Exam 1
1/96
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
97 Terms
Reactive Neutrophilia
Circulating Immature Neutrophils
Left shift/bandemia: ↑ bands, metamyelocytes → bacterial infection
Leukemoid reaction: Left shift and high neutrophil count secondary to inflammation due to infection/sepsis, sterile inflammation, or paraneoplastic G-CSF or cytokine production
Leukoerythroblastic reaction: Left shift plus presence of nucleated RBCs and tear drop RBCs (dacrocytes) → bone marrow–occupying (myelophthisic) lesion with extramedullary hematopoiesis
Agranulocytic Angina
Ulcerative, necrotizing lesions of the gingiva, mouth, pharynx
Ulcers of Skin, Vagina, Anus or GI tract (Cecum)
Risk of severe, life-threatening bacterial (Lungs, Urinary tract) or deep fungal infections caused by Aspergillus or Candida
Skin/mucosal Bacterial colonies may grow with little inflammatory response (Botryomycosis/Bacterial Pseudomycosis)
Neutropenic Fever
Empiric antibiotic therapy should be instituted immediately in all febrile neutropenic patients
Empiric antifungal therapy in patient who do not respond to empiric antibacterial therapy
Chronic myeloid leukemia
BCR-ABL fusion gene due to t(9;22) (Philadelphia chromosome)
Fusion protein has constitutive non-receptor tyrosine kinase activity → uncontrolled myeloid proliferation
Leukocytosis
Granulocytosis with full spectrum of maturation
“Left shift” with ↑ eosinophils and ↑ basophils
Blasts may be present in peripheral blood but <10%
Platelets increased
Leukocyte alkaline phosphatase (LAP) score decreased
Bone marrow hypercellular with increased myeloid-to-erythroid (M:E) ratio
Anemia → fatigue, weakness
Weight loss, anorexia
Splenomegaly due to neoplastic extramedullary hematopoiesis
Causes dragging sensation or fullness in the left upper quadrant
Early satiety
Left upper quadrant abdominal pain due to splenic infarction
Hypermetabolic state from increased cell turnover
Fever and night sweats
Hyperuricemia due to DNA purine catabolism → gout, nephrolithiasis, or uric acid nephropathy
Can progress to acute leukemia
Chronic Lymphocytic Leukemia
Small, mature-appearing lymphocytes on blood smear
“Smudge cells” often present due to fragile lymphocytes
Neoplastic cells are B-cells expressing CD19+, CD20+, and CD23+
Co-express aberrant T-cell marker CD5+
Hypogammaglobulinemia common
Autoimmune hemolytic anemia (AIHA)
Immune thrombocytopenic purpura (ITP)
Transformation to more aggressive malignancy: Prolymphocytic leukemia or Large B-cell lymphoma (Richter transformation)
Acute Lymphoblastic Leukemia
Pre-B:
Leukemic (marrow and blood involvement)
Preschool children (peak age ≈3 years)
Pre-T:
Mass (lymphoma) in the mediastinum (thymus) or lymph nodes
May rapidly progress to a leukemic phase
Adolescent and young adult males
Prognosis worse than pre-B type
Anemia
Thrombocytopenia
Bone marrow is hypercellular and packed with lymphoblasts
>20% of marrow cells to be blasts
Acute Myeloid Leukemia
Mutations in transcription factors for differentiation
Blasts replace normal bone marrow
Anemia, neutropenia, and thrombocytopenia (pancytopenia)
Activating mutations in genes controlling the epigenome (methylation, acetylation)
Disabling mutations affecting TP53
Activation of pro-growth receptor or signaling pathways
Diagnosis requires >20% myeloid blasts in the bone marrow
Auer rods form by abnormal fusion of azurophilic granules
Blasts with azurophilic (Primary) granules (lysosomes containing Myeloperoxidase)
Poor Prognostic Factors:
>50 years
AML arising from prior myelodysplasia or following chemotherapy/radiation (therapy-related AML)
Cytogenetic abnormalities: deletions of chromosomes 5 or 7
TP53 mutations or translocations involving chromosome 11 (KMT2A/MLL)
Therapy-related AML
Very poor prognosis
Following alkylating agent therapy or radiation:
Latency 2–8 years
Associated with deletions of 5q-, 7q-, or 20q-
Following topoisomerase II inhibitor therapy:
Latency 1–3 years
Hallmark abnormality: MLL gene rearrangements
Acute promyelocytic leukemia
t(15;17) → PML-RARα fusion protein
Excellent prognosis if treated promptly before development of DIC
Promyelocytes contain abundant cytoplasmic granules and Auer rods
Release of granules can trigger disseminated intravascular coagulation (DIC) → medical emergency
Normal retinoic acid receptor α (RARA) binds retinoids (vitamin A) to induce maturation of promyelocytes into neutrophils
