KINE 4518 - Midterm #1
1/70
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
71 Terms
What is Cell Signaling
1. A means where by cells can adapt to changing conditions
when you lose this…this is when you get disease
2. A means to regulate cellular behavior and activity
Allows for cells to communicate
3. A mechanism to turn responses off and on
there are some things we don't always want ‘on’
Cell signaling is important for understanding…
vital to proper maintenance of cell function
integral in molecular biology
often involved in diseases
With cancer it is often not cell division that goes wrong…but cell preparation that goes wrong
What Regulates Protein Phosphorylation
1. Protein Kinases (enzyme that adds phosphate by taking off a hydrogen)
reversibly adds a covalent phosphate group to amino acids on target proteins
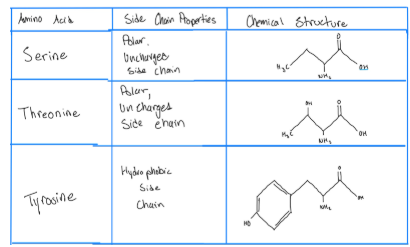
occurs on serine (S), threonine (T) or tyrosine (Y) hundreds of kinases; specific target proteins (target protein is the protein that in the end we want to control)
Out of the 20 amino acids we can only phosphorylate these 3 S,T,Y
Cell signaling typically involves only 3 amino acids
Most common phosphorylation is on S and T and they get phosphorylated by the same protein because although different their side chains are the same
What phosphorylates S and T CAN NOT phosphorylate Y its has a separate protein that does that and vice versa
Tyrosine side chain has a 6 carbon ring which is why it gets phosphorylated by a different protein
phosphate (negatively charged) gets added to side chain (phosphorylation)…takes a hydrogen and adds a phosphate through a covalent bond…once it gets added if the protein kinases gets turned off the phosphate stays on the amino acid
2. Phosphatases (enzyme that removes phosphate)
removes the phosphate group from the protein far fewer phosphatases than kinases
less specific than protein kinases
What Determines Whether a Protein is turned on or off?
Phosphatases more general
Kinases are mor specific
Attributes of Amino Acids: Serine, Threonine, and Tyrosine
polar = likes water
Phosphorylation Affects Protein Function
adds negative charges (when you get phosphorylated) to a modified protein causes conformational changes (activates or deactivates) exposes enzyme catalytic/active site sends original signal “downstream” to other effector proteins
the active site or catalytic might have been hidden or blocked until it gets phosphorylated
‘Downstream’ = something needs to tell the cell it needs to change e.g. the brain
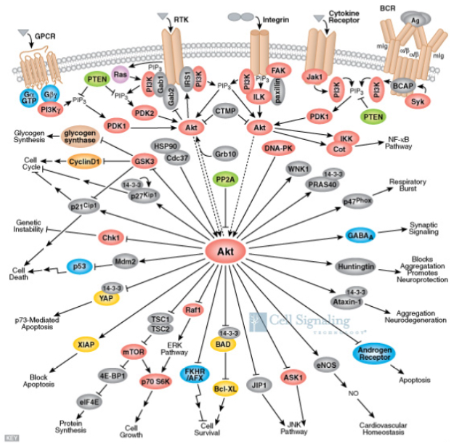
The PKB/AKT Pathway (Example of a Signaling Pathway)
Akt (protein kinase B)= is a kinase and it phosphorylates if it has an arrow going to it
Akt doesn't work alone…other things interact with it to determine an outcome
a cell responds to individual signals by deciding the outcome based on the net input from everything (a cell doesn't know when to grow…it grows when it gets told to grow)...which is why in this example you can see all these different outcomes
Decides outcome based on the NET input from everything
E.g. cells don’t become cancerous because they ‘want’ to…they became cancerous because they are told to
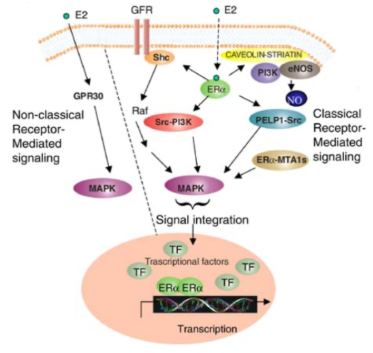
The MAP Kinase Pathway
ER = estrogen receptor…
involved in vaginal, ovarian, breast cancer
The receptors in the membrane are hydrophilic
The JAK/STAT Pathway (Discovered at Sick Kids)
estrogen, testosterone are lipid soluble (cell membrane is made of lipids)
ER is located inside the cell (as composed to the outside of the cell) and that is because it is lipid soluble so they can go through the cell membrane
Something that is water soluble won't dissolve in fat/lipid; something that is lipid soluble won't dissolve in water
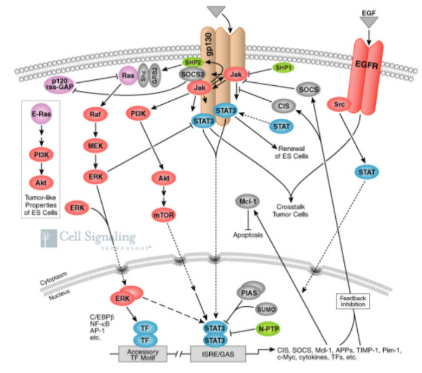
The Estrogen Receptor Signaling Pathway
Muscles and white blood cells have the SAME DNA but they look different because we use different things to tell cells to be a blood cell, hair cell, or muscle cell ect ect
Stem cell differentiate
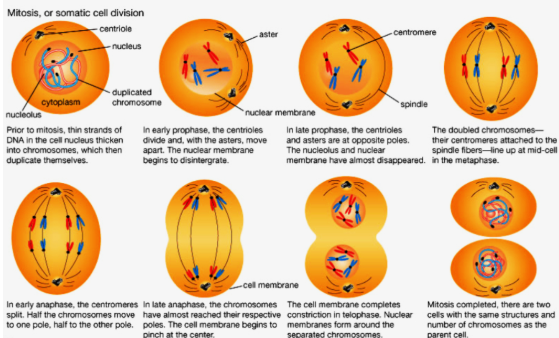
Mitosis
start with a single cell and end with 2 cells (daughter cells— identical to each other and identical to the original)…if the daughter cells are different then the original then it changes functions and this is usually due to a mutation…these mutation can possibly lead to cancer …mutation in cell division is bad (if it leads to cancer)
not all mutations are bad
mitosis is splitting of the cell
most mutations occur before the cell starts to divide (prior to mitosis)…and once the cell commits to dividing its going to divide
not all cells divide and replicate at the same rate and not all cells divide…
e.g. you have never heard of heart cancer…the only ‘heart’ cancer is due to the cells in the SAC around the heart becoming cancerous (pericardial cancer)
e.g. skin cells go through mitosis often as compared to muscles cells
cells that become cancer HAVE to be able to divide
epithelial cells tend to be the cells that turn cancerous…a lot of our structures are lined with epithelial cells
main function of epithelial cells = protection
Our cells don’t want to grow it takes a lot of effort to grow… they also have to be told to grow…and are only told to divided
if you have more pro growth signals than anti growth signals then the cell with grow and vice versa…and this is regulated and specific
cells in a HEALTHY individual ONLY grow when they are told to
2 people that have colon cancer have different cancers because the cause is different so a treatment that works for one person might not work for another which makes it difficult to cure cancer
not all mutations are bad though… a giraffe has mutated to be able to reach vegetation high above
What Initiates Cell Division/Proliferation?
Growth signals: (things that tell the cell to grow)
natural/normal —> development/healing
aberrant (non proper growth)—> diseases/cancer (inability to maintain homeostasis)
Most our signals are External growth factors
something outside the cell tells the cell to grow
examples of a signal could be…make more energy
sometimes the mutation occurs with inside the cell and removes the need for an external growth signal (very bad)
mis regulation of cellular pathways
not a ‘real’ signal; inappropriate growth
Cancer tends to be an older person disease because you need to accumulate a lot of cell division to experience 1 cell division that goes wrong
over the number of years cancer cases have gone up but deaths from cancer has gone up (deaths/cases = %) but this percentage goes down due to the drastic increase in cases
we want this percent number to be 0 (the average right now is about 40%)
cancer treatments want to target the number of deaths and bring it down
we see a better mortality rate but still the number of deaths from cancer continue to increase
sometimes the mutation of cell happens within which ‘removes’ the need for an external growth signal
The Mammalian Cell Cycle
Getting this process STARTED is very difficult
if you remove the growth signal then the cell stops after mitosis and does not enter G1
average cycle is 24 hours in a mammalian cell
we can speed this process up and slow it down depending on what our body TELLS it that we need
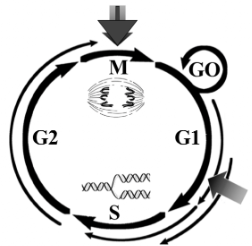
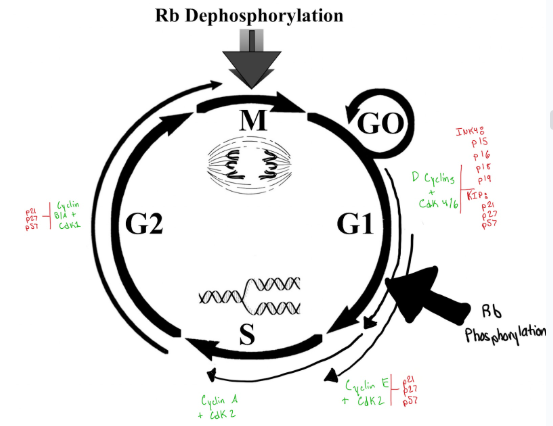
Phases of the Cell Cycle
M Phase (M = mitosis) = cell separation/division (1 hour)
S Phase (S= synthesis) = DNA synthesis/replication (6 hours)
G1 Phase: intrinsic way to make sure the environment can support new cells
Gap 1 Phase
originally thought as a rest phase —> not true
preparation of cells for chromosome replication
cell grows in size
cell prepare for DNA synthesis
cell survey environment to see if conditions are right…in humans we don't need to check this because we regulate these as humans…but if you trick the cell and make it think there no food it would stop growing
cell looks for food/energy because if you from 1 cell to 2 cell to 4 cells it needs enegry…so before it divides it looks for energy sources
it also looks for temperature at 37
the cell also looked for pH (to acidic it wont survive)
if its a plant it looks for sunlight
G2 Phase
Gap 2 phase
also originally thought as a rest phase
preparation of cells for mitosis (prepare for the cell to split)
cell grows in size due to production of more cytoplasm
check integrity of DNA (often broken during cancer)
checks for mutation and gaps
How Does Cancer Therapy Work
basically damages the DNA so much so that the cells commits suicide but it targets your healthy cells as well resulting, rashes, sores in the mouth, hair cell, skin damage, GI thinning, anemia, affects blood and therfor white blood cells making you immune compromised an dall this together can make the cancer work
patients take MTD (maximum tolerant dose)…so the persona takes as much of the drug as they can take so that the drug can ACTUALLY recap the tumour/cancer
therefore cancer is a battle of chemo
Important Factors in G1, S, G2 and M Phase
we get to transition point from G1 and S phase and this transition is represented by the right triangle (3 o’clock)…once it clears the check point (meaning the environment can support a new cell) then the cell can then move into S phase…the white arrows go one direction because you can't go back…if you transition into the next phase without being ready it can cause problems… checkpoint snake sure everything that is going through has been done properly
Our check pints:
Between G1 and S (managed by proteins cyclin E/cdk2)
Between S and G2
Between G2 and M
cyclin E and cdk2 monitor/control the check printer between G1 and S phase and for these proteins to check the cell they need to be FULLY activated to be able to do their job properly
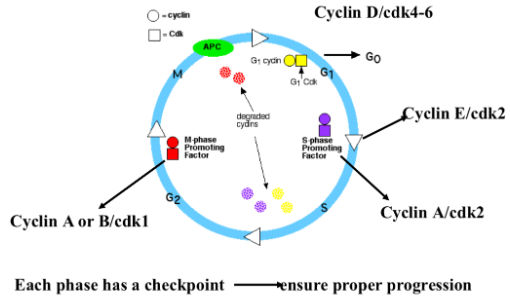
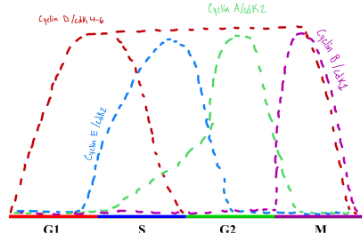
The Cyclins
cyclins are proteins that are expressed cyclically (meaning they go up and go down)…they can’t go up in a unorganized pattern
Cyclins by themselves go up and down according to their role in mitosis
At the beginning and end of the cell cycle we don't have much cyclin D
Cyclin D is a gas pedal
This is sequential…meaning it happens in this order
Cycin D is important for starting the process
Cyclin E is important for the transition between G1 and S phase
Cyclin A is important for the transition between S into G2 phase
Cyclin B is important for the transition between G2 into M phase
why do we NOT want these check point “on” all the time…because if they were then we would lose our cell regulation
Cyclin D is present more prominently in G1…in SOME cells cyclin D stays up throughout S, G2 and part of M phase…and in those other phases it spectates the cell and the regulation but it still MUST come down BEFORE the cell fully divides because we DON'T want them on all the time because if they were then we would lose our regulation of the cell (e.g. more cells being made than we need, mutations can occur)
cyclins on their own don't do much…so they need a partner proteins called cdk (Cyclin Dependent Kinase) [kinase - enzyme that adds phosphate]…to activate the kinase we change the level of their partner (cyclin) [cdk is dependent on the cyclin and won’t do anything if the cyclin isn’t around]
kinase makes the engine go
![<ul><li><p><span style="background-color: transparent;">cyclins are proteins that are expressed cyclically (meaning they go up and go down)…they <u>can’t</u> go up in a unorganized pattern </span></p></li><li><p><span style="background-color: transparent;">Cyclins by themselves go up and down according to their role in mitosis </span></p></li><li><p><span style="background-color: transparent;">At the <u>beginning </u>and <u>end </u>of the cell cycle we don't have much cyclin D</span></p></li><li><p><span style="background-color: transparent;">Cyclin D is a gas pedal </span></p></li><li><p><span style="background-color: transparent;">This is sequential…meaning it happens in this order </span></p></li><li><p><span style="background-color: transparent;">Cycin D is important for starting the process</span></p></li><li><p><span style="background-color: transparent;">Cyclin E is important for the transition between G1 and S phase</span></p></li><li><p><span style="background-color: transparent;">Cyclin A is important for the transition between S into G2 phase</span></p></li><li><p><span style="background-color: transparent;">Cyclin B is important for the transition between G2 into M phase</span></p></li></ul><ul><li><p><span style="background-color: transparent;">why do we NOT want these check point “on” all the time…because if they were then we would lose our cell regulation </span></p></li><li><p><span style="background-color: transparent;">Cyclin D is present more prominently in G1…in SOME cells cyclin D stays up throughout S, G2 and part of M phase…and in those other phases it spectates the cell and the regulation but it still MUST come down BEFORE the cell fully divides because we DON'T want them on all the time because if they were then we would lose our regulation of the cell (e.g. more cells being made than we need, mutations can occur)</span></p></li><li><p><span style="background-color: transparent;"><u>cyclins </u>on their own don't do much…so they need a partner proteins called <u>cdk </u>(Cyclin Dependent Kinase) [kinase - enzyme that adds phosphate]…to activate the kinase we change the level of their partner (cyclin) [cdk is dependent on the cyclin and won’t do anything if the cyclin isn’t around] </span></p><ul><li><p><span style="background-color: transparent;">kinase makes the engine go </span></p></li></ul></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/9ac9631f-c1e5-4fea-878b-65868343fc59.png)
Activates of the Cyclin/cdk Complexes
Cyclin A and E share the same cdk protein
Cyclin D can interact with cdk 4 or 6
The Mammalian Cell Cycle - p Proteins
p = molecular weight…p27 is the most researched
Green means = go
Red means = brake
must overcome ALL the brakes acting on the single cyclin/cdk to move on in the process of cell division which is WHY it takes ALOT to start the cell cycle
7 potentially inhibitory proteins that you have to get rid of or modify in order to get through to go from G1 to S Phase
The Cyclins and cdk’s
Cyclin-dependent kinases (cdks) are the motor that makes the cell cycle go
cdk’s are always around… and this benefit is that it is quick…BUT we dont want them ON all the time
Ser/thr kinases (mentioned back in first class) [NOT Tyrosine kinases]
Unlike cyclins cdk’s are not cyclically expressed meaning cdk is always present at any given time and they only become active when they are told to become active
to turn on cdk’s we increase the level of their partner proteins cyclin
What need to happen to cyclin/cdk’s to move through the cell cycle?
