Tegay - Carbohydrate Metabolsim Defects
1/21
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
22 Terms
Carbohydrates
Carbohydrates (“Hydrated carbon”)
Empiric formula (CH2O)n
Smallest n = 3
Primary source for energy and ATP production
Saccharide
Saccharide = surgar
Monosaccharides
Single sugar molecules
Eg. Glucose, fructose, galactose
Disaccharides
2 linked sugar molecules
Eg. Sucrose, lactose, maltose
Polysaccharides
Long chains of linked sugar molecules
Eg. Glycogen, amylose, amlyopectin, cellulose
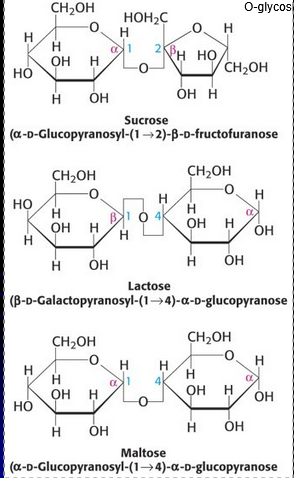
Common Disaccharrides
Digestive enzymes (eg. sucrase, lactase, and maltase) exist on the surface of small intestinal cells to catalyze breakdown of disaccharides to monosaccharides for absorption
Lactose intolerance is due to the loss of lactase activity in the small intestine
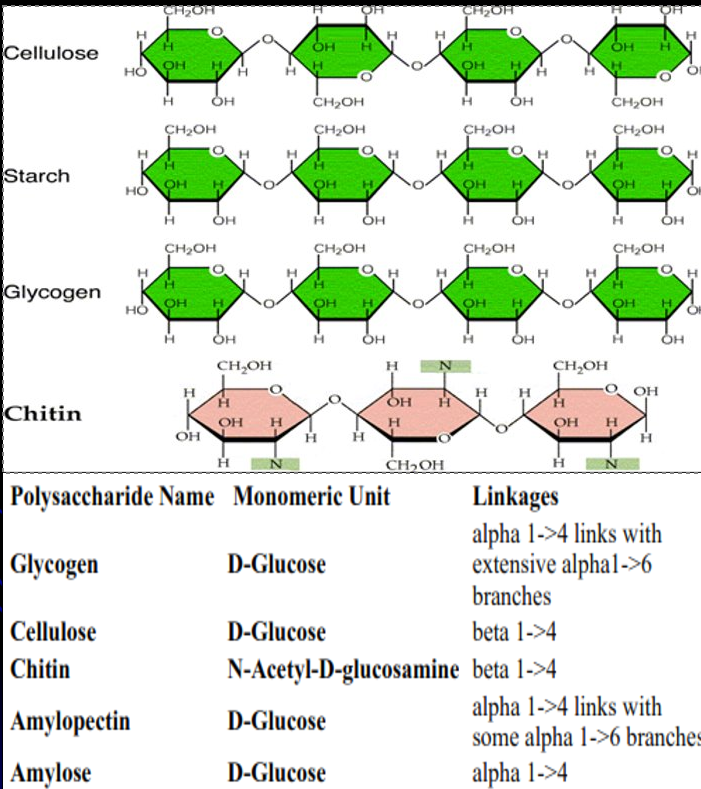
Polysaccharides
Polysaccharides
Long chains of linked sugar molecules
Eg. Glycogen, amylose, amlyopectin, cellulose
Starch
Amylose and Amylopectin
Major plant glucose storage polysaccharide
Amylose is unbranched with alpha-1,4 linkages
Amylopectin is branched with alpha-1,4 linkage (30X) attached to alpha-1,6 linkage
Mechanical breakdown of food by chewing starts digestion of starches but is insufficient alone
Amylase must be secreted by salivary glands and pancreas to hydrolyze amylose and amylopectin to glucose for absorption across intestine
Glycogen
Major animal glucose storage polysaccharide
Highly branched alpha-1,4 and alpha-1,6 glycosidic bonds
Body needs constant supply of blood glucose
Liver synthesizes and stores glycogen
When blood glucose is low, liver glycogenolysis occurs
Glycogen → Glucose-6-Phosphate → Glucose
Glucose can leave liver but not glucose-6-phosphate
Liver glycogen stores provide ~8 - 10 hours of glucose
Fat stores provide days to weeks of energy reserves
Muscle has glycogen but no glucose-6-phosphatase
Provides energy source but only within muscle cell
Brain has NO glycogen stores
Glycolysis
Process of metabolizing glucose in energy intermediaries for ATP production
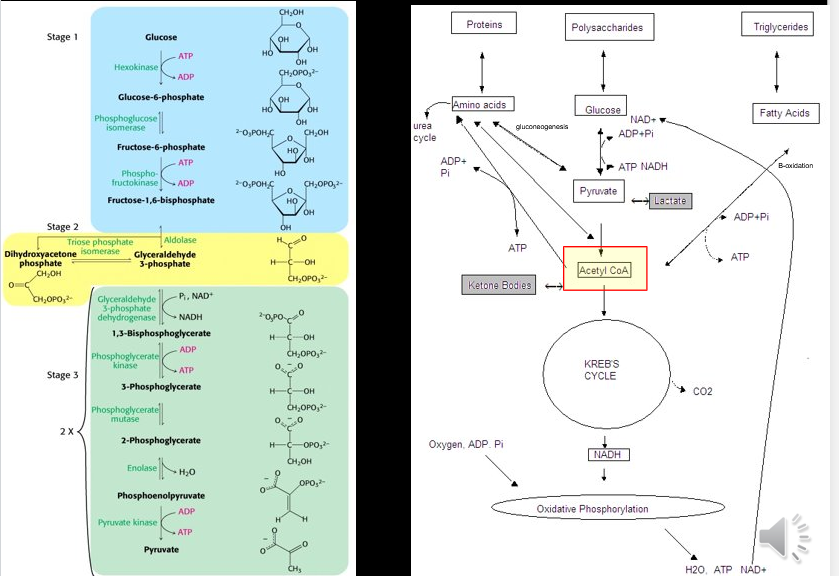
Gluconeogenesis
Process of synthesis of glucose within cells
Glucose can be synthesized from within cells from a variety of substrates
Glucogenic acids can be turned into glucose
Glycerol can enter into gluconeogenesis
Ketones
Ketones