Translocation t(15;17) creates a PML::RARA fusion receptor that fails to respond to physiologic vitamin A levels
Resulting receptor dysfunction blocks differentiation at the promyelocyte stage
Polycythemia vera
Clonal proliferation of myeloid stem cells with predominant increase in red cell mass
Activating JAK2 V617F mutation
Increased RBC production independent of erythropoietin
EPO low
Leukocytosis and thrombocytosis
Hyperviscosity due to increased hematocrit
Plethoric (ruddy) complexion, headache, dizziness, pruritus (especially after hot shower), hypertension, and thrombosis or bleeding
Splenomegaly due to extramedullary hematopoiesis
Bone Marrow: Erythroid hyperplasia
Clotting\thrombosis lead to Stroke and MI
Spent phase: progressive marrow fibrosis with cytopenias
May transform into acute myeloid leukemia (AML)
Treatment:
Phlebotomy to maintain normal hematocrit
JAK2 inhibitors
Essential thrombocythemia
Chronic myeloproliferative disorder with sustained thrombocytosis
Clonal proliferation of megakaryocytes; often with JAK2, CALR, or MPL mutation
Platelet production increased independent of thrombopoietin regulation
Persistent elevated platelet count
Episodes of thrombosis and bleeding due to dysfunctional platelets
Microvascular occlusion may cause erythromelalgia (burning pain in hands/feet)
May transform into myelofibrosis or AML
Giant platelets on peripheral smear
Dysplastic Megakaryocytes in bone marrow
Primary myelofibrosis
Clonal myeloproliferative neoplasm with marrow fibrosis and extramedullary hematopoiesis
JAK2, CALR, or MPL mutations
Neoplastic megakaryocytes release PDGF and TGF-β → fibroblast activation → progressive marrow fibrosis
Hematopoiesis shifts to spleen and liver
Anemia and fatigue
Massive splenomegaly with early satiety and abdominal discomfort
Leukoerythroblastic blood picture with teardrop RBCs and nucleated precursors
Infection, hemorrhage, and cachexia in advanced disease
May evolve into acute myeloid leukemia (AML)
Bone marrow hypocellular, diffusely fibrotic marrow with numerous dysmorphic Megakaryocytes (some clustered) & enlarged trabecula (osteosclerosis)
Multiple myeloma
Plasma cell proliferation and survival—MYC, cyclin D, or IL-6
Clonal malignant proliferation of plasma cells in bone marrow producing a monoclonal immunoglobulin
Major translocations involve IgH locus
High IL-6 levels promote plasma cell growth and survival
Bone marrow features:
Sheets of plasma cells with eccentric nuclei, clock-face chromatin, and perinuclear hof
Increased osteoclast activation via RANKL and reduced osteoblast activity → lytic bone lesions
Diagnostic criteria (CRAB): Hypercalcemia, Renal failure, Anemia, Bone lesions
Lab findings: M-spike on SPEP, monoclonal light chains in urine (Bence Jones protein), and >10% clonal plasma cells in marrow
Rouleaux on blood smear (stack of coins)
↑ Beta 2 microglobulin
Imaging: multiple “punched-out” lytic bone lesions on skull or spine
Bone pain
Anemia and fatigue (marrow replacement)
Infections (norm Ig not produced)
Renal failure (light chain cast nephropathy, hypercalcemia, or amyloidosis)
Neurologic symptoms (Hyperviscosity from M protein)
Anisocytosis
Variation in RBC size (↑ RDW)
Microcytosis
Decreased MCV (small cells)
Macrocytosis
↑ MCV (large cells)
Hypochromia
Decreased MCH, ↑ area of central pallor
Polychromatophilia
Bluish tint, young RBCs (Reticulocytes)
Poikilocytosis
Abnormally shaped RBCs
Spherocyte
Spherical, without area of central pallor
Schistocyte
Fragmented; irregularly shaped
Intravascular mechanical destruction of RBCs (fragmentation hemolysis)
Sickle cell
Curved, banana shaped
Target cell
Mexican hat
Spleenectomy
Dacrocyte
Tear drop shape
Extravascular hematopoiesis
Howell-Jolly Body
Remnants of RBC nuclei that are normally removed by the spleen
Basophilic Stippling
Remnants Ribosomal RNA in RBC
Coarse: lead and heavy metal poisoning
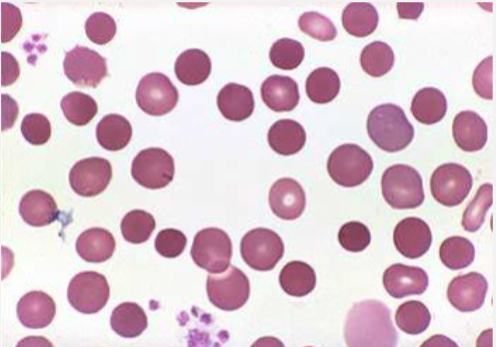
Spherocytes
Sign of Extravascular Hemolysis
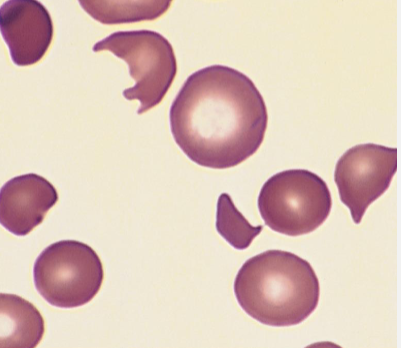
Schistocytes
Sign of Intravascular Hemolysis
Extravascular Hemolysis
Immune: due to antibodies bound to RBCs
Macrophages bind Fc tails & Complement of opsonized RBCs -> Phagocytosis (partial phagocytosis of bits of membrane -> Spherocytes)
Non-immune: Intrinsic RBC membrane abnormalities (Hereditary or Acquired)