1. Cdk’s need to be bound with their associated cyclin to become functional
Increasing the presence of the cdk’s corresponding cyclin will help activate the cdk
when cdk’s and cyclins bind together they expose the active site on the enzyme (confirmation change)
both the cyclin and cdk change shape
kinase (cdk’s) is what is the ‘gas’ and makes the system ‘go’
2. Activated/deactivated by phosphorylation
Kinases are deactivated by phosphorylation (we have deactivating phosphorylation and activating phosphorylation)
Often activated by phosphorylation, cyclin, and being removed from cdk inhibitors
3. Cyclins and cdk’s need to be removed from their cdk inhibitors
are inhibited by cdk inhibitors (CKIs)
their are ‘breaks’ on cdk’s that we need to remove
ALL 3 of these NEED to happen to get advancement through the cell cycle
Cyclin D- G1 Cyclin
Known as the G1 cyclin
you can't start cell Division WITHOUT increasing cyclin D
Three isoforms: Cyclin D1, D2, D3
all very similar but have slightly different 3D shape but all do the same thing
D1 is more common especially in cancer
depends on the tissue you are looking at
we get these to go up through transcription…transcription factors get activated, they go into the nucleus and make cyclin D1,D2, or D3 by taking mRNA translating it into protein that we can use
Cycling D become active through transcription (shown in image —> c-myc and MAPK
if you knock out either of the 3 isoforms at a time we still have 2 mechanisms to activate cyclin D…having 3 isoforms also allows that if something goes wrong with one of them you still have 2 more to fall back on
each is somewhat dispensable…you don't need all 3 isoforms
pros of having all 3
reducnecey…if something goes wrong with cyclin D3 you have something else to rely on and the outcome is the same
even of you knock out ⅔ of them the out come is the same
binds to either cdk4 or cdk6

2 Main Functions of Cyclin D
2 Main Functions: change shape of Rb + change location of kip proteins
1. Phosphorylate Rb (retinoblastoma)
Rb is another protein
very important function
1 target but…5 options
3 cyclins
2 cdk’s
2. Sequester Kip Proteins
sequester (bring it somewhere else) kip (‘break’/slow down) protein [inhibitor protein]
sequesters break proteins
takes something away from where it’s supposed to be
Kip proteins are in the nucleus…if you take kip proteins out of the nucleus then they can’t do their job…because then they would have been removed from where they need to be to keep the ‘brake’ on cyclin D+ cdk4/6
Since cyclin D is in the nucleus it can easily remove KIP from the nucleus
1) and 2) are ‘breaks’ / stops cell division and by REMOVING the ‘brakes’ it allows the process of cell division to proceed
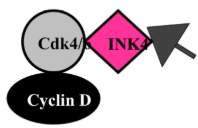
INK4 Proteins
INK4 (Inhibitor of cdk4)
inhibits cdk4
must get rid of INK4 to activate cyclin D/cdk4
4 family members: (4 isoform acting on 1 kinase)
p15
p16
p18
p19
binds only to cdk4 or 6
sole inhibitor of Cyclin D/cdk4 or 6
even if cyclin D is present as long as the INK4 is still around it keeps the protein in an inactive form
Cyclin E
Similar to cyclin D in function
multiple forms of cyclin E
Integral to G1/S phase transition
FULL ACTIVATION to be able to move between phases
Transcribed by E2F transcription factor…they make mRNA that makes proteins
mRNA is transcribed by a transcription factor called E2F transcription factor and this makes mRNA that makes proteins
Binds to cdk2 to form active complex
broader spectrum of proteins phosphorylated than cyclin D
cyclin D/cdk4-6 phosphorylated Rb only (changes shape of retinoblastoma ONLY)
Cyclin E/cdk2 Phosphorylates:
cyclin E/cdk2 phosphorylates: (changes shape of 3 things NOT just Rb)
1. Rb —> activates its own transcription
cause the production of more cyclin E (positive feedback)
cyclin E/cdk 2 also phosphorylates Rb because Rb inhibits cyclin E transcription and when Rb gets phosphorylated it helps increase the production of cyclin E (positive feedback)
helps prepare DNA to be replicated
2. Histone (H1)
Histone winds DNA (makes it smaller) and puts it in the nucleus
DNA is about 6 feet long and a cell is 50 ųm…DNA is 40,000x larger than the length of your cell
when histones gets phosphorylated (by cdk 2) it unwinds the DNA(change in shape) so that we can duplicate it in S phase…if we don't unwind it we don’t duplicate all the DNA which could result in inaccurate DNA and IF it doesn't kill it self and survives the cell cycle it could lead to cancer
3. p27 —> removes its inhibitor, enhances its own activity
p27 NATURALLY is an inhibitor
removes more inhibitor proteins which helps the production of cyclin E
p27 - KIP1 (Kinase Inhibitor Protein 1)
P 27 is a Cell Cycle Inhibitor Inhibits: inhibits cell cycle progression (break in cell cycle)
1. Cyclin E/cdk2 → G1-S Transition
2. Cyclin A/cdk2 → S Phase
3. Cyclin A-B/cdk1→ G2-M
*p27 can cause cell cycle arrest not matter where we are in the cell cycle since it can stop it at any stage of the cell cycle…suggested that it is a universal cell cycle inhibitor which is important to understand cancer progression
If p27 is present in any of these cell cycle stage it will cause cell cycle arrest
***P27 is Highly regulated by phosphorylation; while cyclins are regulated mostly by mRNA transcription
prevents cell cycle progression
assembles Cyclin D/cdk4-6 —> cell cycle progression
***when p27 interacts with Cyclin D/cdk4 it brings the complex together which helps cdk4 become active so that we can phosphorylate Rb…and in this case instead of being an inhibitor it actually acts as a ‘gas pedal’
P27 has a binding site for cyclins and cdk
P27 changes configuration when certain proteins are bound to it which allows it to have these 2 different functions…depending on its shape
since INK4 is the primary inhibitor of cyclin D/cdk4 (specifically inhibits the cdk) when the cell is undergoing mitosis that will be the primary inhibitor BUT when the cell is in REST with NO growth signal p27 WILL inhibit cyclin D/cdk4 [depends on where the cell is within the cycle]
levels high in G1
regulated at the protein level (not transcribing cyclins)
Protein expression of p27
In cell rest p27 levels are high and right at the transition between G1 and S phase p27 levels drop off rapidly and it stays constant and as the cell starts to divide p27 levels shots up again (cyclins have a opposite trend)
When a protein goes flat at any given point we call it ‘steady state’ which means the level of input = the level output…what you make is the same as what it getting used/getting rid of
When p27 level drop synthesis either has to go down or degradation of p27 has to go up…in the cell cycle it is caused by p27 degradation
Cyclin E/cdk 2 phosphorylates p27 which causes p27 degradation
As cyclins and cdks go up we stars reduce the ‘breaks’ in the cell cycle
as we reach S phase there is no inhibition of cell division at least by the KIP proteins which makes transitioning to the next phase easy
![<ul><li><p><span style="background-color: transparent;">P 27 is a Cell Cycle Inhibitor Inhibits: inhibits cell cycle progression (break in cell cycle)</span></p><ul><li><p><span style="background-color: transparent;">1. Cyclin E/cdk2 → G1-S Transition</span></p></li><li><p><span style="background-color: transparent;">2. Cyclin A/cdk2 → S Phase</span></p></li><li><p><span style="background-color: transparent;">3. Cyclin A-B/cdk1→ G2-M</span></p></li><li><p><span style="background-color: transparent;">*p27 can cause cell cycle arrest not matter where we are in the cell cycle since it can stop it at any stage of the cell cycle…suggested that it is a universal cell cycle inhibitor which is important to understand cancer progression</span></p></li><li><p><span style="background-color: transparent;">If p27 is present in any of these cell cycle stage it will cause cell cycle arrest </span></p></li></ul></li><li><p><span style="background-color: transparent;">***P27 is Highly regulated by phosphorylation; while cyclins are regulated mostly by mRNA transcription </span></p></li><li><p><span style="background-color: transparent;">prevents cell cycle progression </span></p></li><li><p><span style="background-color: transparent;">assembles Cyclin D/cdk4-6 —> cell cycle progression </span></p><ul><li><p><span style="background-color: transparent;">***when p27 interacts with Cyclin D/cdk4 it brings the complex together which helps cdk4 become active so that we can phosphorylate Rb…and in this case instead of being an inhibitor it actually acts as a ‘gas pedal’ </span></p><ul><li><p><span style="background-color: transparent;">P27 has a binding site for cyclins and cdk </span></p></li><li><p><span style="background-color: transparent;">P27 changes configuration when certain proteins are bound to it which allows it to have these 2 different functions…depending on its shape </span></p></li></ul></li><li><p><span style="background-color: transparent;">since INK4 is the primary inhibitor of cyclin D/cdk4 (specifically inhibits the cdk) when the cell is undergoing mitosis that will be the primary inhibitor BUT when the cell is in REST with NO growth signal p27 WILL inhibit cyclin D/cdk4 [depends on where the cell is within the cycle] </span></p></li></ul></li><li><p><span style="background-color: transparent;">levels high in G1 </span></p></li><li><p><span style="background-color: transparent;">regulated at the protein level (not transcribing cyclins)</span></p></li><li><p><span>Protein expression of p27</span></p></li><li><p><span>In cell rest p27 levels are high and right at the transition between G1 and S phase p27 levels drop off rapidly and it stays constant and as the cell starts to divide p27 levels shots up again (cyclins have a opposite trend) </span></p></li><li><p><span>When a protein goes flat at any given point we call it ‘steady state’ which means the level of input = the level output…what you make is the same as what it getting used/getting rid of </span></p></li><li><p><span>When p27 level drop synthesis either has to go down or degradation of p27 has to go up…in the cell cycle it is caused by p27 degradation </span></p><ul><li><p><span>Cyclin E/cdk 2 phosphorylates p27 which causes p27 degradation </span></p></li><li><p><span>As cyclins and cdks go up we stars reduce the ‘breaks’ in the cell cycle</span></p></li></ul></li><li><p><span>as we reach S phase there is no inhibition of cell division at least by the KIP proteins which makes transitioning to the next phase easy </span></p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/5df7a1c8-2945-4df6-978a-a1d206fbae4f.png)
The Retinoblastoma (Rb) Protein
Tumour suppressor gene (meaning it is a cell cycle inhibitor)...BUT is a cell cycle inhibitor always a tumor suppressor? …no
tumor suppressor means if you remove it there is 100% that the cell will get cancer
if you knock out Rb the chances of you getting cancer is drastically increases
