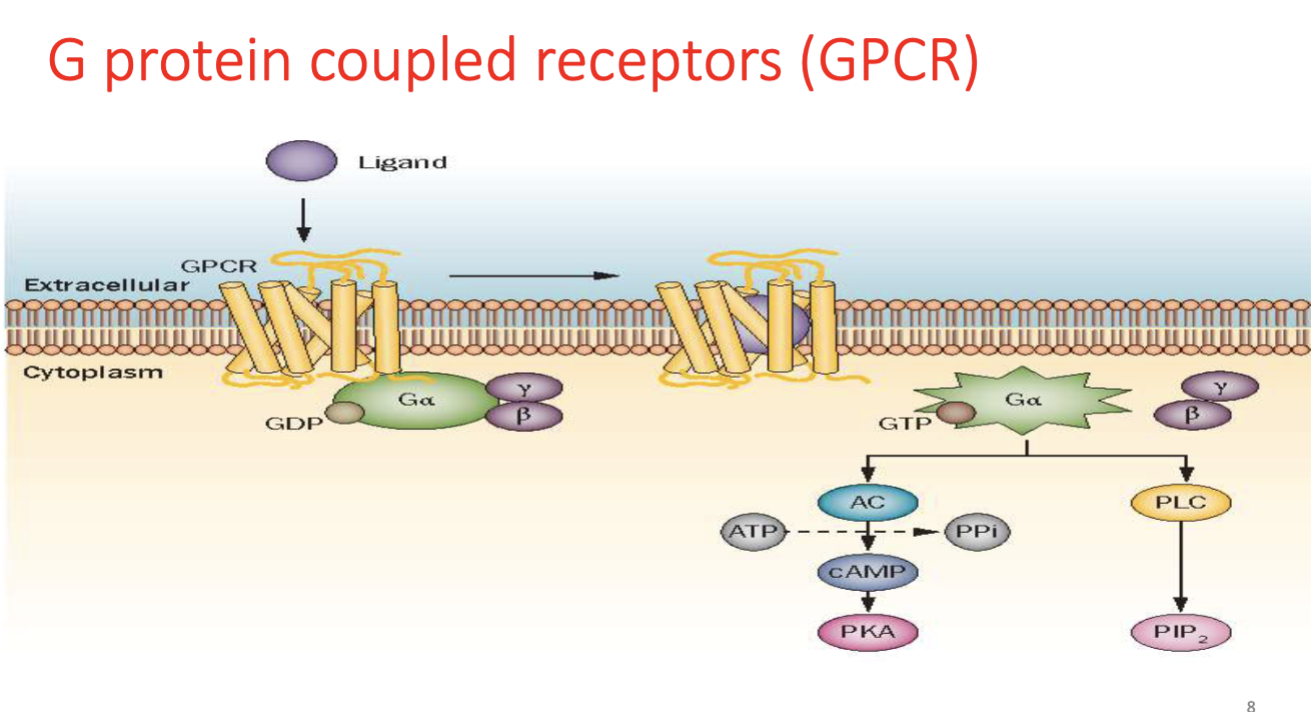
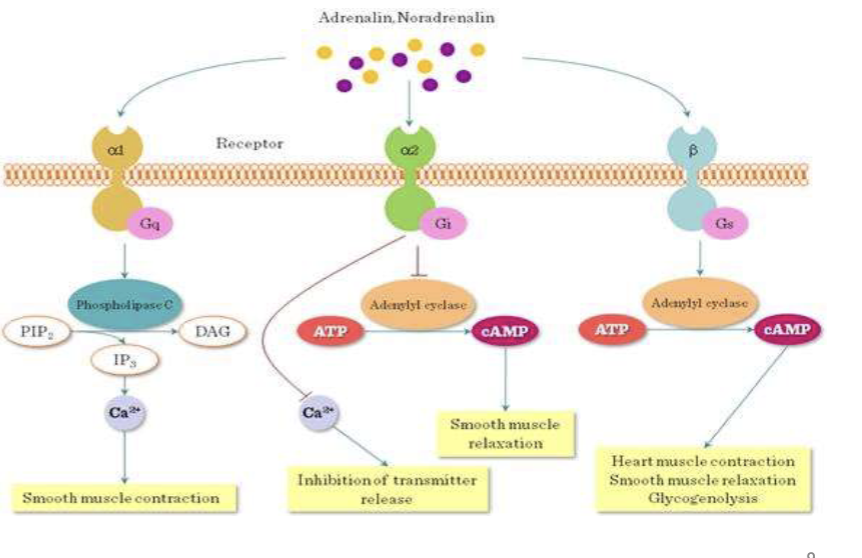
GPCR Subtypes (Gs, Gi, Gq)
G-protein subtypes that mediate different signaling pathways: Gs stimulates adenylyl cyclase, Gi inhibits it, and Gq activates phospholipase C.