Macrophage recycling of heme groups
Protein Globin chains recycled; Fe saved & stored as Ferritin or Hemosiderin
Heme group -> Bilirubin (unconjugated)
Anemia, splenomegaly & jaundice
↑ Bilirubin (unconjugated/indirect)
Blood smear: Spherocytes
Intravascular Hemolysis
RBCs lyse inside vessels -> free Hb into plasma -> binds to Haptoglobin -> Hapto/Hgb cleared by Macrophages; excess free Hgb filtered by kidneys -> Hemoglobinuria
Fe saved as Ferritin or Hemosiderin
Anemia, Hemoglobinemia, Hemoglobinuria (brown urine from oxidized Hb/methemoglobin), Hemosiderinuria & Jaundice (↑ unconjugated bilirubin)
Massive hemolysis chills, fever, hypotension, hemoglobinuria, renal failure, back pain, or DIC
↓ Haptoglobin
↑ Bilirubin (unconjugated/indirect)
↑ LDH
Schistocytes if due to trauma (micro/macro angiopathic)
Hemoglobinuria (may result in Acute Renal failure)
Warm Autoantibody Autoimmune hemolytic anemia
IgG against RBC protein Ag
Half are idiopathic (primary); others are secondary to autoimmune disease (SLE), drugs or lymphoid neoplasms (CLL or Lymphoma)
Auto-Ab acts as opsonin → Hemolysis mostly EXTRAVASCULAR by Macrophages → Spherocytes
+DAT
Drugs may bind to RBC surface & act as Antigens (penicillin, cephalosporins), Haptens (Quinidine) or break self-tolerance (α-methyldopa)
No blood is compatible for transfusion
Cold Autoantibody Autoimmune hemolytic anemia
IgM binds to individual RBC polysaccharide Ag I/i in cold areas (fingers, toes, ears) & deposits opsonizing C3b → rewarming at core organs disassociates IgM but leaves behind C3b → extravascular hemolysis (DAT + for Complement only)
May appear transiently after infections (Mycoplasma pneumonia, IM or other viruses) or persist as chronic hemolysis from B-cell neoplasms (Waldenstrom) or idiopathic
IgM also Agglutinates RBCs in cold, exposed, parts of body (Cold Agglutinin Disease) inducing Acrocyanosis (necrosis if severe), Raynaud phenomenon, or Livedo Reticularis
Paroxysmal Nocturnal Hemoglobinuria
Common Myeloid Progenitor stem cell
Acquired LOF mutation in PIGA on X chromosome which makes a phospholipid that protects myeloid cell membranes (RBC, Platelets & Neutrophils) from lysis by Complement
Loss of CD55 & CD59
RBC: Hemolytic Anemia w/ episodes of intravascular hemolysis
Complement activation of platelets: thrombosis leading cause of death
Thrombi in large veins and Thrombocytopenic
Neutrophils: lysed; neutropenia (infections)
From pre-existing Aplastic Anemia and can progress to Myelodysplasia or AML
DAT (-)
Normocytic or macrocytic anemia with normal or ↑ Retic count
Over time may become Fe deficient due to Hemosiderinuria
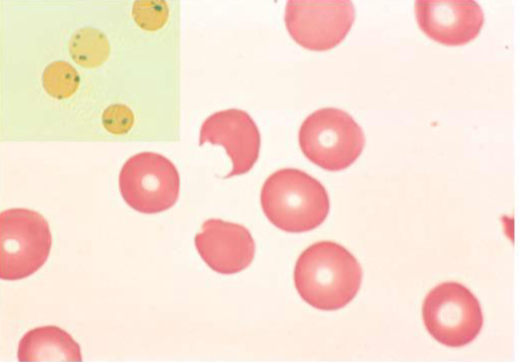
G6PD Deficiency
Heinz bodies
Bite cells
Spherocytes
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY (G6PD)
↓ NADPH → ↓ Glutathione peroxidase
Oxidative damage
Most are asymptomatic
Inherited as X-linked recessive
Triggers: Infections (pneumonia, viral hepatitis, etc.), drugs (anti-malarials, sulfonamides, others) & foods (Fava beans)
Presents as Hemoglobinuria (dark urine)
Self-limited
Blood smear: Denatured Globin chains precipitate (Heinz bodies) & damage membrane
Severe damage -> intravascular hemolysis
Less severe damage -> extravascular hemolysis
"Bite cells": removal of Heinz bodies by Spleen
Spherocytes
Jaundice, fatigue, back pain, tachypnea, tachycardia & dark urine
↓ hemoglobin, ↑ reticulocytes, ↑ LDH, ↑ unconjugated bilirubin, ↓ Haptoglobin, Hemoglobinuria
PYRUVATE KINASE DEFICIENCY HEMOLYTIC ANEMIA
Autosomal recessive
Mutations in RBC enzyme in glycolytic pathway producing ATP
Chronic extravascular hemolysis
RBC maturation also affected (ineffective erythropoiesis) -> results in Fe overload
+/- Anemia
↑ Retic count
Negative DAT
Hyperbilirubinemia, gallstones, Fe overload
Hx of transfusions
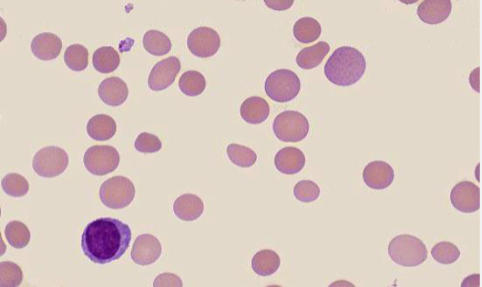
Hereditary spherocytosis
Spherocytes
Hereditary spherocytosis
Autosomal dominant
Intrinsic Defect in RBC Membrane cytoskeletal tethering proteins → make RBCs spheroid ↓ lifespan to 10-20 days due to sequestration & destruction in spleen (Extravascular hemolysis)
Anemia, Splenomegaly & Jaundice
Osmotic fragility test (RBCs rupture more easily in a hypotonic solution)
Aplastic Crisis due to Parvovirus B19 infection which destroys RBC progenitors
Artefactual Pseudohyperkalemia (K+ leaks out of cells with cooling after draw)
Bilirubin gallstones