Under hypoglycemic states, fatty acid B-oxidation produces excess acetyl-CoA which is shunted from energy production to ketone body formation
Acetoacetyl CoA, Acetone, B-OH butyrate
Ketone bodies cross cell membranes and are an alternative energy source for brain and muscle
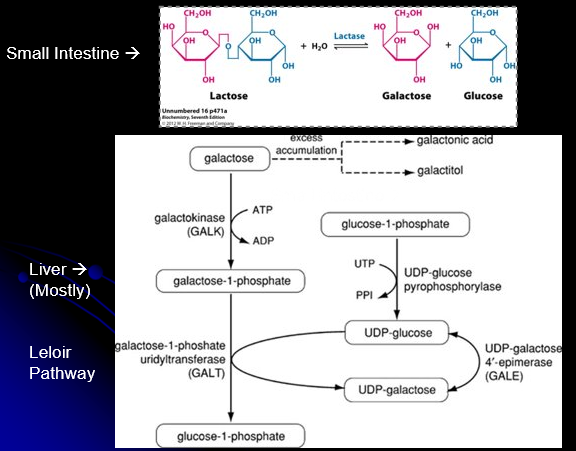
Galactose Metabolism
Lactose is a disaccharide consisting of galactose and glucose
Must be broken down via galactase to be absorbed
Galactose must be metabolized into usable glucose forms via Leloir pathway
When Leloir pathway is blocked, there will be accumulation of certain substrates and accumulation of alternative metabolic products
Galactose Metabolism Errors
Autosomal recessive
3 inborn errors of Galactose Metabolism
Galactokinase Deficiency
Untreated leads to cataracts
Diagnosed via high galactose, red cell enzyme, mutation
GALT Deficinecy (Galactosemia)
Mild or severe
Diagnosed via high galactose-1-phosphate and uridine diphosphate galactose, red cell enzyme, mutation
GALE Deficiency
Treatment
Life-threatening illnesses usually quickly resolved, although long term issues
Reduced IQ and growth, ataxia, ovarian dysfunction
Dietary galactose restriction
Classic Galactosemia (GALT Deficiency)
Autosomal recessive
Symptoms
Neonatal onset
Elevated liver enzymes and bilirubin, coagulopathy, gram negative bacteria
Lethargy, poor feeding, vomitting, diarrhea, death
Liver failure, jaundice, bleeding, hepatomegaly
Renal dysfunction
Cataracts
E-coli sepsis
Mutation anaylsis allows PGD / Prenatal
Treatment
Dietary galactose restriction
Urine / Blood Galactose Measurements
Urine
All monosaccharides are reducing sugars that reduce inorganic ions such as Cu++
Called Fehling’s reagent
Urine testing detects presence of reducing sugars in urine (normally negative)
Thin layer chromatography (TLC) can distingusih specific sugars
Blood
Newborn screening often measures blood spot Galactose and Gal-1-P levels
Detect all 3 galactose metabolsim defects but false negatives if on lactose-free formulas
NY state measures GALT blood spot
Detects only Galactosemia
Transfusiosn cause false negatives
Fructose Disorders
Autosomal recessive
3 Main Disorders
Fructokinase Deficiency (Fructosurea)
Hereditary Fructose Intolerence (Fructosemia)
Fructose 1,6-Biphosphatase Deficiency
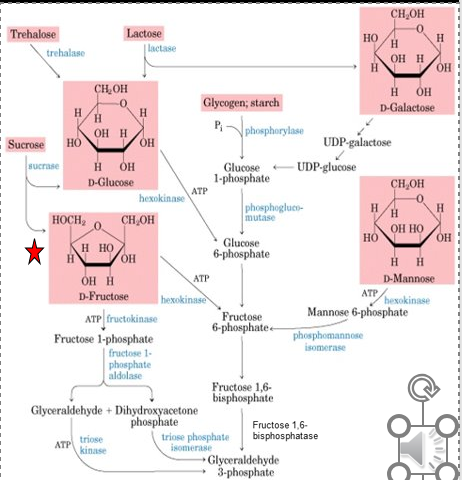
Fructokinase Deficiency (Fructosurea)
Autosomal recessive
Symptoms
Benign condition
Usually detected incidentally