Mutations in Rb are rare and if they happen they aren't necessarily passed down genetically
If you get a problem or mutation with a cell cycle inhibitor there is no guarantee you will get cancer
3 isoforms BUT they don’t compensate for each other which is not good
inhibit the E2F transcription factors (main function of retinoblastoma proteins)
Mutation in retinoblastoma effects the ability to inhibit E2F transcription factor
Retinoblastoma
In a normal person when you flash a light at someone and take a photo their eyes look red because the light is absorbed
If you shine a light and its foggy looking its because the eye is reflecting the light which suggest that the retina should be ONE layer thick to allow light through and if the eye is reflecting the light its because the retina has multiple layer
Retinoblastoma is Often a Young Childs Disease as the Retina is Formed
If you let retinoblastoma go untreated it can grow a tumor and you can notice it has vasculature (red)...for it to grow so large it need blood vessels to deliver stuff to it … thought process → most cancer treatments target the tumor but the problem with this is tumour growth is different in everyone so MAYBE if we can target the tumors blood supply it wouldn’t be allowed to grow any more because you're also then targeting the tumour nutrients and oxygen with it (could be a protein cancer treatment)...might be able to shrink it…die with it but not die from it
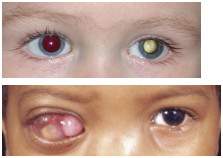
Jeff Healy
Jeff Healey is a musician who plays his guitar on his lap because he had retinoblastoma but has been treated for it but his vision is gone because it kept coming back and got into his systems and died
Retinoblastoma Function Regulation
Rb Function is regulated by phosphorylation on multiple sites
Post transcriptional regulation
Phosphorylation causes a change in shape
Originally phosphorylated by cyclin D/cdk 4 and as this happens we start to get more cyclin E/cdk2 will also start to phosphorylate it
phosphorylated by active Cyclin E and Cyclin D complexes
hypo-and hyper-phosphorylated forms
hypo - to little or less; less phosphorylated
hyper - to many or more; more phosphorylated
Rb is NEVER not in its dephosphorylated form so we use reactive terminology
Hyperphosphorylated Retinoblastoma
hyperphosphorylated Rb (Cyclin D/cdk4 and Cyclin E/cdk2):
1. Dissociates from E2F transcription factors
Lots of Rb in G1
E2Fs become active
must get rid of Rb to move through the cell cycle
2. Is targeted for degradation by proteasome (degradation of proteins)
more phosphorylation —> more change in shape —> more degradation of Rb —> more inhibition of E2F factor
Cyclin D/cdk4 can stay active through S and G2 phase BECAUSE Rb is not present during those stages and Rb is cyclin D/cdk4 ONLY target…we want cyclin D/cdk4 to come down though before the cell divides because if it stays up from start to finish then that can be dangerous
cyclin D/cdk 4 goes down when Rb goes up is what we want to happen
results in S-phase entry
Mitogenic Signal + Retinoblastoma
Mitogenic signals = growth signals
the box represents a divider…it is not a nucleus
K4 = cdk 4
k2 = cdk 2
Rested Cell conditions:
No cyclin D
Cdk4 (bound to INK4) [inactive] (inhibitor)
Lots of kinase inhibitor proteins → p21, p57, p27 [inhibitor]
cyclin E/cdk 2 and cyclin D/cdk4 can BOTH phosphorylated Rb (specifically the cdk’s do) and separate Rb from E2F the cyclin E binds to cdk2 to phosphorylate MORE Rb (positive feedback)… BUT there is SO MANY KIP/p27 proteins that it inhibits cyclin E/cdk2 so how does cyclin E/cdk 2 ever become active if p27/KIP proteins are so abundant that it inhibit cdk 2…p27 in its pool when cyclin D/cdk 4 comes around will take some of that pool and bring it over and once there is a growth signal it will no longer be an inhibitor like it is for cyclin E/cdk2 it actually brings cyclin D/cdk4 together (protein complex) and when they are bound together it is not inhibited (like cyclin E is) it is active which can then phosphorylate Rb
Goal
Hyperphosphorylate Rb to get rid of it inhibition on E2F so they can make more cyclin E and we can build up the cyclin E over the amount of inhibitors
More cyclin E binds to cdk2 which can phosphorylate Rb
Removal of p27 is something we need to do and we do that by phosphorylating cyclin E/cdk 2
Cyclin E/cdk 2 phosphorylate p27 and causes its degradation
BUT how do we phosphorylate p27 when p27 inhibits the kinase
everyone of these steps have room for something to go wrong and all it takes is one mistake to potentially cause a mutation that causes cancer
![<ul><li><p><span style="background-color: transparent;">Mitogenic signals = growth signals </span></p></li><li><p><span style="background-color: transparent;">the box represents a divider…it is not a nucleus </span></p></li><li><p><span style="background-color: transparent;">K4 = cdk 4 </span></p></li><li><p><span style="background-color: transparent;">k2 = cdk 2 </span></p></li><li><p><span style="background-color: transparent;">Rested Cell conditions:</span></p><ul><li><p><span style="background-color: transparent;">No cyclin D</span></p></li><li><p><span style="background-color: transparent;">Cdk4 (bound to INK4) [inactive] (inhibitor)</span></p></li><li><p><span style="background-color: transparent;">Lots of kinase inhibitor proteins → p21, p57, p27 [inhibitor]</span></p></li></ul></li><li><p><span style="background-color: transparent;">cyclin E/cdk 2 and cyclin D/cdk4 can BOTH phosphorylated Rb (specifically the cdk’s do) and separate Rb from E2F the cyclin E binds to cdk2 to phosphorylate MORE Rb (positive feedback)… <u>BUT </u>there is SO MANY KIP/p27 proteins that it inhibits cyclin E/cdk2 so how does cyclin E/cdk 2 ever become active if p27/KIP proteins are so abundant that it inhibit cdk 2…p27 in its pool when cyclin D/cdk 4 comes around will take some of that pool and bring it over and once there is a growth signal it will no longer be an inhibitor like it is for cyclin E/cdk2 it actually brings cyclin D/cdk4 together (protein complex) and when they are bound together it is not inhibited (like cyclin E is) it is active which can then phosphorylate Rb </span></p></li><li><p><span style="background-color: transparent;">Goal</span></p><ul><li><p><span style="background-color: transparent;">Hyperphosphorylate Rb to get rid of it inhibition on E2F so they can make more cyclin E and we can build up the cyclin E over the amount of inhibitors </span></p></li></ul></li><li><p><span style="background-color: transparent;">More cyclin E binds to cdk2 which can phosphorylate Rb </span></p></li><li><p><span style="background-color: transparent;">Removal of p27 is something we need to do and we do that by phosphorylating cyclin E/cdk 2</span></p><ul><li><p><span style="background-color: transparent;">Cyclin E/cdk 2 phosphorylate p27 and causes its degradation </span></p></li><li><p><span style="background-color: transparent;">BUT how do we phosphorylate p27 when p27 inhibits the kinase </span></p></li></ul></li><li><p><span style="background-color: transparent;">everyone of these steps have room for something to go wrong and all it takes is one mistake to potentially cause a mutation that causes cancer </span></p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/454affaf-1a75-4d00-b850-685dec2a79e1.png)
The E2F Transcription Factors
Family of transcription factors…they make mRNA
8 different isoforms (all involved in the cell cycle)….redundancy meaning it is very important…they also all compensate for one another and Rb is the only inhibitor of the E2F factor which is why we need to get rid of Rb
E2F is important for cell cycle progression
Bind to ‘pocket’ region on Rb —> inhibits E2F activity
hyperphosphorylation of Rb dissociates E2F complex (cdk2/cdk4)
E2F can become active —> binds to other transcription factors to help enhance its activity (DP-1)….in action to removing inhibitor we can also bind to other proteins that help other proteins become more active
E2F Transcription Factors Important Structural Domain
1. DNA binding (all transcription factors have this…needed to transcribe DNA) [DNA binding site]
2. Pocket protein binding domain (section of protein that interact with other proteins and these proteins are called pocket proteins and Rb is an example of a pocket protein - specific to Rb)[we still want to remove Rb]
A pocket protein is a member of a small family of tumor suppressor proteins that share a conserved “pocket domain” — a structural region that binds transcription factors of the E2F family and other regulatory proteins.
3. Dimerization domain (area where other proteins interact…where transcription enhancers bind to resulting in a change in shape of the E2F and therefore change its function)
E2F Transcription Factor: Promoter Binding Site
Promoter binding site: (T/C)TT(C/G)(G/C)CG(G/C)
this is DNA and DNA is basically sugar
have 3 phosphates
anything that is in blue it can be either or
the black letter have to be in that order so that when it folds on itself it can fit into the DNA binding site…this is how cdk4/6 know their targets since they only have one…and that's because the amino acid sequence fits into their active region and all the other proteins don’t …and this is specific to each protein and/or enzyme
How does the promotor binding site interact with the DNA binding domain on E2F?
The promoter binding site (the consensus DNA motif in the gene) interacts directly with the E2F DNA-binding domain, which locks onto the DNA via sequence-specific contacts. Stability is reinforced by DP partners, and activity is controlled by whether Rb/pocket proteins are bound.
E2F Transcription Factors: Gene Targets Involved in:
E2Fs are master regulators of the cell cycle, so their gene targets fall into several big groups:
1. Cell cycle (Cyclin E)
2. DNA replication (DNA polymerase alpha)[enzyme that copies DNA]
3. DNA damage repair (BRCA1)
BRCA 1 and 2 genes is the only hereditary mutation which means you are at higher risk of cancer (breast, uterine, female estrogen linked cancer)[defective DNA repair check]
common in ovarian and breast cancer
activates p53
if BRACA1 is damaged it wont activate p53
Normally, p53 is kept at low levels because MDM2 ubiquitinates and degrades it.
BRCA1 can interfere with MDM2–p53 interaction, reducing p53 degradation.
BRCA1 has a transactivation domain that can directly bind p53.
This binding enhances p53’s ability to recruit co-activators (like CBP/p300) to promoters of target genes.
Result: stronger transcription of p53 target genes (e.g., p21 → cell cycle arrest, BAX → apoptosis).
When BRCA1 is mutated, the feedback control on E2F1 is lost.
E2F1 may then overactivate replication genes without adequate DNA repair.
E2F activates BRCA1, and BRCA1 helps regulate E2F1’s activity.
Together, they synchronize cell cycle progression and DNA repair — ensuring replication happens safely.