Splenectomy is definitive Rx
↑ MCHC (because of shape)
MCV low or normal (mix of small spherocytes + large Retics)
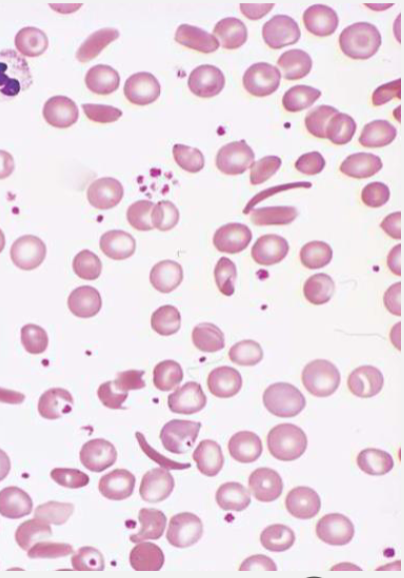
Sickle Cell Disease
Sickle Cell Disease
Autosomal recessive point mutation in β-globin gene -> HbS = both β-globins mutated
HbS prone to polymerization into needle-like crystals when deoxygenated
Distorted RBCs become “sticky” & adhere to endothelium -> results in episodes of microvascular obstruction & tissue ischemia (Vaso-Occlusive Crisis)
Inflammation
Extravascular hemolysis
Normocytic Anemia, ↑ Bilirubin (unconjugated/indirect), ↑ Retics
Smear: irreversibly sickled cells
Pre & Post-capillary microvascular occlusions cause the hallmark of the disease: Recurrent, unpredictable episodes of Microvascular occlusion: Vaso-occlusive (“Pain”) Crises
Bones: painful bone crises common, particularly in children
Bone pain is due to bone marrow infarction in bones with the most bone marrow activity
X-ray appearance: frontal & parietal bossing & prominent maxilla due to marrow hyperplasia expanding the bone
X-ray of skull -> “Hair-on-end” pattern
BM biopsy: Erythroid hyperplasia; foci of infarction (necrosis) during crises
Lungs: Acute Chest Syndrome
Sickle Cell Disease
Fever, cough, chest pain
Ischemia due to pneumonia -> Sickling -> more ischemia-> more sickling
Most common cause of death
Sickle Cell Complications
Priapism
Liver: multiple infarcts → failure
Kidney: Papillary Necrosis is a common renal manifestation of vaso-occlusion → isosthenuria (inability to concentrate urine) → prone to dehydration → kidney failure
Brain: Stroke (common in children), retinopathy (loss of visual acuity/blindness); loss of cognitive function over time
Salmon-patch hemorrhage in retina
Gallbladder: Cholelithiasis (bilirubin stones) common in children
Avascular necrosis of Femoral or Humeral heads
Leg Ulcers: impaired healing of minor trauma
Growth delay: Children small & thin; delayed sexual development
Aplastic Crisis with Parvovirus B19
Autosplenectomy due to infarcts
Sequestration Crisis: In younger children (usually < 5) with still intact spleens, massive entrapment of sickled cells by the spleen → rapid splenic enlargement, hypovolemia & even fatal shock
Β-THALASSEMIA
No Beta production
β-Thalassemia Minor:
Mild microcytic hypochromic anemia with Target cells; ↑ RBC count
β-Thalassemia Intermedia: Intermediate
β-Thalassemia Major:
6-9 months of age
↓ RBC production
Unpaired α chains precipitate in RBC precursors & damage membrane → apoptosis (ineffective erythropoiesis)
↑ Splenic hemolysis.
Severe anemia
Smear: Microcytic, hypochromic with Anisocytosis, Poikilocytosis (Targets, fragmented RBCs, basophilic stippling), variable Retics, NRBCs
BM: Erythroid hyperplasia
Hair on end pattern on skull X-ray caused by longstanding hyperplasia of the bone marrow
Extramedullary Hematopoiesis (skeletal abnormalities/Hepatosplenomegaly)
Facial bone hyperplasia
Iron Overload (↑ Ferritin): ineffective erythropoiesis suppresses Hepcidin (↑ Fe absorption from gut) + multiple transfusions → severe Fe overload which damages liver, heart & pancreas
Α-THALASSEMIA
Decrease production of α-chains due to deletion of 1, 2, 3 or all 4 alleles
1 Silent Carrier
2: Microcytosis with mild or no anemia
3: Hemoglobin H Disease- Moderate or Severe microcytic anemia; may be iron overloaded
4: Hydrops Fetalis (Nonimmune Hydrops)- Lethal in utero or shortly after birth
Corrected Reticulocyte Count
Reticulocyte % adjusted for patient’s degree of anemia
< 2% → Inadequate marrow response (hypoproliferative anemia)
• 2% → Appropriate marrow response (blood loss or hemolysis)
Aplastic anemia
Chronic failure/injury of Common Myeloid Progenitor Stem Cell
Pancytopenia
Most commonly Autoimmune, but deficiency of stem cell Telomerase
1/3 have associated expanded PNH clones
Markedly Hypocellular or Acellular bone marrow
↓ Reticulocytes
Pancytopenia; mild macrocytosis
↑ Erythropoietin level (EPO)
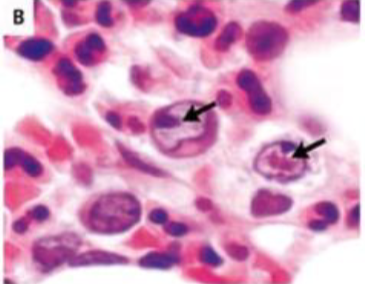
Parvovirus intranuclear inclusions
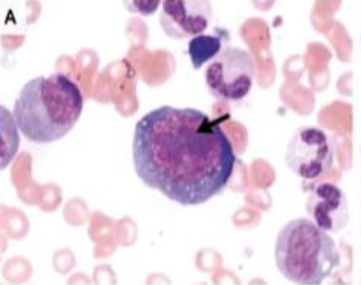
Parvovirus intranuclear inclusions
Pure red cell aplasia
Selective suppression of committed marrow erythroid precursors
Parvovirus B19
Diamond-Blackfan Anemia:
1st year
Mutations in ribosomal protein genes; associated with numerous/characteristic physical abnormalities (thumbs); ↑ risk MDS, AML, other tumors