Elevated urine reducing substance: fructose
NO treatments necessary
Hereditary Fructose Intolerance (Fructosemia)
Autosomal recessive
ALDOB gene
Fructose-1-P Aldolase B Deficiency
Symptoms
On consuming fructose
Vomitting, lethargy, irritability, seizures
Postprandial hypoglycemia, liver/kidney disease
Liver failure, acidosis, growth failure, death
Diagnosed via suspect with elevated urine reducing substance for fructose
Confirm with enzyme activity and/ or mutation analysis
Treatment
Eliminating fructose typically prevents further symptoms
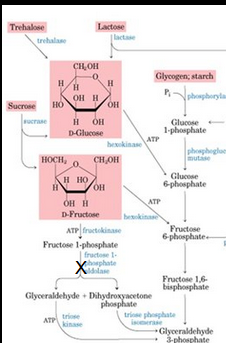
Fructose 1,6-Bisphosphatase Deficiency
Autosomal recessive
Symptoms
Sudden early life-threatening episodes of
Fasting hypoglycemia with lactic acidosis
Hyperventilation, vomiting, lethargy
May be lethal
Diagnosed via no urine fructose, can check enzyme on liver biopsy, urine glycerol-3-phosphate level, molecular testing
Treatment
Acute: correct hypoglycemia and acidosis, IV glucose
Chronic: avoid fasting with frequent glucose feeding, limit fructose
Good outcomes once treatment is initiated
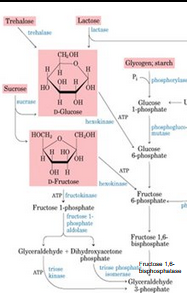
Glycogen Storage Disease
Almost all autosomal recessive
Primarily effect liver and/or muscle
Liver involvement
Hepatomegaly and hypoglycemia
Muscle involvement
Muscle breakdown and weakness
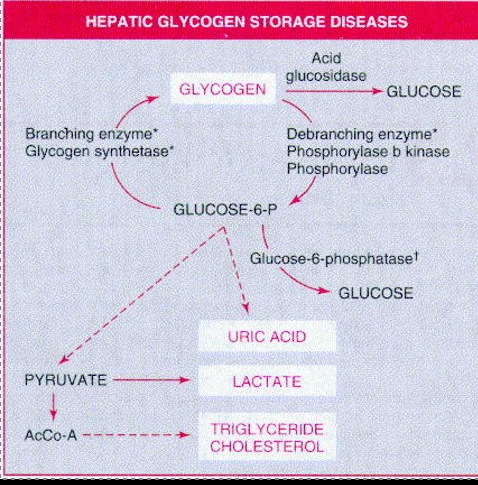
Von Gierke (GSD 1)
Autosomal recessive
Type 1a
Glucose-6-phosphatase enzyme is defective
GSD1a: Glucose-6-phosphate translocase (SLC37a4) gene
Type 1b
Translocase that transports glucose-6-phosphate across the microsomal membrane is defective
Both types mess up glucose-6-phosphate conversion to glucose and make individuals susceptible to fasting hypoglycemia
Symptoms
Sometimes hypoglycemia and lactic acidosis during neonatal period
Usually present at 3 - 4 months of age with hepatomegaly, hypoglycemia, hyperuricemnia, hyperlipidemia, and lactic acidosis developing after short fasting
Often “doll-like” faces with fat cheeks, relatively thin extremities, short stature, and protuberant abdomen
Type 1b also have neutropenia
Diagnosed after suspicious signs of:
Hypoglycemia with minimal fasting
Lactic acidosis, hyperuricemia, hyperlipidemia
Confirmed via
Enzyme analysis (requires liver biopsy)
Mutation analysis
GSD1a: Glucose-6-phosphate translocase (SLC37a4) gene
Similar to GSD III but different gene, myopathy, milder
Treatment
Avoid fasting, maintain blood glucose