4. DNA synthesis (thymidine kinase)
Thiamine starts out as thymidine kinase…we add 3 phosphates to it to become thiamine to be incorporated into DNA
5. Apoptosis (pro-caspase 3)
programmed cell death
protein that is responsible for this is pro-caspase 3
*1,2,4 are parts of S phase which is why we want to activate them BEFORE we get to S phase
3 Main Functions of E2F Transcription Factor
1. DNA replication in S-phase
we don't want extra copies of genes
2. Ensure DNA integrity
e.g. BRAC1
3. Prepare for cell destruction/death if DNA isn't intact
usually in the form of apoptosis
E2F Transcription Factor Continuation
Phosphorylated by Cyclin —> increases affinity for Rb A/cdk2
E2F gets phosphorylate by cyclin A/cdk2
Prepares E2F to bind new hypophosphorylated Rb
inhibits its activity
once we get into S phase we get degradation (phosphorylation) of E2F by cyclin A/cdk2 which phosphorylates E2F transcription factors for degradation (occurs in S phase) or change the shape of E2F factors so that they aren’t active anymore (can't interact with DNA)
remember when E2F returns to its original shape (after E2F gets phosphorylated) it wants to bind Rb but in late G1/S phase there is no Rb but it prepares it for end of G2/M phase where Rb in the cell starts to rise
E2F transcription factors get shut off during late G1/S phase…if E2F stayed active you’d get all 3 functions BUT the problem would be that if you continue to make DNA after you make the one copy you need you are then gonna get extra copies and that can be harmful and could lead to disease
p53 Discovery
(+/+) = 2 copies of the gene (wild type)
(+/-) = 1 copy of the gene
(-/-) = no copies of the gene
Discovered ≈ 38 years ago (40 years ago ish)
Discovered as a cancer protein
screens all cancers and found that in 80% of cancers p53 was elevated
When proteins cause cancer we refer to it as oncogene/oncoprotein
Scientist then tested this on animals and made a knockout aniaml… with the assumptions that without p53 the animal wouldn't get cancer because at this point with elevated p53 levels in 80% of cancers it was assumed p53 causes cancer…mice with (+/+) lived for about 500 days which is normal given mice only live to 2-3 years…but without p53 (-/-) scientist found that the animals died faster/quicker…
showed p53 is not an oncogene and that is a tumour suppressor…if you knock its out 100% die quicker and they get cancer everywhere and they die from cancer NOT WITH cancer
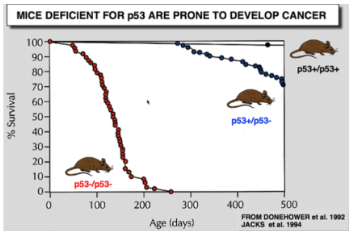
p53
P53 is a family of proteins (p63 and p73)
these isoforms don’t compensate for one another
tumour suppressor
Involved in cell cycle and apoptosis (cell death)
most diseases are multifactorial meaning there is not no one cause and we call this sporadic and anything that is associated with it is referred to as a risk factor….it may make you more susceptible to cancer but doesn't mean anything one thing causes it…
causes of cancer:
DNA mutation (can happen through radiation e.g. sunlight is radiation)[radiation causes DNA damage]…but yet radiation is a form of cancer treatment with the idea that it causes enough DNA damage that the cell kills itself
p53 is Responsible for Decision of Cell to “Live or Die”
p53 helps the cell kill itself
the reason it was elevated in the cancer cells is because those cells had so much DNA damages that they were trying to kill its self
in 60 to 70% of cancers p53 is mutated
therefor any therapy invoking DNA damage in a cancer with p53 mutation that treatment wont work or be more harmful
people with cancer now one of the first things they do is get tested to see if they have a p53 mutation
for some reason p53 is in a location where it gets skipped over that allows it to be a common mutation
cell knows when to die is there are high levels of p53
P53 is a Transcription factor…meaning it has a DNA binding domain
gene targets induce cell cycle arrest or apoptosis
protein level regulated post-translationally
p53 levels/activity low in unstressed cells
2 Main Functions of p53
Cell Cycle Arrest
induces cell cycle arrest during DNA damage (UV)
1. Transcribes p21 —> inhibits Cyclin E/cdk2 G1 arrest
cell cycle inhibitor…causes G1 arrest
2. Transcribes Gadd45 Inhibits cdk1 (G2/M)
causes G2 arrest
3. Inhibits c-myc transcription (G1)
binds DNA and inhibits transcription
inhibits protein called c-myc
c-myc is also a transcription factor that transcribes cyclin D1 mRNA…resulting in G1 arrest
p53 stops this from happening resulting in a G1 arrest
Apoptosis (programmed cell death)[if DNA is not salvageable]
during DNA damage p53 up regulates pro-apoptosis genes
Including:
Death - pro apoptotic
Bax
PUMA
Nox
survival - Anti apoptotic
Counteract Bcl-2
we have lots of this signal in the cell but as time goes on we make more death signals if there is DNA damage
as long as you have more cell survival signal then the cell will survive…but if the death signals surpass the survival signals then the cell will die
p53 Function Regulated by:
1. Regulation of p53 protein levels
specifically through degradation
2. Cellular localization of the protein
p53 is a transcription factor which means it needs to be in the nucleus
3. Modulation of activity
changes 3D shape (phosphorylation, proteins binding)
the protein responsible for 1.,2.,3. Is called mdm2 (mouse double minute protein 2)
p53 and mdm2
1. Degrades p53
2. Shuttles p53 out of nucleus
3. Binds with p53 and inhibits p53 —> prevents interaction with other transcription factors
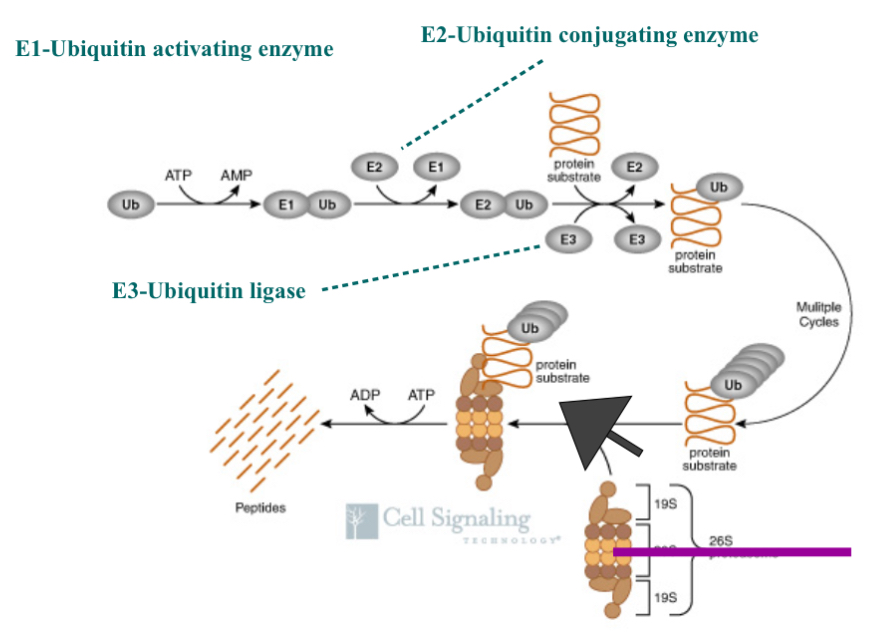
The Ubiquitin Proteasome Pathway
Mechanism for targeting proteins for degradation
proteasome break down proteins…identifys the cell we need to get rid of it
ubiquitin is a protein and when we add it to a protein it breaks sit down (degradation)
identifies these to the cell that we need to get rid of it
Addition of ubiquitin to target proteins on lysine (amino acid)
we add Ub to proteins on a lysine amino acid
lysine ONLY (1/20 amino acids)
changes function or signals degradation
we can add one Ub or multiple Ub
Ubiquitin —> 76 amino acids protein (8 kDa)
proteasomes are always there for degradation BUT proteins MUST be TARGETED by multiple or one ubiquitin for degradation
Multiple ubiquitin molecules added in a chain (polyubiquitination)
can also just add 1 ubiquitin (monoubitquitination)
modifies protein function by changing shape…acts as phosphorylation
adding only 1 Ub does not signal degradation it signals the protein to change shape and therefore change function
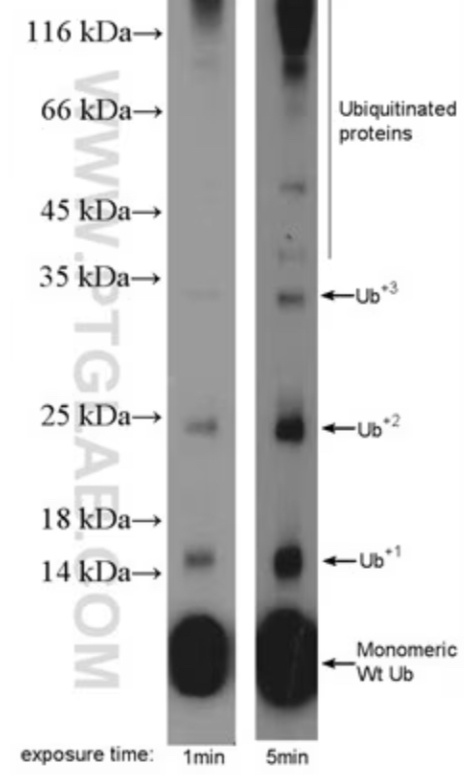
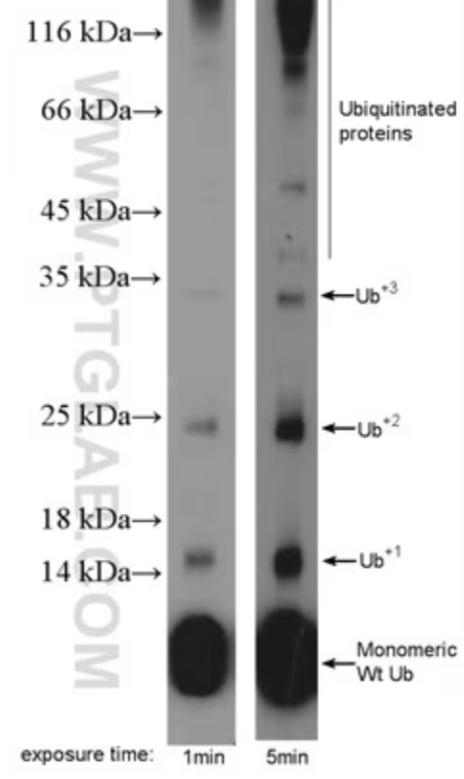
Explain the Image
in the 1 minutes exposure time you can see there are already Ub+2 and then after 5 minutes you can see that Ub+3 if not more than that…eventually the levels kind drop off as that the protein starts to get degraded…this shows that Ub gets ADDED to the protein…this shows how we can understand Ub and degradation
this is how the cell knows that a protein needs to get degradation
Ubiquitinization Pathway
Ub = ubiquitin (in high amounts in every cell)
it is not specific…can be added to everything as long as there is a lysine
not rate limiting in this process as we always have enough ubiquitin
protein substrate = protein of interest
1. Use energy from ATP to link Ub to E1 (ubiquitin activating enzyme)
By doing this you activate Ubiquitin
E1 is not specific, not many of them in the cell, and they are always there and ready to go so they are not rate limiting
2. Take Ub and add it to E2 (Ub conjugating enzyme) that mediates the preparation to be add Ub to the protein substrate
E2 are no specific,only a few in the cell, are always there in the cell, and are always ready to go
3. Once E2 and Ub are together it can be added to the protein of interest by the help of E3 (Ub Ligase)
join Ub to the target
E3 goes up and down, and they are specific…E3 gives the process specificity
if you can prevent a specific protein from being degraded you have a way to intervene a pathway that may be disrupted in cancer
Steps 1., 2, and 3. Continue and create a chain of Ub on the protein of interest
4. 26S chromosome (garbage can with a lid thing) the part on the top and bottom is what interact with Ubiquitin and that’s how the Ubiquitin targets the protein to be degraded…chain of ubiquitin molecules interacts with the lid of the garbage can and brings the protein into the garbage
5. The protein inside the garbage can there are enzymes in the garbage can that break the amino bonds between the amino acids and breaks the protein down into smaller peptides and/or the individual amino acids…this changes the structure and therefore changes the amino acids function…this broken off amino acids can then either be used in metabolism or used to make new proteins
What Happens if you are Energy Deprived?
Let’s say your energy is deprived and you're not taking in enough protein or calories to maintain daily function…we typically will take it from fat…or we can break down proteins in our body because when one amino acid is linked to another there is energy in that bond and we then use that energy…if you go on a low calorie diet amend use up your fat stores you then start to get into crisis mode and your body will start to break down proteins…a by product of breaking these amino acid bonds are heat (heat is a by product of metabolism)…your body will slow down metabolism when your energy intake is restricted resulting is a lower resting heart rate (since a lower RH requires less energy) which gives the impression that they individual is ‘fitter’…the other thing that will happen is the body will redirect blood to the core and away from the extremities because we need to maintain blood temperature of 37 degrees for the organs and brain because your metabolism is now slowed resulting in a decrease in heat production because the by product of metabolism is heat…and that often why people with anorexia are cold and have a lower resting heart rate
intermediate fasting is the best way to lose weight because you don't allow your body to go into crisis mode…not enough data on the longer term effects
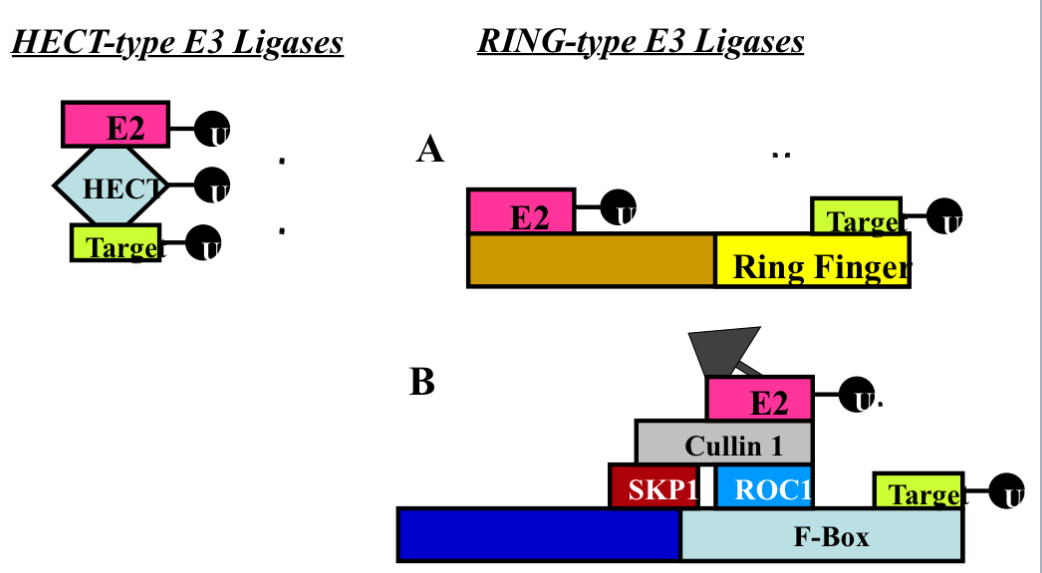
2 Main Classes of E3 Enzymes (UbI quit in Ligases)
1. HECT-Domain
single protein…protein binding domain (Interactionist with other proteins)
the domain is a spot on the protein that allows it to bind other proteins
HECT domain is approx 350 amino acids (40 kDa) [pretty big proteins]
forms thiol-ester bond with Ub before transfer of Ub to target protein
E3 ligase forms a permanent bond with Ub before it transfers it to the target protein…take Ub off E2 and holds it and then gives it to the target
thio-ester bond means permanent bond (the type of bind it uses)
Smurf2 (86 kDa), Nedd4 (115 kDa), and E6AP (100 kDa) [all these are big in size and that because it is one protein that does everything] {examples of E3 Ligase}
the firsts 2 (Smurf2, Nedd4) we have in our cells currently BUT we don't have E6AP in our cells
E6AP is a viral protein and is in the Human Papillomavirus (HPV)…there are 3 variations of HPV that have the gene for E6AP (there are other variations of HPV)
E6AP is a E3 ligase that degrades p53
BUT P53 IS TYPICALLY REGULATED BY MDM2
since E6AP is a viral protein we need to be infected by one of its variants of HPV that carries that gene…if you get infected with the version that contain E6AP you get degrade on of p53
is in infected cells and you have a higher degradation rate of p53 in those cells that have E6AP…and if cells don't have p53 the cell will more likely become cancerous (specifically cervical cancer because it is a sexually transmitted disease)
females who get infected by HPV have a good chance of getting cervical, vaginal, anal, and ovarian cancer for females
high-risk types of HPV significantly increases the risk of several cancers.