↑ Erythrocyte Adenosine Deaminase, Macrocytic anemia
Autosomal Dominant
Normocytic/Macrocytic anemia; Low retic count; high EPO
BM: normocellular with absent/arrested erythroid precursors
In Parvovirus infection, intranuclear inclusions
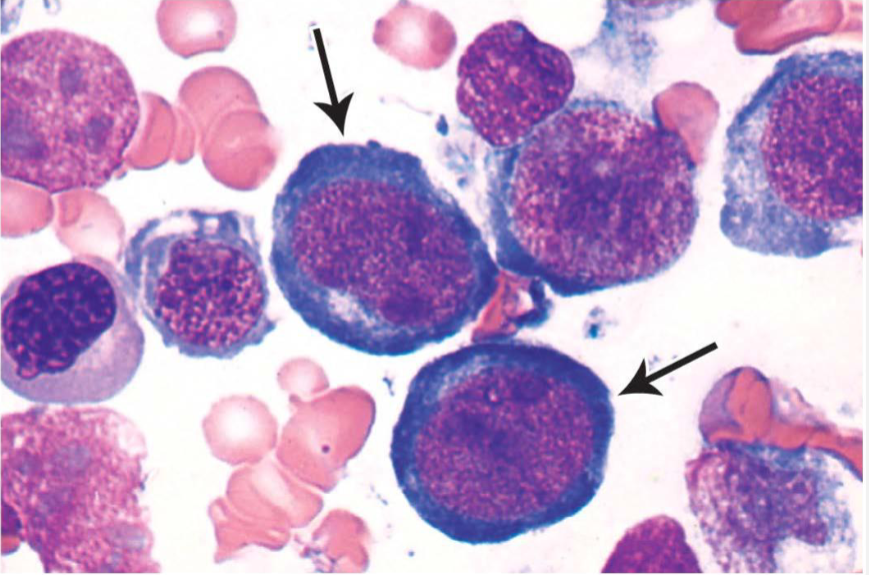
Megaloblastic Anemia showing megaloblastic erythroid precursors
Megaloblastic anemias
Vit B12 (Cobalamin) or Folic Acid deficiency
Both are coenzymes required for synthesis of Thymidine in DNA
Abnormally large erythroid precursors (Megaloblasts) & macrocytic RBCs (Macrocytes)
Hypersegmented neutrophils
Macro-ovalocytes
Thrombocytopenia; Pancytopenia
↓ Reticulocyte count
Bone Marrow: Hypercellular marrow with hyperplasia of erythroid series; ↓ M:E ratio
Megaloblastic erythroid, myeloid & megakaryocytic series
↑ EPO
For B12: ↑ Methylmalonic acid (MMA) best for early deficiency
For folate: Increased Homocysteine
Pernicious Anemia
Megaloblastic Anemia caused by Vit B12 deficiency due to Autoimmune atrophic gastritis
Autoimmune atrophic gastritis destroys parietal cells which produce Intrinsic Factor (IF) required for Vit B12 absorption from the gut
Antibodies to Intrinsic Factor, Parietal Cells
Gastric Bx: fundic gland atrophy; intestinal metaplasia (↑ risk of gastric cancer)
Atrophic glossitis
Subacute Combined Degeneration of spinal cord: demyelination of the Dorsal + Lateral spinal tracts with loss of voluntary movement, stiffness, sensory ataxia, severe paresthesia in lower limbs
Tingling, "pins and needles" sensation in the hands or feet
Lost sense of touch
Wobbly gait and difficulty walking
Clumsiness & stiffness of the arms & legs
Dementia
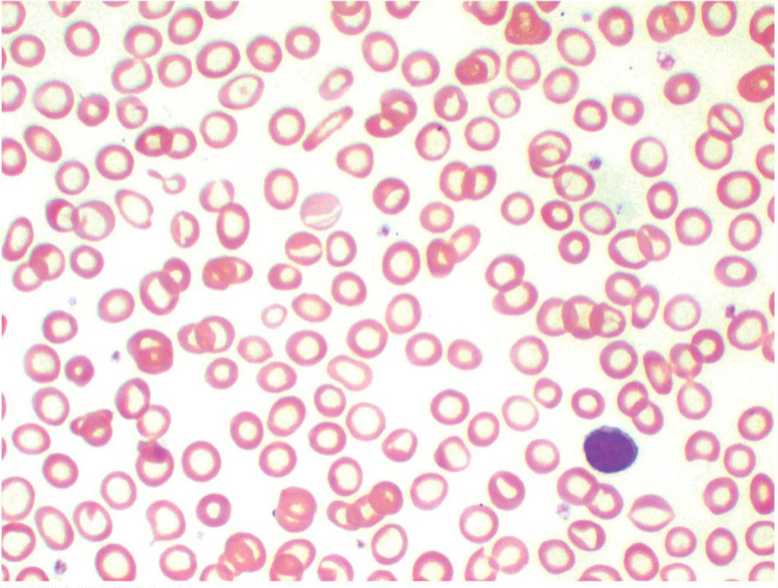
Iron deficiency anemia
Microcytic, Hypochromic cells
Iron deficiency anemia
Functional Fe is carried in the blood by TRANSFERRIN
TIBC (Total Iron Binding Capacity) is an indirect measurement of Transferrin
Iron stored intracellularly as protein-iron complexes FERRITIN & HEMOSIDERIN (which is aggregated Ferritin)
Ferritin levels in blood correlate well with total body iron store
Iron balance is regulated by ABSORPTION in the duodenum
Hepcidin ↓ Duodenal Fe absorption
MICROCYTIC HYPOCHROMIC ANEMIA
Low serum Iron + Ferritin
High TIBC (Total Iron Binding Capacity), ↑ Transferrin
Low per cent saturation of serum Transferrin
↑ RBC distribution width (RDW) → variation in RBC size (anisocytosis)
↓ Reticulocyte count
↑ EPO
Thrombocytosis
BM: erythroid normoblastic hyperplasia with ↓ or absent stainable Fe
Koilonychia: Spoon nails
Angular cheilitis
PLUMMER-VINSON SYNDROME
Severe Fe deficiency anemia + esophageal webs + atrophic glossitis
↑ risk of Esophageal Squamous Carcinoma
Anemia of chronic disease
Decreased RBC production seen with chronic inflammatory conditions (infections/autoimmune) or malignancy due to ineffective use of iron stores
IL-6 -> ↑ liver production of Hepcidin → ↓ Fe absorption from the gut, & ↓ Fe transport from
Macrophage stores to erythroid precursors (deprive bacteria of iron)
EPO response muted
Normocytic or mildly Microcytic anemia
↓ serum iron, ↓ Saturation, but ↓TIBC + Ferritin
Mildly ↓ Retic count
PAPPENHEIMER BODIES IN RBCS
Abnormal granules of iron found inside red blood cells on routine blood stain
Sideroblastic anemia, sickle cell disease, post-splenectomy, & some
hemolytic anemias
Immune thrombocytopenia purpura
Destruction of platelets by Anti-platelet IgG autoantibody
Chronic ITP (> 12-month duration):
SLE, CLL (secondary) or primary (Idiopathic)
Autoantibodies opsonize platelets → Destroyed in the Spleen
Spleen normal size; BM mildly ↑ Megakaryocytes, some immature (single, non-lobulated nuclei), large platelets
Adult women (3:1 F:M)
Petechial bleeding (Skin & mucosa); ecchymoses (usually in dependent areas), epistaxis, gingival bleeding, easy bruising & bleeding with minor trauma, melena, hematuria, heavy periods
Acute ITP (< 12-month duration):
Mainly in childhood 1-2 weeks usually after a self-limited viral illness
Self-limited
Treatment:
Steroids
Intravenous Immunoglobulin
Splenectomy
Heparin-induced thrombocytopenia
Type I HIT: Thrombocytopenia immediately after start of Heparin Rx
Direct platelet-aggregating effect of Heparin
Not clinically significant
Type II HIT:
Onset typically between 5-10 days after the start of Heparin therapy
Defined as a decreased platelet count of >50% from baseline.