protein w/ 7 transmembrane helical units w/ extracellular ligand binding domain and intracellular trimer
Cyclic AMP causes Smooth muscle relaxation, Calcium = contraction
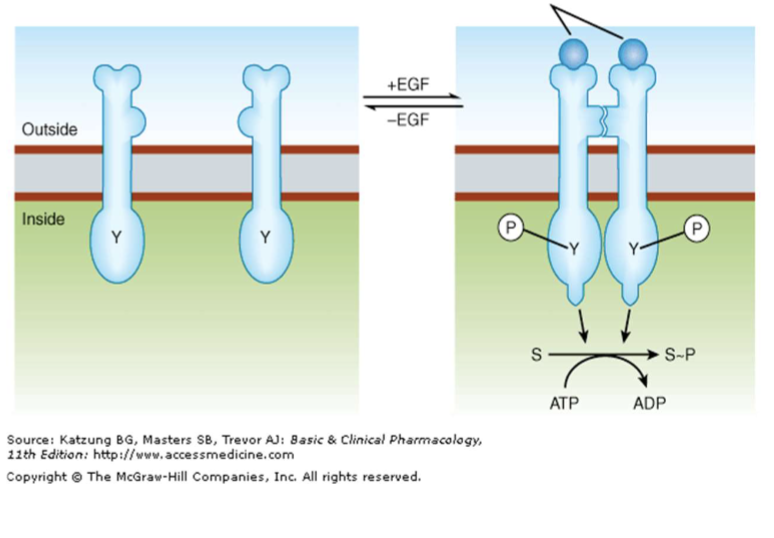
Tyrosine Kinase Receptor
Receptors that dimerize and activate tyrosine kinase signaling (e.g., insulin, EGF, PDGF); targeted by TK inhibitors like Imatinib.
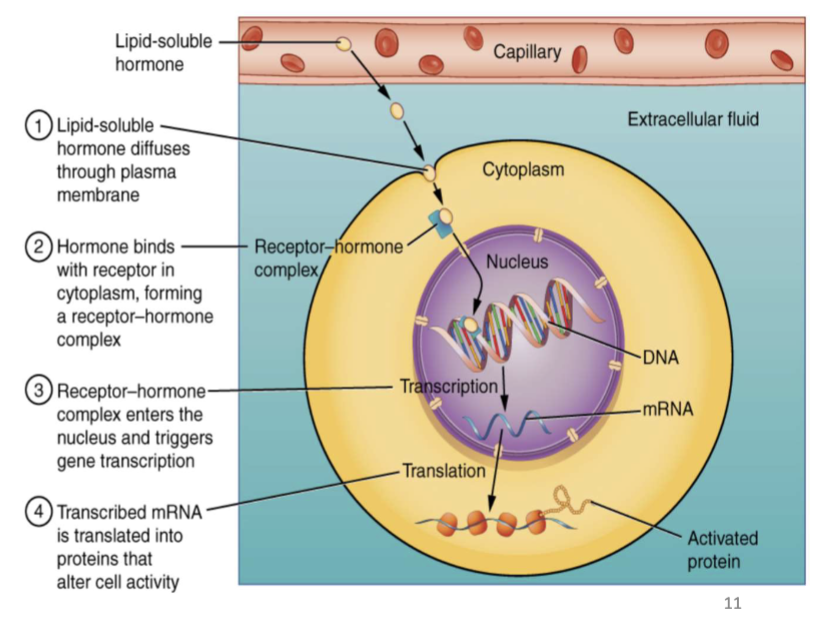
Intracellular (Nuclear) Receptors
Receptors for lipophilic ligands (e.g., thyroid hormones, adrenal cortical steroids, glucocorticoids, vitamin D) that regulate gene transcription and protein synthesis; effects are delayed.
Drug Affinity
ability of a drug/ligand to bind to it’s receptor to form the DR complex
governs receptor occupancy
can be measure as the dissociation constant (Kd)
Drug Intrinsic Activity
Ability of drug to trigger a pharmacologic response after binding to a receptor forming a DR complex
Governs receptor activation
HMG-CoA Reductase
Enzyme inhibited by statins; an example of an enzymatic drug target.
SGLT2
Glucose transporter in the proximal tubule inhibited by canagliflozin; an example of a transport protein target.
Tubulin
Structural protein inhibited by colchicine; an example of a non-receptor drug target.
ED50
Effective dose at which 50% of subjects exhibit the specified therapeutic (quantal) effect.
TD50
Toxic dose at which 50% of subjects exhibit a specified toxic effect.
LD50
Lethal dose at which 50% of subjects die; commonly used in toxicology but less used clinically.