GOOD NEWS THOUGH…since E6AP is foreign in our immune system we prime our body to the antibodies through vaccines
HPV vaccines protect you before infection by teaching your immune system to recognize the HPV outer shell (L1 protein).
Because there’s no E6 or E7 gene, the vaccine:
Never produces the E6 protein that would hijack E6AP.
Leaves E6AP and p53 functioning normally.
2. RING-Finger Ligases - RING Domain (same Principle as HECT domain but instead of a HECT domain it’s domain is the RING domain)…
2a. Single Protein
RING domain binds target proteins
transfer Ub to target protein (takes E2 and target protein and bring them together THEN the Ub goes from E2 into target)[same end result just slightly different way of doing it as compared to the HECT domain]
Example of these: MDM2 (90kDa), Cbl (110 kDa) (E3 ligase for epidermal)
2b.Multi-Protein Complex
SKP1/Cullin 1/F-box protein (E3 ligase - multiple protein complex that does the same thing)[same thing but many proteins each take a step]
the F-BOX protein gives the E3 complex specificity
also includes ROC1 —> RING-finger protein
acts as a cord to hold everything together
every compares has SKP1/Cullin 1/ and ROC1
F-Box protein gives E3 complex specificity —> bind to target
F-box creates the difference in each complex
transferred Ub to target protein
SKP2 (F-Box of p27) [48 kDa] , FBW7 (F-box protein for cyclin E)[74 kDa]
![<ul><li><p>1. HECT-Domain</p><ul><li><p>single protein…protein binding domain (Interactionist with other proteins)</p></li><li><p>the domain is a spot on the protein that allows it to bind other proteins </p></li><li><p>HECT domain is approx 350 amino acids (40 kDa) [pretty big proteins] </p></li><li><p>forms thiol-ester bond with Ub before transfer of Ub to target protein </p><ul><li><p>E3 ligase forms a permanent bond with Ub <u>before</u> it transfers it to the target protein…take Ub off E2 and holds it and then gives it to the target </p></li><li><p>thio-ester bond means permanent bond (the type of bind it uses) </p></li></ul></li><li><p>Smurf2 (86 kDa), Nedd4 (115 kDa), and E6AP (100 kDa) [all these are big in size and that because it is one protein that does everything] {examples of E3 Ligase}</p><ul><li><p>the firsts 2 (Smurf2, Nedd4) we have in our cells currently BUT we don't have E6AP in our cells </p></li><li><p>E6AP is a viral protein and is in the Human Papillomavirus (HPV)…there are 3 variations of HPV that have the gene for E6AP (there are other variations of HPV) </p><ul><li><p>E6AP is a E3 ligase that degrades p53 </p><ul><li><p>BUT P53 IS TYPICALLY REGULATED BY MDM2</p></li></ul></li><li><p>since E6AP is a viral protein we need to be infected by one of its variants of HPV that carries that gene…if you get infected with the version that contain E6AP you get degrade on of p53 </p></li><li><p>is in infected cells and you have a higher degradation rate of p53 in those cells that have E6AP…and if cells don't have p53 the cell will more likely become cancerous (specifically cervical cancer because it is a sexually transmitted disease) </p></li><li><p>females who get infected by HPV have a good chance of getting cervical, vaginal, anal, and ovarian cancer for females </p><ul><li><p>high-risk types of HPV significantly increases the risk of several cancers.</p></li></ul></li><li><p>GOOD NEWS THOUGH…since E6AP is foreign in our immune system we prime our body to the antibodies through vaccines </p></li><li><p>HPV vaccines protect you before infection by teaching your immune system to recognize the HPV outer shell (L1 protein).</p><p>Because there’s no E6 or E7 gene, the vaccine:</p><p></p><ul><li><p>Never produces the E6 protein that would hijack E6AP.</p></li><li><p>Leaves E6AP and p53 functioning normally.</p></li></ul><p></p></li></ul></li></ul></li></ul></li><li><p>2. RING-Finger Ligases - RING Domain (same Principle as HECT domain but instead of a HECT domain it’s domain is the RING domain)…</p><ul><li><p>2a. Single Protein</p><ul><li><p>RING domain binds target proteins</p></li><li><p>transfer Ub to target protein (takes E2 and target protein and bring them together THEN the Ub goes from E2 into target)[same end result just slightly different way of doing it as compared to the HECT domain]</p></li><li><p>Example of these: MDM2 (90kDa), Cbl (110 kDa) (E3 ligase for epidermal)</p></li></ul></li><li><p>2b.Multi-Protein Complex </p><ul><li><p>SKP1/Cullin 1/F-box protein (E3 ligase - multiple protein complex that does the same thing)[same thing but many proteins each take a step] </p><ul><li><p>the F-BOX protein gives the E3 complex specificity </p></li></ul></li><li><p>also includes ROC1 —> RING-finger protein </p><ul><li><p>acts as a cord to hold everything together </p></li></ul></li><li><p>every compares has SKP1/Cullin 1/ and ROC1</p><ul><li><p>F-Box protein gives E3 complex specificity —> bind to target </p></li><li><p>F-box creates the difference in each complex </p></li></ul></li><li><p>transferred Ub to target protein </p></li><li><p>SKP2 (F-Box of p27) [48 kDa] , FBW7 (F-box protein for cyclin E)[74 kDa] </p></li></ul></li></ul></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/4065afb1-580f-4d21-934f-529ffbc415a8.jpg)
Summary of E3 Enzymes
target ends up with ubiquitin
HECT summary
Ub goes from E2 → HECT → Target protein
RING summary
RING domain acts with target protein
Rest of the protein interact with E2
Ub goes form E2 directly to the target and NVEVERR interacts with the E3 (RING protein)
F-BOX summary
Instead of a single protein there are multiple protein that do the same job
F-Box gives specificity
Most our E3 ligases are in the form of the F-Box protein
Gaol = target protein has at least 1 if not more Ub attached to it
The Cell Cycle and Cancer
Cancer is a disease of the cell cycle
Starts out as a normal cell
our immune cells can’t recognize cancer cells because they STARTED out as a normal cell
our immune systems recognizes KAPOSI Sarcoma (why does our immune system only recognize this…still in research)
Discovered through brown bumps found on people with AIDS in the 80’s and the brow bumps came on because their immune system was fighting the cancer resulting in these brown cancerous bumps
Cell Cycle and Cancer Can be Caused by:
1. Genetic mutation or deletion in something that regulate the cell cycle
Environmental impact; mutagens/carcinogens, Pollution, UV radiation
2. Misregulation of normal cell cycle processes
hit the gas or take foot off the brakes
What Exactly Happens in Cancer
Cell will often interpret continuous growth signals (happens in cancer)
1) Lose the ability to regulate normal growth
2) Lose the ability to get rid of damaged cells (apoptosis)
increase the chance of further mutation
apoptosis occurs in G2 and is highly mutated in cancer
There is another form of cell apoptosis in G1 (we talk about later one in the course)
3) Lose the ability to detect/repair damaged DNA