Development of Ab against the Heparin-Platelet Factor 4 complex
Binding of Ab to this complex ACTIVATES the platelets → Thrombosis & develop a consumptive thrombocytopenia
Venous & arterial thrombosis dominant effect despite the thrombocytopenia
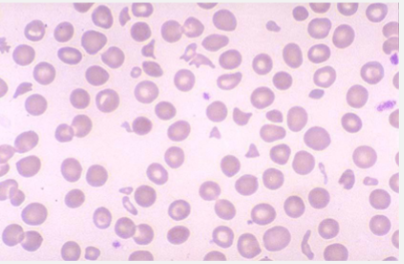
Thrombotic microangiopathies
Fibrin strand- “Piano wire”
Schistocytes
Thrombotic microangiopathies
Thrombotic Thrombocytopenic Purpura (TTP) & Hemolytic Uremic Syndrome (HUS)
Excessive activation of platelets forming small vessel platelet rich thrombi (microthrombi composed primarily of platelets & Von Willebrand Factor) → Microangiopathic
Hemolytic Anemia causing widespread organ dysfunction & a consumptive Thrombocytopenia
Thrombotic Thrombocytopenic Purpura
ADAMTS13 deficiency- vWF not cleaved
Thrombocytopenia + Microangiopathic Hemolytic Anemia
Renal failure, fever & CNS involvement
Young adult; F>M; ↑ incidence in HIV
Fatigue, dyspnea, petechiae, or other bleeding
Weakness, dizziness, abdominal pain, easy bruising, or nausea & vomiting
Thrombocytopenia
Anemia due to mechanical hemolysis with Schistocytes (MAHA)
Gastrointestinal
Bleeding or purpura due to the consumptive thrombocytopenia
Neurologic: Coma, stroke, seizure, transient focal abnormalities, headache, confusion)
Hemolytic Uremic Syndrome
Microangiopathic Hemolytic Anemia + Thrombocytopenia + Renal impairment
Shiga-like Ecoli
Bloody diarrhea → sudden onset of bleeding manifestations
Atypical: Excessive Complement deposition & activation
Bernard-Soulier Syndrome
GpIb deficiency → defective adhesion
Giant platelets, thrombocytopenia
Glanzmann Thrombasthenia
GpIIb/IIIa deficiency → defective aggregation
Normal platelet count, absent aggregation
Hemophilia A
Factor VIII deficiency
X-linked recessive
Spontaneous hemarthroses, muscle hematomas
Hemophilia B
Factor IX deficiency
X-linked recessive
Clinically indistinguishable from Hemophilia A
Disseminated Intravascular Coagulation (DIC)
Systemic activation of coagulation
Widespread microthrombi + consumption of clotting factors & platelets
Thrombosis and bleeding
Causes:
Sepsis (endotoxin, cytokine storm)
Trauma
Obstetric complications (amniotic fluid embolism, placental abruption)
Malignancy (acute promyelocytic leukemia, adenocarcinoma)
Nephrotic syndrome
Transfusion reactions
Prolonged PT and aPTT
Thrombocytopenia
Low fibrinogen
High D-dimer
Schistocytes on peripheral smear
Bleeding: petechiae, ecchymoses, oozing from IV sites/surgical wounds
Microthrombi: organ dysfunction (renal failure, respiratory distress, CNS changes)
Acute hemolytic reactions
Within minutes to hours of transfusion
Preformed recipient antibodies (usually anti-A or anti-B) bind donor RBCs → complement-mediated intravascular hemolysis
Fever, chills, flank pain, hypotension, hemoglobinuria
May progress to DIC, shock, acute renal failure
Positive direct antiglobulin (Coombs) test
Hemoglobinemia, low haptoglobin, elevated LDH and bilirubin
Delayed hemolytic reactions
Days to weeks after transfusion
Anamnestic IgG response to minor RBC antigens → extravascular hemolysis
Mild jaundice, low-grade fever, unexpected drop in hemoglobin
Usually less severe than acute reaction
Positive direct antiglobulin (Coombs) test
Elevated bilirubin and LDH, no hemoglobinemia or hemoglobinuria
Transfusion-Related Acute Lung Injury (TRALI)
Immune-mediated reaction from donor anti-HLA or anti-neutrophil antibodies
Acute non-cardiogenic pulmonary edema within 6 hrs of transfusion
Hypoxemia, dyspnea, bilateral infiltrates, normal BNP
Stop transfusion, supportive O₂, avoid diuretics
Prognosis: Usually recovers in 48–96 hrs
Transfusion-Associated Circulatory Overload (TACO)
Volume overload or rapid transfusion exceeding cardiac capacity
Dyspnea, hypertension, JVD, pulmonary edema, elevated BNP
Worsens with fluids; improves with diuretics
Give diuretics, O₂, slow future transfusions
Infectious mononucleosis
EBV in saliva
Infects oropharyngeal and nasopharyngeal epithelial cells and mature B cells (via CD21)