Therapeutic Index (TI)
Ratio TD50/ED50 (or LD50/ED50); a higher TI indicates a wider safety margin, safer drug
Therapeutic Drug Monitoring (TDM)
Monitoring plasma drug concentrations to keep levels within a therapeutic window and avoid toxicity.
Therapeutic Window
Range of drug doses between the minimal effective dose and the dose that causes unacceptable toxicity.
ie lithium, digoxin, warfarin, phenytoin
Therapeutic Drug Monitoring (TDM)
Quantal Dose-Response Curve
All-or-nothing responses in a population; results are expressed as % of subjects responding at each dose.
ie prevention of arrhythmia, convulsions or death
Useful in quantifying an outcome in a big population to assess the variability in drug response such as ED50, Median ED50, Median TD50, LD50, Therapeutic Index (TI or PI)
Graded Dose-Response Curve
Continuous, measurable response in individuals across different doses; used to determine EC50/ED50 and maximal efficacy.
ie serum lipid levels
Y axis is average response at each dose
Median effective dose (ED50/EC50) dose concentration of drug needed to produce 50% of drug’s max effect
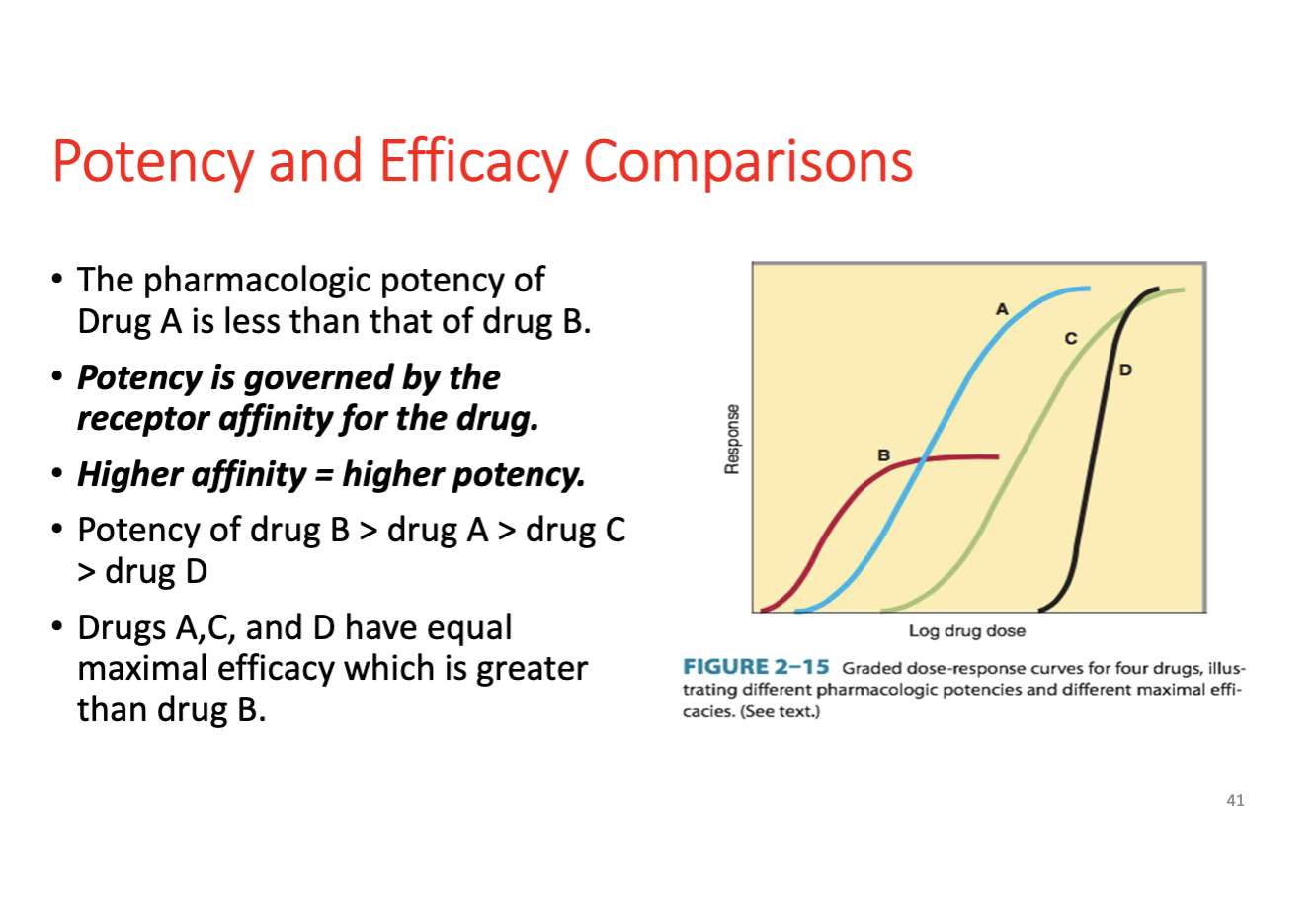
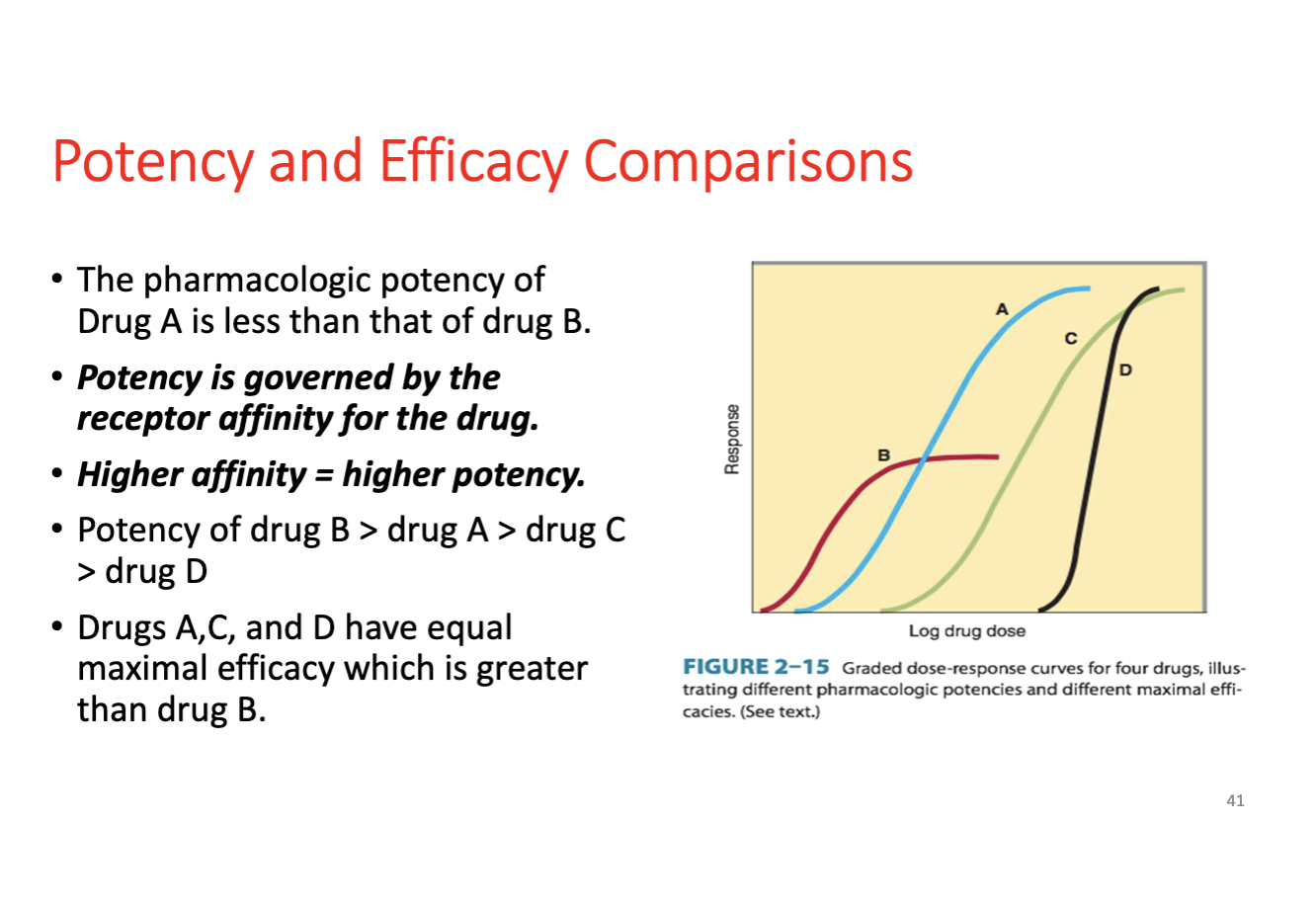
Potency
Dose or concentration required to produce a given effect; higher potency means a smaller dose and a leftward shift in the curve; does not reflect maximal efficacy.
Efficacy (Intrinsic Activity)
Maximal response a drug can produce once bound to its receptor; reflects the degree of receptor activation.
.
.
KD (Dissociation Constant)
Affinity measure for drug–receptor binding; lower KD indicates higher affinity.
Receptor Occupancy
Proportion of receptors bound by drug at a given concentration; depends on affinity and drug concentration.
Dose-Response Curve Shift (Concepts)
Competitive antagonists cause a rightward shift (higher dose needed) without reducing max; noncompetitive antagonists reduce max response.
Quantal Dose Response Curves - shows frequency of occurrence of a specified response to a drug
Graded Dose Response Curves
Used to determine maximal drug efficacy/response