Big problem with cancer is when there is NO growth signal but the cell is still be activated and dividing…and it will continue to divide until its told to stop but if it interprets a continuous growth signal then we have a big problem
Once you go down these steps…it becomes very difficult to reverse
Fun Fact!
We actually have the tools to repair DNA but it doesn't guarantee that it gets done properly… we don't have the technology to repair DNA/genes and ensure it gets place in the exact sport it should be on the chromosome that it has to be…and if you put that gene in the wrong spot and it is not regulated like it should be it can cause trouble and possibly make it worse
Cell Cycle and Cancer End Result
End result: Unwanted proliferation of cells
tumour development
numerous potential underlying causes
meaning they should have different therapies
60% of our current cancer therapies work but treatment is harsh and can be too hard on patients…
New thought process is instead of one high concentrated drug…they are doing multiple drugs all together but all at lower doses
Problem 2 is that even if you have a new drug that works on animals to bring it to humans means that they get to worse of the worse cancers because we let our current treatments that works 60% of the time run its course first before the patient is allowed to test any new and upcoming cancer treatments…this harms our ability to make new drugs especially for the mild cancers before the progress to much
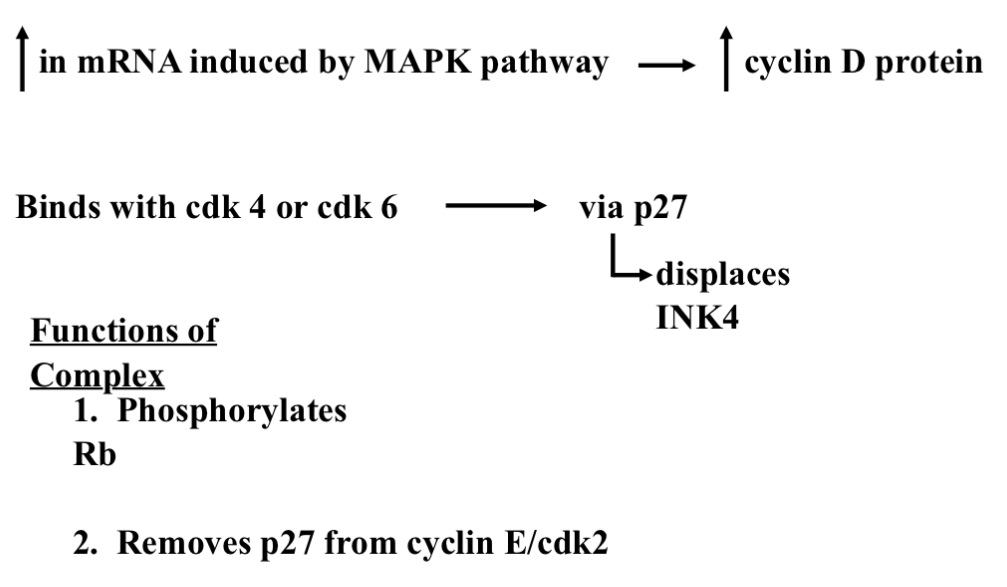
Cyclin D —> on Switch
Transcriptionally up-regulated in response to mitogenic stimuli (growth signal)
Initiated by MAP kinase pathway —> c-myc (end product- protein)
MAP kinase activates c-myc
When MAP kinase activates c-myc we get an increase in cyclin D (enter into G1/S phase)
P53 inhibits c-myc which prevent transcription of cyclin D
Translates to increase in protein
Must combine with cdk4 or cdk6 for effect
Cdk must be phosphorylated by Cyclin Activating Kinase (CAK) on threonine 172
Cyclin D and cdk4/6 Assembled by Kip proteins (p21 or p27)[p21 and p27 are in the nucleus of all cells]
Displaces INK4 protein
Complex translocates to nucleus and phosphorylates Rb
Very specific to phosphorylating Rb (no other targets)[very specific…very important- directed response]
What tells us it's important…
Fewer targets
More redundancy
Phosphorylation of Rb dissociates it from E2F proteins
E2F binds with DNA and then transcription factors become active that advance the cell cycle
Transcribe genes that advance the cell cycle
when it binds with DNA
Summary of Cyclin D
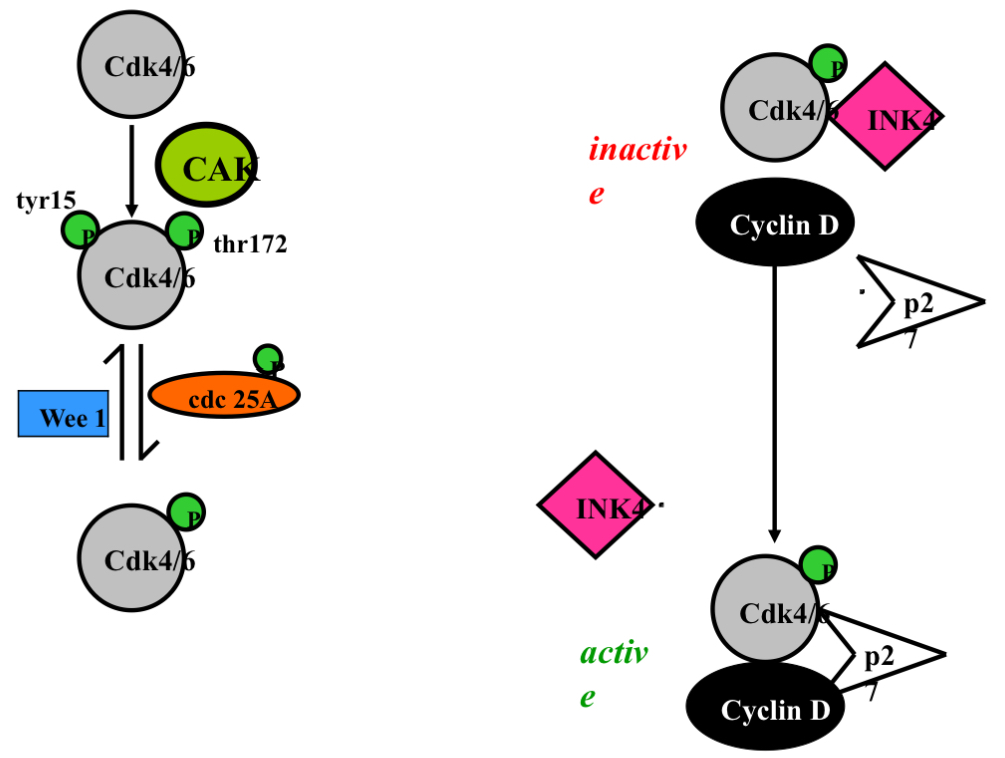
Summary Cyclin D and p27
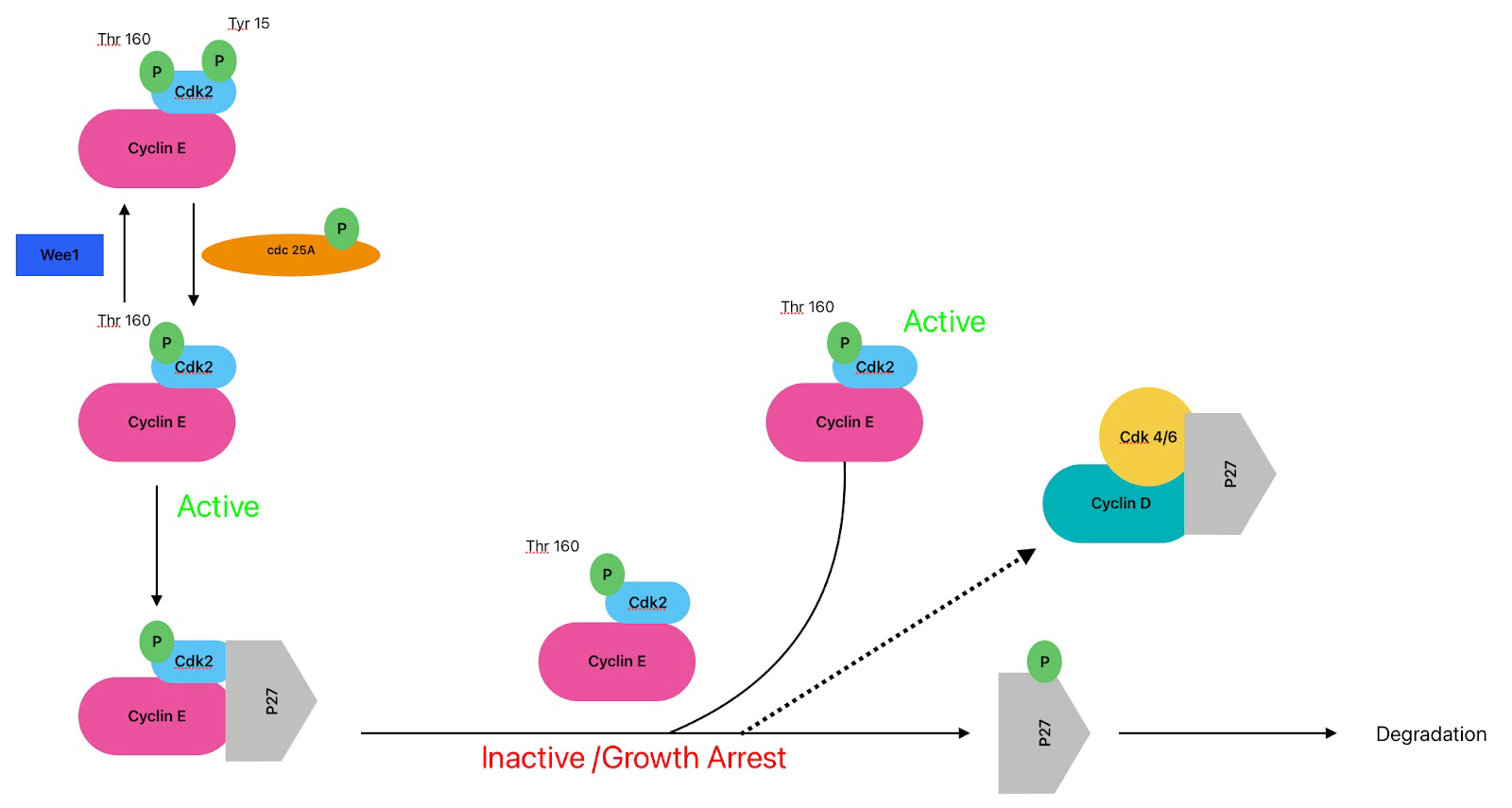
Tyr = tyrosine
Thr = threonine
Left Image
each little green circle is a phosphate
cdk4/6 are always present
in a arrested cell we want to turn off the cell via phosphorylation
cdk4/6 is phosphorylated at tyrosine 15 (inhibitory phosphorylation-making it not active ) and the kinase that does that is Wee 1 which turns it off (way cdk 4 is inhibited in a arrested cell)
wee 1 = break signal (turn off by MAPK)
Top get cdk4 active we need cyclin D and to get cyclin D we need to remove the inhibitory phosphorylation on cdk4…the phosphatase that does this is cdc 25A in a rested cell is inactive once there is a growth signal cdc25A becomes phosphorylated by MAPK and it now gets turned on
Cdc25A phosphatase removes those same inhibitory phosphates that Wee1 added.
to make cdk 4/6 active it (cdc25A) gets phosphorylated by thr 172 by CAK (cdk activating kinase)
CAK adds phosphorylating at Thr172
MAPK activates cyclin D and removal of inhibition on cdk 4 (does 2 things in the same pathway - this is how responses are coordinated and amplified)
Growth signal:
1. Removal of tyr 15 inhibition phosphorylation
2. Addition of thr 172 phosphorylation
These two steps now allow cdk4 to phosphorylate retinoblastoma
Still needs removal of INK4 and cyclin D (p21 or p27)
Right Image:
We need to get rid of INK4
When cyclin D comes in INK4 comes off making the complex active and go phosphorylate Retinoblastoma
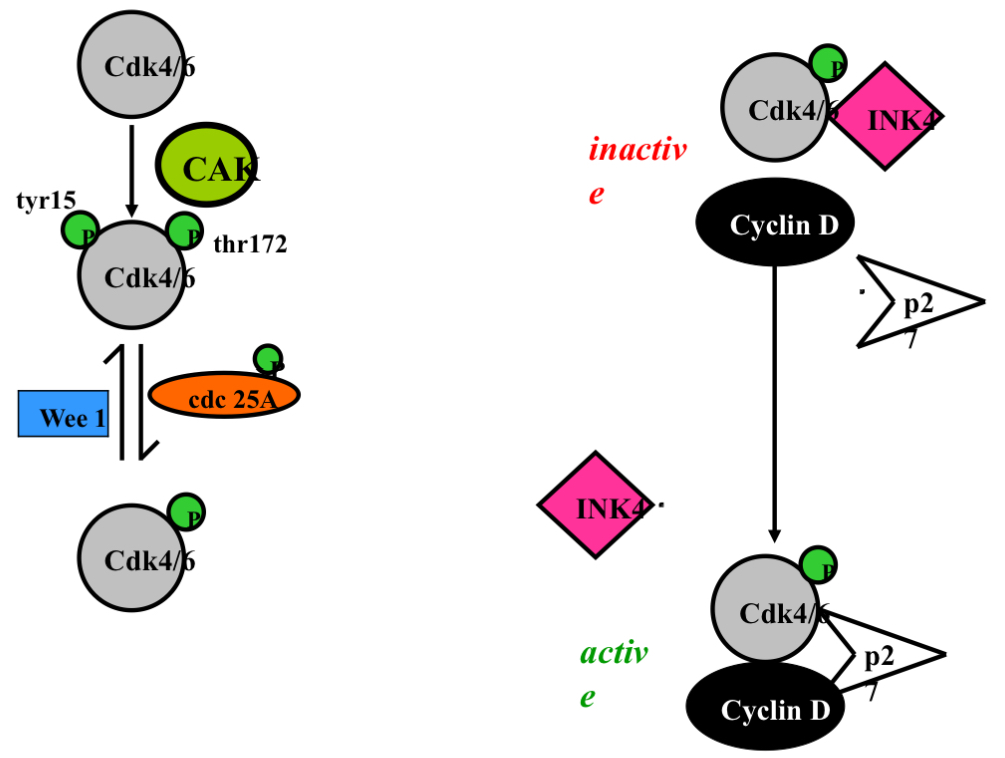
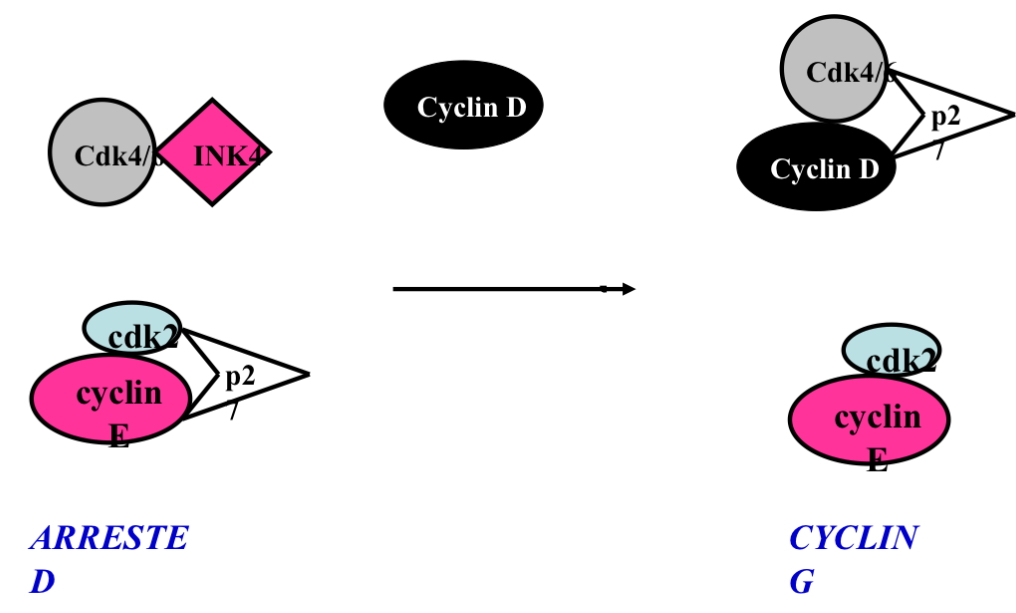
Summary. Cyclin E and p27 and INK4 and Cyclin D
growth signal —> increase in cyclin D
When cyclin D comes around INK4 breaks off cdk4 …after the arrival of the growth signal the cdk4/6/cyclin D/p27 complex is now active
This binding of cyclin D and cdk 4/6 would still happen without p27 just at a much slower rate
Question to answer: what does p27 in a arrested cell inhibits the complex but once a growth signal occurs it does not inhibit the complex….to be answered later on

Activation of cdk4/6
order isnt important just as long as all 3 are there

Cyclin D —> Off Switch
Cyclin D is Deactivated by phosphorylation by GSK-3b (glycogen synthase kinase-3 beta) on thr 286 → results in a change in a change in shape which Induces cyclin D (and its associated cdk) nuclear export (make it leave the nucleus)
Rb is in the nucleus and if you move cyclin D/cdk4 out of the nucleus it can no longer phosphorylated Retinoblastoma which is located in the nucleus
GSK-3b causes:
Removal of cyclin D/acdk 4 from the nucleus and bring it to the cytoplasm as well as degradation of cyclin D
Targets protein for degradation by the proteasome (ubiquitin) [this happens after it leaves the nucleus]
Decreases protein stability (half-life 25 min down to 10 minutes)
stability = how long it takes fora protein to stay around measure by half life (time it takes to get rid of 50% of the protein)
Proteins that are stable are around for a while and their levels stay relatively constant…..if we want to decrease a protein we want to decrease the time that it is around
Cyclin D —> Off Switch —> E3 Ligase for Cyclin D
E3 ligase for Cyclin D degradation recently identified and referred to as Fbx4 (F-Box protein)
Fbx4 adds ubiquitin Cytoplasmic degradation of cyclin D
Fbx4 is a group of proteins and 1 fbx4 protein is not enough to cause ubiquitination you need more to interact with it (you need 2) and they come together in the N-terminus (beginning of the protein sequence) [closest to N group - Amino Group] (C-terminus is the end of the protein sequence)[closest to carboxyl end]
Contains N-Terminal dimerization domain
Dimerization = refers to the process by which two molecules (often proteins) bind together to form a complex called a dimer
Phosphorylation changes the shape so Fbx4 can “shake hands” with another Fbx4, forming the active dimer…allowing for more cyclin D degradation
Dimerization regulated by phosphorylation on Ser12
Fbx4 must be phosphorylated on ser 12 in order to be able to interact with another fxb4…why ? to change shape and become active
Mutations of Fbx4 (in N terminal) decrease dimerization which decrease ubiquitination of cyclin D1 (pro cancer)
Meaning cyclin D goes up and doesn't come down
*Mostly increased expression of cyclin D1 (not D2 or D3)
In cancer it is mostly cyclin D1 we are talking about D2and D3 don't seem to be too involved in the progression of cancer
What Happens is Cyclin D Doesn’t get Turned Off?