Reactivation may occur via EBV Latent Membrane Protein (LMP-1), which mimics co-stimulatory CD40 B-cell receptor
Induces division and production of numerous polyclonal antibodies (Monospot test)
Specific antibodies against EBV are also produced
CD8+ T lymphocytes, NK cells, and specific antibodies control disease; usually self-limited
The lymphocytosis and “atypical lymphocytes” in peripheral blood are mostly CD8+ (cytotoxic) lymphocytes, with some NK cells
Residual small number of latently infected B cells persist for life
Atypical lymphocytes: increased cell size, abundant cytoplasm, indented or folded nucleus
Lymph nodes: paracortical expansion by atypical lymphocytes
Spleen: enlarged due to infiltrates of atypical lymphocytes
Liver: hepatitis with atypical lymphocytes infiltrating portal and sinusoidal areas, with foci of apoptosis or parenchymal necrosis
EBV-associated malignancies include Burkitt lymphoma, some large B-cell lymphomas, some Hodgkin lymphomas, nasopharyngeal carcinoma, extranodal NK/T-cell lymphoma, and gastric carcinomas
Infectious mononucleosis Diagnosis
Acute infection: positive VCA IgM ± VCA IgG; negative anti-EBNA
Past infection: positive VCA IgG and anti-EBNA
Infectious mononucleosis Clinical Presentation
Young adults: fatigue, ± fever, sore throat
Enlarged tonsils; generalized lymphadenopathy (neck, axilla, groin)
Enlarged spleen: rapid enlargement with capsular stretching increases risk of rupture with minor trauma
Complications: autoimmune hemolytic anemia, thrombocytopenia, hepatitis. involve nervous system, eyes, kidneys, lungs, or heart
Infection more serious in T-cell immunodeficiencies
Follicular Lymphoma
Painless generalized lymphadenopathy
From Germinal center B-cell (Centrocyte)
CD10, 19, 20, BCL-6 & CD5
t(14;18) juxtaposing Ig Heavy chain genes (14) to BCL-2 -> ↑ BCL-2 expression -> ↓ Apoptosis
Transformation to aggressive B-cell lymphoma (DLBCL or sometimes Burkitt)
Nodes enlarged, often form matted masses
Small lymphocytes with cleaved nuclei
Follicle-like nodules of neoplastic cells without true germinal centers
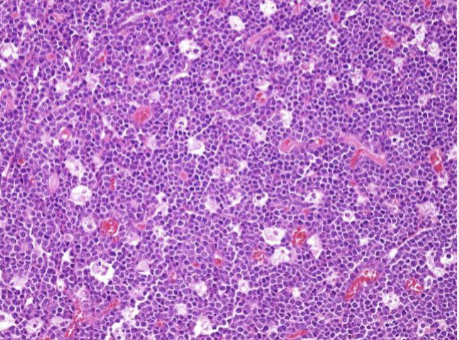
Starry Sky
Burkitt lymphoma
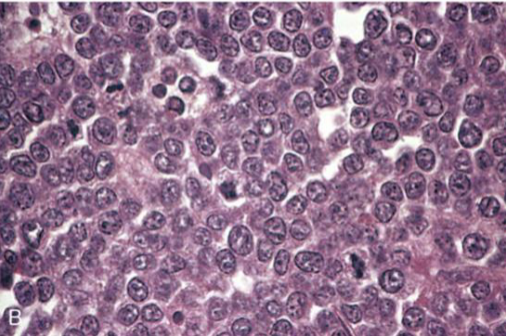
Starry Sky
Burkitt lymphoma
Burkitt lymphoma
Highly aggressive B-cell neoplasm characterized by the overexpression of the Myc gene on Chromosome 8
Translocation is the IgH heavy chain locus t(8;14)
Starry Sky appearance due to evenly interspersed macrophages with apoptotic debris in cytoplasm
Intermediate to small cells with noncleaved nuclei, coarse chromatin, small multiple nucleoli & very high mitotic index
African (endemic) Burkitt:
Very strong association with EBV
Extranodal, facial; tends to involve mandible & abdominal visceral organs
Burkitt lymphoma in HIV:
Intermediate association with EBV
Extranodal sites (GI, Bone marrow)
Sporadic Burkitt lymphoma in the US:
Weak association with EBV
Usually presents as mass of ileocecum/peritoneum
Both endemic & sporadic tends to occur in children or young adults
Mantle cell lymphoma
Have t(11;14) -> Cyclin D1 gene overexpression promotes cell cycle progression
B-cell markers & CD5
Intermediate grade
Painless generalized lymphadenopathy
Blood involvement
Spleen, liver & gut
Extranodal marginal zone lymphoma
GI mucosa → Mucosal-Associated Lymphoid Tissue Lymphoma (MALT Lymphoma)
Memory B-cell origin
Extra-nodal tumors often arise in the context of chronic inflammation (autoimmune or infectious) such as Sjogren syndrome (salivary glands), Hashimoto Thyroiditis, H. pylori gastritis
Polyclonal inflammatory proliferation
B-cell clone emerges that still depends on Ag stimulated T-cell signaling
Over time, additional mutations acquired that render neoplastic cell growth Ag independent (usually translocations which ↑ MALT-1, BCL-10, NFκB)
Usually remains localized for long periods, but with further clonal evolution may transform to DLBCL
Mature (Peripheral) T-cell lymphoma
Post-thymic T cells (outside of the Thymus) in lymph nodes, gut, spleen or skin
TdT negative
NK Cell tumors due to HTLV-1