May need continuous nasogastric glucose
Uncooked cornstarch = slow release glucose every 4 hours for infants younger than 2 years
With age can go to every 6 hours by mouth as a liquid
Restricted fructose and galactose
Dietary supplemenets
Allopurinol and uric acid, stains for cholesterol
Watch for hepatic adenomas
G-CSF for neutropenic immunodeficiency
Pompe Disease
Autosomal recessive
Lysosomal Acid-akpha-Glucosidase (Acid maltase)
GAA Deficiency → Lysosomal Glycogen Storage
Forms
Infantile, Juvenile, Adult onset
Symptoms
Organs affected by glycogen accumulation
Cardiac, skeletal and smooth muscle
Muscle and organ enlargement via glycogen storage
Tongue (macroglossia), Heart (cardiomegaly), Liver and spleen
Progressive muscle breakdown and weakness
Elevated CPK, hypotonia, hypertrophic cardiomyopathy, cardio-respiratory failures
Diagnosed via muscle biopsy with glycogen staining, enzyme, and DNA
Pseudodeficiency = low enzyme but asymptomatic → check DNA
New born screening = <10 days in IOPD
Treatments
Without treatment → Death by 1 year / variable death
With treatment outcomes are variable but significant improvement
Administer acid alpha-glucosidase algucosidas alfa
Treatment depends on infantile onset or later onset
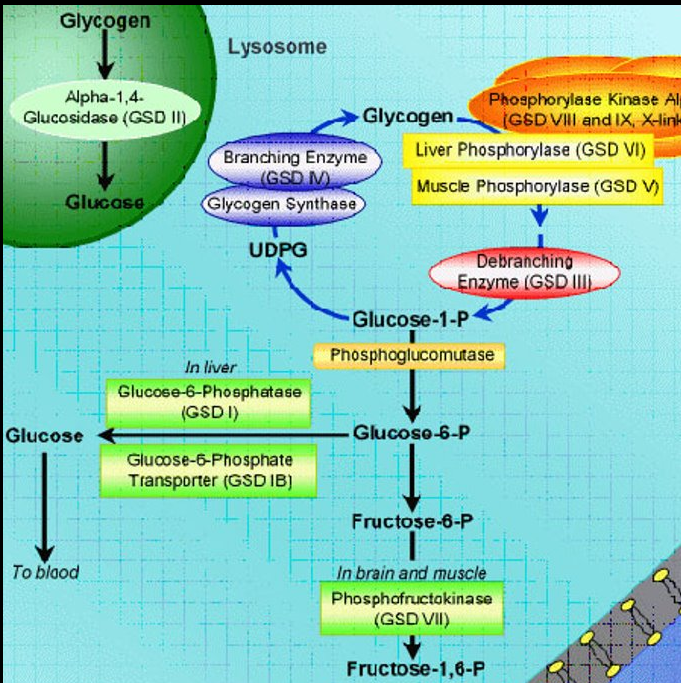
McArdle Disease (GSD V)
Autosomal recessive
Mutation of muscle myophosphorylase (PYGM)
Decreased glycogenolysis
Symptoms
Initially recurrent exercise induced muscle cramps and pain that is relieved by rest
Recurrent episodic myoglinurea that can lead to renal failure
Lifelong poor exercise capacity, fatigue, stamina
Chronic proximal muscle weakness
Diagnosed
Elevated serum muscle creatine kinase (CK) level post exercise
Urine myoglobin levels
Confirmed via:
Enzyme analysis (Myophosphorylase activity on muscle biopsy)
Molecular analysis (PYGM gene sequencing and deletion / duplication)
Similar to GSD VII (Tarui’s)
Treatment
Sub-maximal aerobic exercise
Avoid medications that promote myopathy
Diet and supplements
GLUT1 Deficiency
Autosomal dominant
Disorders of glucose transporters
Classic Symptoms
Seizures (before 2 years in 90%), DD, acquired microcephaly, complex movement disorder and cognitive impairment
Symptoms increase with fasting fever, and infection
Diagnosed via low CSF glucose with normal blood glucose
Treatment
Results in significant improvement
Keto diet, L-Carnitine, avoid glucose intake