1. Chromosomal Translocations (gene is moved/partial moved)
Can cause “new” improper regulation/expression (cancer)
lymphoma, parathyroid adenoma, myeloma (examples of cancer that have this-chromosomal translocation of cyclin D1
2. Gene Amplification
gene transcribed too often
Have more than 2 copies of a gene…usally happens during duplication process…uslaly the extra copy will get filters out the problem is when it doesn't get filtered out
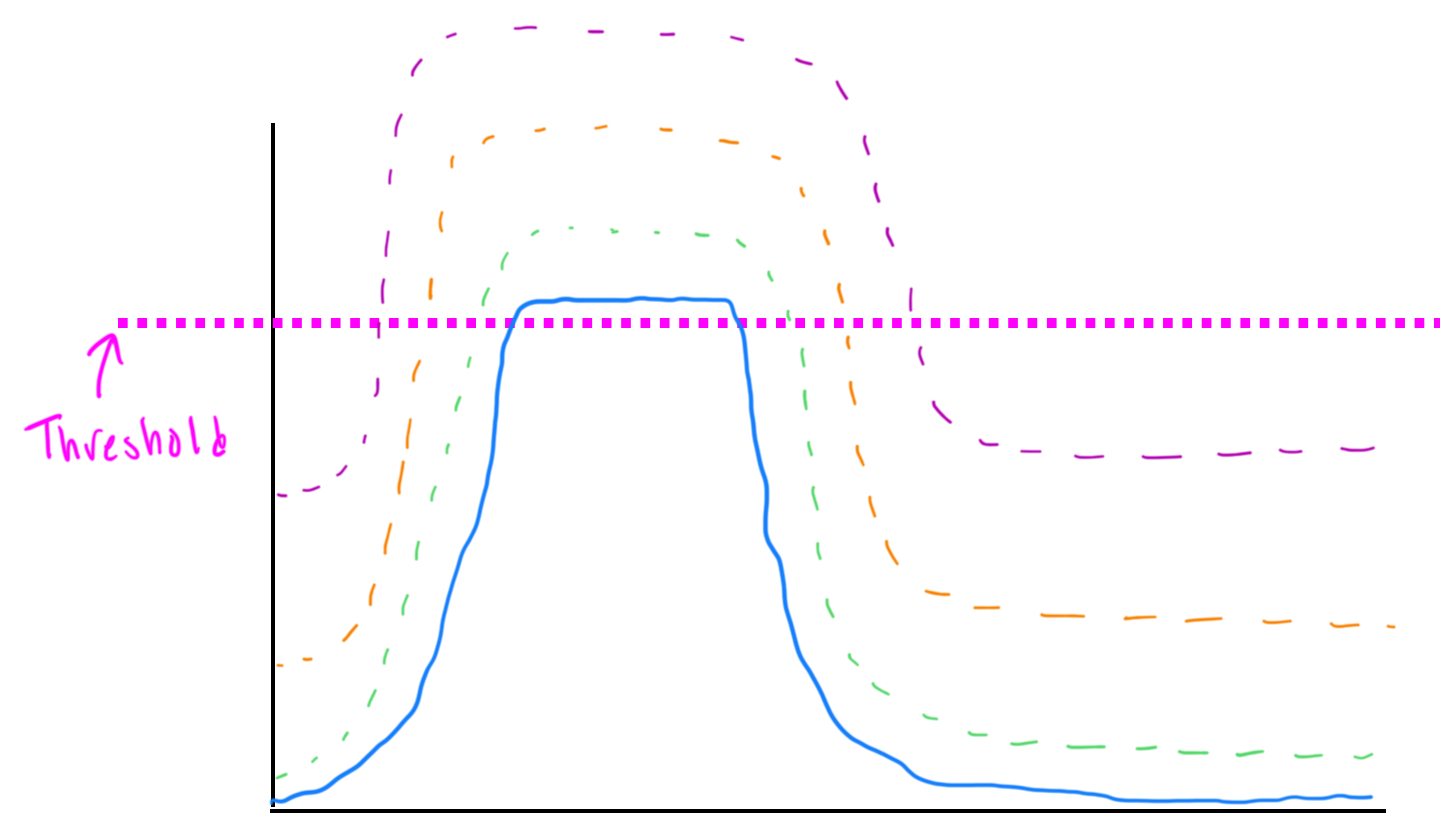
since now there's more mRNA we make more proteins and when the cell cycle comes around again cyclin doesn't drop down to 0 anymore the way it is suppose to…and we wont be blur to get rid of Cyclin D below the threshold resulting in the cell entering the cell cycle on its own
removal of growth signal
lung, head and neck, pancreatic, bladder, pituitary and breast
3. Mutation
Cyclin D is Deactivated by phosphorylation by GSK-3beta (also phosphorylates Fbx4 on ser12→ f box protien for cyclin D) on thr 286 [allows it to be exported into the cytoplasma] (exported to cytoplasma = cant do its job)
thr 286 is not mutated in any cancers
G870 —> A [found a mutation here in this amino acid]
gene sequence mutation
3-d structures change may interfere with thr 286 phosphorylation
mutations (mutation of ser 12) of Fbx4 discovered:
ser8, ser12, pro13, lys23, pro76 —> lowers/decrease degradation of cyclin D
what if we can find away that interacts with this mutate protein and it does not change the amino acid but changes the 3D shape back to normal
preventing phosphorylation of thr 286 could prevent nuclear export and interact with fbx4 to be polyubiquitionated
What happens if we don’t phosphorylate fbx4 on ser12?
that phosphorylation is needed for fbx4 to bind to another fbx4 and you need that to get ubiquitin and degradation of cyclin D…so if we mutate fbx4 nd change ser to something else then we inhibit cyclin D degradation which can lead to loss of cell cycle regulation as cyclin D would now always be present
2 Cases:
1. Amino acids can be mutated
2. Everything around it can be mutated
4. Misregulation
Can be a mutation but if it is its a mutation thats not directly in the pathway
Tumours can have activated Ras
ras is a kinase
Ras is activated by membrane proteins
Ras activates ferc1 or MAP kinase
if you have a mutation that results in more activation of ras that leads to increase in c-mec which leads to an increase in cyclin D
ras has to activated by a external force (lots of external factors that affect ras)
even if you figure out exactly what the problem is since there is so many possibilities something else is probably already gone wrong
2 possibilities: (is it s a primary or secondary affect)
Is the cell cycling because ras is activated?
is ras activated because the cell is dividing?
Ras activities the MAP kinase pathway
Ras is activated in every cell cycle
ras is activated in a lot of cancers….ras is activated every time a cell divides
MAP kinase pathway regulates transcription of cyclin D
by effecting c-myc which transcribes cyclin D
increase cyclin D transcription and protein level
perpetual cell cycle entry (you’ve lost the ability to turn it off)
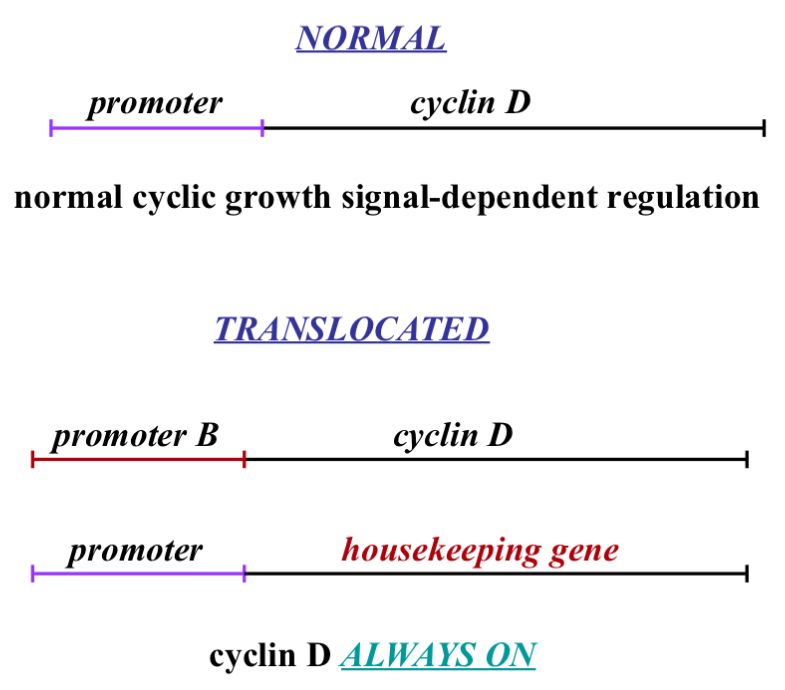
Cyclin D translocation Explained more in Depth
Normal
C-myc binds to promoter and causes transcription of cyclin D mRNA and this only happens if c-myc is activated which occurs after a growth signal is detected
Translocated
In the process of DNA duplicating we have enzymes in our nucleus that cut and move DNA around..this can be a good thing (a way of adapting)...
Cell copies DNA but modifies it so now you have 2 proteins and lets say one is a housekeeping gene (meaning the promoter for it is always on/available) because with housekeeping genes we want to always be making it (me make it slowly and get rid of it slowly - related to half life - talked about earlier)
If during DNA duplication and we take the promoter from cyclin D and we put it on this housekeeping gene (which is on a different chromosome - possibly) and we take that promoter and put it in the cyclin D…this means the housekeeping gene is regulated by growth signals meaning it can be turned on and off the way cyclin D is suppose to be… the problem in this case is that cyclin D then becomes active all the time because it no longer needs a growth signal to start a cell cycle…that is cancer
This can be done with other proteins e.g. p27 or E2f or cyclin E
Can we do this switch with a 'brake’ pedal and prevent cancer or slow it down
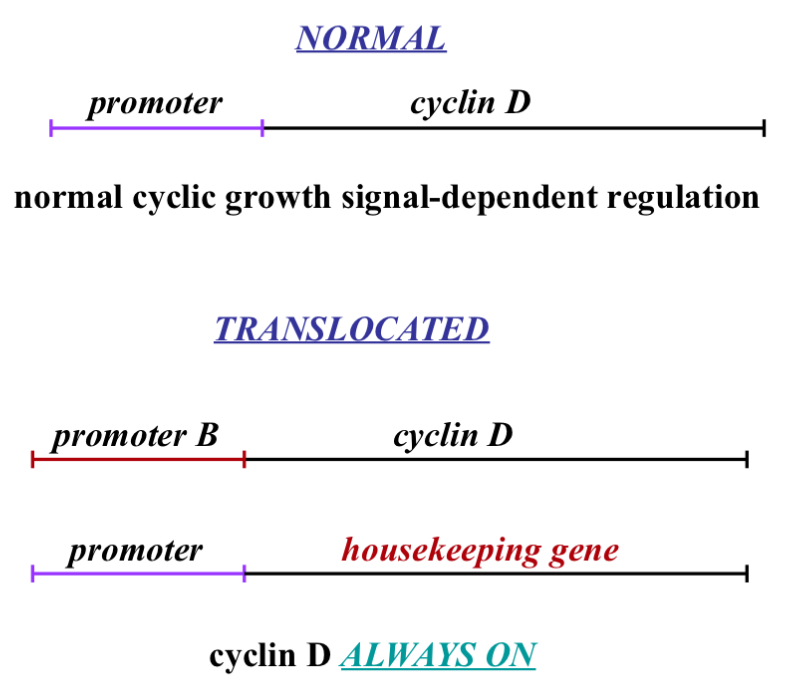
Cyclin D Gene Amplification Explained more in Depth
Protein level is dependent on the level of mRNA…cyclin D is regulated at the protein level…our cell are designed to degrade protiens…but there is a maximal level of protein that they can degrade…and if we have an extra copy of the gene (mRNA) what happen is when we get that cell to enter the cell cycle because we have more mRNA we are going to make more protein and our cells can only degrade as much as they are set to so whatever extra protein is leftover it will carry into the next cell division and cyclin D won't return to 0 before the next cell division and then the in D levels star higher in the next cell division and it keeps increasing
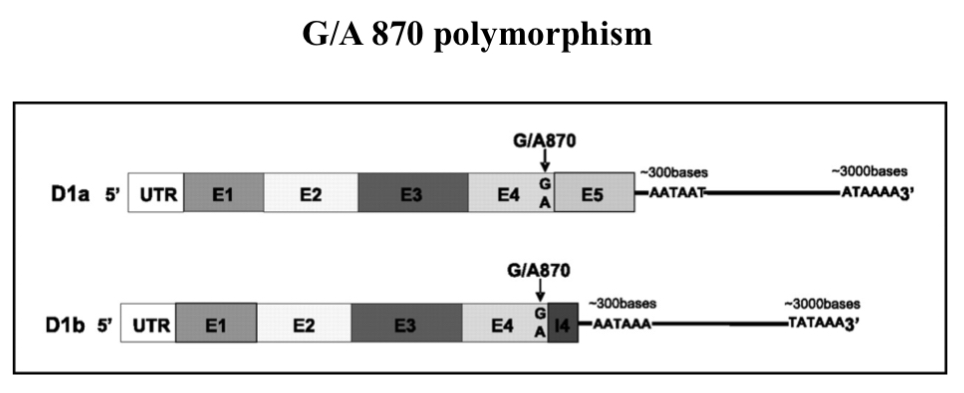
Cyclin D Mutation Explained more in Depth
UTR = un translated region
tail give mRNA stability and prevents it form be degraded
E =2.718 Exons —> what go into coding region for protein
I = introns —> things that get cut out…they are in DNA sequence but dont code for anything and they get cut out in the end product
we want all E…introns should all be gone
if you mutate G870 to A it changes the way it is read by other enzymes and other translation machinery and once you get to 870 what happens is instead of cutting out the Intron the enzyme that cuts out Introns mis reads the cut site and puts an end sequence/terminal sequence (meaning translation stops)…this changes the proteins shape meaning it possibly can't be phosphorylated on thr 286 and the protein might not be able to be exported and degraded properly and it could result in gene amplification
at 870 I erased of cutting out interns it cuts something out creating a end sequence resulting in translation stops…meanin you miss protein that it needs to code for
2 studies found difference
1. That this causes cancer
2. That it doesn't
Cyclin D Therapy Ideas
Specific inhibitor of cdk4 and cdk6 —> PD 0183812 (this number means the drug is in trials) [this drug work BUT it inhibits cdk4/6 in EVERY cell…doenst distinguish between healthy cells and cancer cells]
this inhibition is a specific Category cancer therapy/drugs for when you have cyclin D mutations
Binds competitively to ATP-binding region in the cdk
you have where a site where ATP binds the kinase then cdk cleaves off one of the phosphate to leave you with ADP phosphate and then we add that phosphate to the target protien…the process needs energy (in the form of ATP occasionally GTP)
If the drug prevents ATP from binding it means the kinase can never cleave off the phosphate group to add it to the target protein (in the case of cyclin D…it can to retinoblastoma)
Inhibits growth of tumour cells that are cdk4-6 dependent
Nothing specific for cyclin D yet
we know what wrong we just cant do anything about it
a drug that targets cyclin D damages all cells because cyclin D is present in every cell that divides
How do we reduce the side effects of these drugs?