Diffuse effacement of nodes with infiltrates of variably sized T-cells admixed with eosinophils & macrophages
Loss of CD45, CD2,3,5,7, CD4/CD8
Monoclonal T-cell Receptor gene rearrangements by PCR
Generalized lymphadenopathy, fever, weight loss +/- eosinophilia & pruritus
Worse Px than B-cell lymphomas
ANAPLASTIC LARGE CELL LYMPHOMA
Mature T-cell tumor that involves nodal & extranodal sites, often skin
CD30 (Ki-1)
ALK (Anaplastic Lymphoma Kinase) translocation
ALK-negative tends to occur in older adults & has a worse Px
Large cells with horseshoe or “embryoid” nuclei & abundant cytoplasm
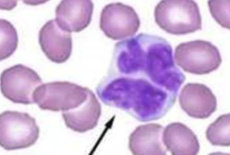
Sézary cells
Sézary syndrome
Typically present with a generalized pruritic exfoliative Erythroderma + circulating malignant T-cells (Leukemia of “Sézary” cells)
Sézary cell leukemia: absolute Sézary cell count of > 1000 with a clonal T-cell Receptor rearrangement with characteristic hyperconvoluted, “cerebriform” nucleus
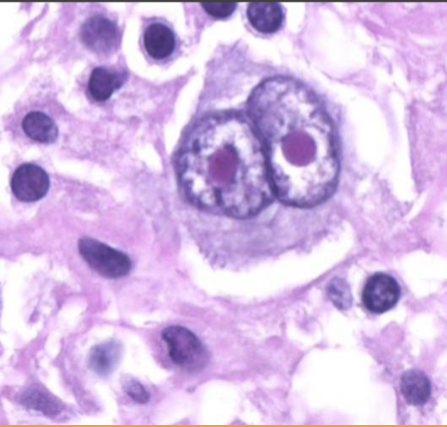
Classic R-S Cell
Hodgkin Lymphoma
Classic R-S Cell
Hodgkin Lymphoma
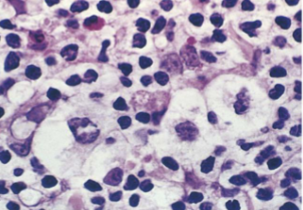
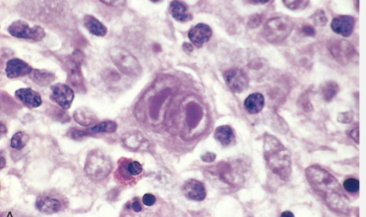
Lacunar R-S cell in Nodular Sclerosis Hodgkin Lymphoma
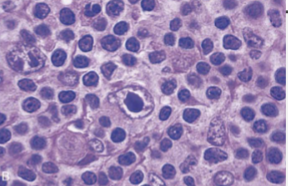
Mononuclear R-S cell in Mixed Cellularity Hodgkin Lymphoma
Hodgkin Lymphoma
B-cell germinal/post-germinal center cell derived tumors
Vast bulk of the tumor is made up of inflammatory & reactive cells (small lymphocytes, plasma cells, histiocytes, eosinophils & variable fibrosis) which harbor the sparsely scattered, malignant giant cells (Reed-Sternberg cells)
R-S cells are the malignant cells in HL
Large Binucleated/bilobed nucleus with prominent eosinophilic nucleoli
Show activation of NF-kB by EBV proteins
Cytokines produced by the R-S cells attract the background infiltrate
CD15 + CD30
Arises in a single node or group of nodes
Classical subtypes:
Nodular Sclerosis (NS)- Lacunar R-S variants
Mixed Cellularity (MC)- Mononuclear variants
Lymphocyte rich
Lymphocyte Depletion
Hodgkin Lymphoma Clinical Presentation
Non-tender adenopathy in a single or group of axial nodes
Cervical neck nodes → Horse collar- symmetric, bilateral node enlargement
Mediastinal nodes- common in NS type → cough, dyspnea
Less commonly, non-axial nodes: Axillary nodes, inguinal nodes, retroperitoneal
Initially spreads predictably to contiguous node groups
Late: spread to Liver, Spleen, & finally to BM & other tissues
“B” symptoms:
Fevers
Night sweats
Weight loss
Worse prognosis
Nodular Sclerosis Hodgkin Lymphoma
Prognosis excellent
Young adults or adolescents
Frequently involves mediastinum, lower cervical & supraclavicular nodes
Usually EBV negative
Histology: Collagen bands form nodules; Classic R-S cells rare, Lacunar RS cells
CD15 + CD30
Background benign lymphs, plasma cells, eosinophils, macrophages
Mediastinal nodes → cough, dyspnea
Mixed Cellularity Hodgkin Lymphoma
Biphasic age incidence (peaks in young & older adults)
M > F
Likely older age, B symptoms & advanced stage at Dx
Diagnostic RS cells + Mononuclear variants plentiful in background of benign lymphs, plasma cells, eosinophils, & macrophages
Usually EBV related
Abundant classic RS cells + mononuclear variants (CD15+, CD30+)
Lymphocyte rich Hodgkin Lymphoma
Predominantly small lymphocytes, but diagnostic R-S cells & Mononuclear variants present
EBV + sometimes
CD15+, CD30+
Very good to excellent Px
Lymphocyte Depletion Hodgkin Lymphoma
Paucity of lymphocytes with numerous atypical R-S cells
Older adults, HIV infected, or in third world; almost all EBV+
CD15+, CD30+
Px: least favorable
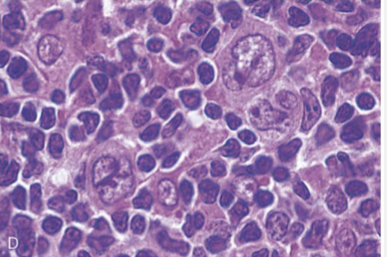
Popcorn cells
Nodular lymphocyte-predominant Hodgkin Lymphoma
Lymphocyte-predominant Hodgkin Lymphoma
D20+
NOT associated with EBV
Cervical, axillary, or inguinal lymphadenopathy
Tends to skip anatomic node regions (non-contiguous spread)
B-symptoms less common
More likely to recur
Indolent with Excellent prognosis
Can progress to Diffuse Large B-cell Lymphoma
Most common in men 30-50 y/o