reduce the dose
maybe:
cyclin E and cdk 2 phosphorylated p27 and causes its degradation…bit we need lots of cyclin E/cdk2
the cdk4/6 cyclin D couples can take some p27 to reduce the levels of p27 to make going through G1 more efficient
if we can stop the cyclin E/cdk2 complex (inactive) we can stop cancer
Cyclin E
Many cancers overexpress (more than there should be) cyclin E mRNA or protein (way more than 50% of cancers)
Breast, lung, cervix, endometrial, GI, lymphoma, leukemia
Anything that affects Rb may increase cyclin E by E2F
we need to take INK4 off cdk 4 and we do this through p27 making the complex active…take p27 away from cyclin E allows cdk4/6 and cyclin D to become active…eventually you will get more cyclin D/cdk4 which will over come that and p27 bumping INK4 off means that it is easier to get cyclin D/cdk4 activate meaning you can start to phosphorylate retinoblastoma easier…but p27 and cyclin E fit into the next step…. Both cyclin E and D are activated by MAP kinase
We get activated cdk2 once we start phosphorylating retinoblastoma that causes the E2F transcription factors to become active and E2F makes cyclin E which binds to prime cdk2 but there is so much p27 once you start to make this it is immediately inhibited…so we need to get rid of p27 to get more cyclin E/cdk2…cyclin E/cdk2 phosphorylate p27 causes its degradation…so we want more active cyclin E/cdk2 to allow it to degrade p27 to make the process of going through G1 more efficient
How does cyclin E/cdk 2 become active to degrade p27?
1. Gene Amplification [cyclin E]
infrequency (2-20%)[only 2 to 20% of cancers that have elevated cyclin E]
endometrial, ovarian, colorectal, breast and gastric
2. Disrupted Degradation (this happens in both cyclin D and E)
2a: loss of Fbw7 (F-BOX protein of cyclin E); 2 step process
A. Loss of one allele (gene copy)[2 Alleles for every gene]
This means when we activate Fbw7 we get half the amount protein leads to half the amount of cyclin E ubiquitination and we get half the amount of cyclin E degradations…affects degradation that affects how cyclin E builds up slowly over time eventually resulting. In 2.
B. Point mutations of remaining allele (one of the amino acids have changed)
cyclin E will now go up and then go down at the beginning…. And cyclin E when it goes up will activate E2F which will transcribe DNA (perpetual activation of E2F0 and then if cyclin E stays up you make more DNA resulting in more genes and now we have lost cell regulation and more mutations get through and we get a mutation in Fbw7 (it could also be that Fbw7 is in a spot that commonly gets skipped over)
2b: Cyclin E Phosphorylated at T380 by GSK3-beta
GSK3-beta causes cyclin E export and degradation
mutation of this site (T380) would prevent phosphorylation, degradation and accumulation of cyclin E would occur
not found yet (in forms of mutation)
GSK3-beta phosphorylates cyclin D and cyclin E and turns it off
Clinical marker
Predictive of patient outcome: Breast, lung, laryngeal, adrenocortical
May be indirect effect
Overexpression may cause genetic instability
Elevated cyclin E activity impairs proper S-phase progression
Impaired replication, DNA breakage and premature entry into M
Effects more dramatic when coupled with p53 loss
Therapy:
Cdk2 inhibitors (not cyclin E)
Problem: If cyclin E overexpression leads to genetic instability……..
inhibiting cdk2 won’t treat what is wrong, unless…….
Cyclin E overexpression detected before genetic problems start
![<ul><li><p><span>Many cancers overexpress (more than there should be) cyclin E mRNA or protein (way more than 50% of cancers)</span></p></li><li><p><span>Breast, lung, cervix, endometrial, GI, lymphoma, leukemia </span></p></li><li><p><span>Anything that affects Rb may increase cyclin E by E2F</span></p></li><li><p><span>we need to take INK4 off cdk 4 and we do this through p27 making the complex active…take p27 away from cyclin E allows cdk4/6 and cyclin D to become active…eventually you will get more cyclin D/cdk4 which will over come that and p27 bumping INK4 off means that it is easier to get cyclin D/cdk4 activate meaning you can start to phosphorylate retinoblastoma easier…but p27 and cyclin E fit into the next step…. Both cyclin E and D are activated by MAP kinase </span></p></li><li><p><span>We get activated cdk2 once we start phosphorylating retinoblastoma that causes the E2F transcription factors to become active and E2F makes cyclin E which binds to prime cdk2 but there is so much p27 once you start to make this it is immediately inhibited…so we need to get rid of p27 to get more cyclin E/cdk2…cyclin E/cdk2 phosphorylate p27 causes its degradation…so we want more active cyclin E/cdk2 to allow it to degrade p27 to make the process of going through G1 more efficient </span></p></li><li><p><span>How does cyclin E/cdk 2 become active to degrade p27? </span></p></li><li><p><span>1. Gene Amplification [cyclin E]</span></p><ul><li><p><span>infrequency (2-20%)[only 2 to 20% of cancers that have elevated cyclin E]</span></p></li><li><p><span>endometrial, ovarian, colorectal, breast and gastric </span></p></li></ul></li><li><p><span>2. Disrupted Degradation (this happens in both cyclin D and E)</span></p><ul><li><p><span>2a: loss of Fbw7 (F-BOX protein of cyclin E); 2 step process</span></p><ul><li><p><span>A. Loss of one allele (gene copy)[2 Alleles for every gene] </span></p><ul><li><p><span>This means when we activate Fbw7 we get half the amount protein leads to half the amount of cyclin E ubiquitination and we get half the amount of cyclin E degradations…affects degradation that affects how cyclin E builds up slowly over time eventually resulting. In 2.</span></p></li></ul></li><li><p><span>B. Point mutations of remaining allele (one of the amino acids have changed) </span></p><ul><li><p><span>cyclin E will now go up and then go down at the beginning…. And cyclin E when it goes up will activate E2F which will transcribe DNA (perpetual activation of E2F0 and then if cyclin E stays up you make more DNA resulting in more genes and now we have lost cell regulation and more mutations get through and we get a mutation in Fbw7 (it could also be that Fbw7 is in a spot that commonly gets skipped over)</span></p></li></ul></li></ul></li><li><p><span>2b: Cyclin E Phosphorylated at T380 by GSK3-beta</span></p><ul><li><p><span>GSK3-beta causes cyclin E export and degradation </span></p></li><li><p><span>mutation of this site (T380) would prevent phosphorylation, degradation and accumulation of cyclin E would occur </span></p></li><li><p><span>not found yet (in forms of mutation) </span></p></li><li><p><span>GSK3-beta phosphorylates cyclin D and cyclin E and turns it off </span></p></li></ul></li></ul></li><li><p><span>Clinical marker</span></p></li><li><p><span>Predictive of patient outcome: Breast, lung, laryngeal, adrenocortical</span></p></li><li><p><span>May be indirect effect</span></p></li><li><p><span>Overexpression may cause genetic instability</span></p></li><li><p><span>Elevated cyclin E activity impairs <em>proper</em> S-phase progression</span></p></li><li><p><span>Impaired replication, DNA breakage and premature entry into M</span></p></li><li><p><span>Effects more dramatic when coupled with p53 loss </span></p></li><li><p><span>Therapy:</span></p><ul><li><p><span>Cdk2 inhibitors (not cyclin E)</span></p></li><li><p><span>Problem: If cyclin E overexpression leads to genetic instability……..</span></p></li><li><p><span>inhibiting cdk2 won’t treat what is wrong, unless…….</span></p></li><li><p><span>Cyclin E overexpression detected before genetic problems start </span></p></li></ul></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/f29c111e-cf03-44d3-ac0e-7bf1fb9a728a.jpg)
Legend (map shapes/colors in the image)
Pink oval = Cyclin E
Blue rounded shape = Cdk2
Green circles = phosphate (P) on specific amino acids (labels: Thr160, Tyr15)
Grey hexagon = p27 (CDK inhibitor)
Teal + yellow = Cyclin D : Cdk4/6 complex
Blue box “Wee1” = kinase that adds inhibitory phosphate on Tyr15
Orange “Cdc25A” = phosphatase that removes the inhibitory phosphate
Dotted arrow / solid arrows = regulatory flows (activation, phosphorylation, degradation)
Step-by-step explanation (focus on Cyclin E)
1) Inactive / growth-arrest state (left / bottom of the image)
Cyclin E is present and associated with Cdk2, but the kinase is kept OFF by two mechanisms shown in the diagram:
Inhibitory phosphorylation on Cdk2 at Tyr15 (green “P” labeled Tyr15). Wee1 is the kinase that adds this inhibitory phosphate. An inhibitory phosphate on Tyr15 blocks Cdk2 catalytic activity even when Cyclin E is bound.
Binding of p27 to the Cyclin E–Cdk2 complex (grey shape attached) — p27 physically inhibits the complex’s activity.
Result: Cyclin E–Cdk2 activity = low → cell remains in G1 / growth arrest.
2) Triggering activation (upper left → center)
When mitogenic signals arrive (growth factors), regulatory changes occur:
Cdc25A (orange) dephosphorylates Tyr15 (removes the inhibitory phosphate).
Separately, CAK (CDK-activating kinase) (not shown explicitly on the picture) phosphorylates Cdk2 at Thr160 — this phosphorylation is required for full catalytic competence.
The net effect in the diagram: Cdk2 loses the Tyr15 inhibitory phosphate and keeps (or gains) the Thr160 activating phosphate → the complex becomes competent/active.
3) Active Cyclin E–Cdk2 (middle / labeled “Active”
Once Cdk2 has Thr160 phosphorylated and Tyr15 dephosphorylated, Cyclin E–Cdk2 is an active kinase.
Active Cyclin E–Cdk2 can now phosphorylate downstream substrates required for G1 → S transition (examples: Rb, components that promote DNA replication, and importantly p27).
4) Cyclin E–Cdk2 phosphorylates p27 → marks it for destruction (center → right)
Active Cyclin E–Cdk2 phosphorylates p27 on a specific residue (classically Thr187 — the diagram shows a green P on the grey p27 shape).
Phospho-p27 is recognized by the SCF^Skp2 E3 ubiquitin ligase (plus Cks1), becomes ubiquitinated, and is sent to the proteasome for degradation (rightmost arrow to “Degradation”).
The image shows p27 being phosphorylated and then an arrow to degradation — that is the key event that reduces cellular p27 levels.
5) Consequences of p27 degradation (far right)
Lower p27 means less inhibition of Cyclin E–Cdk2 and also less inhibition of other cyclin–CDK complexes (Cyclin A–Cdk2 later, and influences on Cyclin D/Cdk4/6 interactions).
The diagram also shows a Cyclin D–Cdk4/6 complex with p27 attached — p27 can bind Cyclin D complexes as well, sometimes acting as an assembly/stabilizing factor. When p27 is removed by degradation, dynamics between Cyclin D and Cyclin E shift so the cell commits to S phase.
The dotted arrow in the image denotes positive feedback: once a little Cyclin E–Cdk2 becomes active it accelerates p27 removal and Rb phosphorylation → more E2F → more S-phase gene expression (including Cyclin E/A and Skp2), which amplifies the response.
Key molecular control points to remember
Cdk2 activation requires:
• Phosphorylation at Thr160 (activating, by CAK).
• Removal of inhibitory phosphates at Tyr15 (and Thr14 in some contexts) — by Cdc25A.
• Absence or low levels of p27 (p27 is a direct inhibitor).Wee1 adds the inhibitory Tyr15 phosphate (keeps Cdk2 off).
Active Cyclin E–Cdk2 phosphorylates p27 (Thr187) → SCF^Skp2 recognizes phospho-p27 → ubiquitination & proteasomal degradation.
Degrading p27 is a critical step that removes a brake on CDKs and commits the cell to DNA replication.
One-sentence summary
The figure shows how Cyclin E–Cdk2 is kept off in G1 by Tyr15 phosphorylation (Wee1) and p27 binding, how Cdc25A and Thr160 phosphorylation turn Cdk2 on, and how active Cyclin E–Cdk2 then phosphorylates p27 to trigger its ubiquitin-mediated degradation — a feed-forward loop that removes inhibition and drives the G1 → S transition.