heme lecture exam 5
1/69
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
70 Terms
What are the 2 main categories of anemia classified as macrocytic?
megaloblastic and Non-megaloblastic/ normoblastic
megaloblastic anemia
Refers to an impairment of DNA synthesis that results in large, abnormal, immature erythroid precursors in the bone marrow.
Hallmarks: Macro-ovalocytes, hypersegmented neutrophils, and ineffective erythropoiesis.
Causes: Vitamin B12 or folate deficiency.
Nuclear-cytoplasmic asynchrony:
A condition where the nucleus matures slower than the cytoplasm due to impaired DNA synthesis.
Main causes of normoblastic macrocytic anemias:
Alcoholism, reticulocytosis, hypothyroidism, aplastic anemia, and drugs
Classic blood smear morphologies for megaloblastic anemia and next tests:
Morphologies: ovalocytes, hypersegmented neutrophils, and Howell-Jolly bodies.
Next tests: Serum B12, folate levels, and methylmalonic acid (MMA) and homocysteine levels.
MCH ↑ (due to the size of the cell); MCHC normal
Relative reticulocyte count is usually normal
All three cell lines are affected
WBCs↓due to neutropenia
Platelets↓, not below 100 × 109/L
BM: Hypercellular
↓ M:E ratio
1/2 of RBC precursors show megaloblastic changes
Folate structure
Folate is a general term for any of the vitamin folic acids.
Folates have the pteridine ring attached to para-amino-benzoate with one or more glutamate residues.
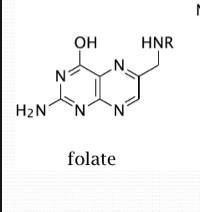
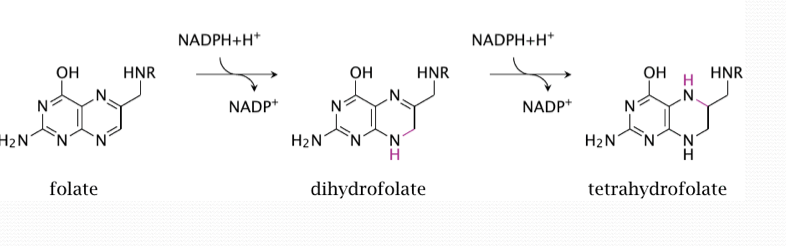
folate vs tetrahydrofolate
Folate circulates in the blood as 5-methyl tetrahydrofolate (THF).
It has to be de-methylated to become THF (tetrahydrofolate) the active form used in metabolic reactions.
What is folate used to make (what’s its function)?
vital for nucleotide and amino acid metabolism
purine and pyrimidine production
foods with folate
beans, dark leafy vegetables, eggs
B12 structure
Corrin ring with central cobalt ion/ crystalline cobalamin with cyanide ligand
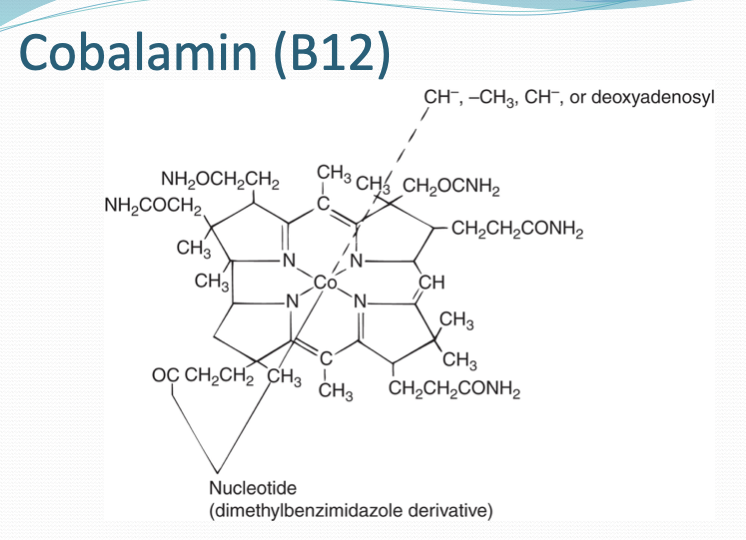
B12 function
DNA synthesis and myelin formation
B12 is man-made form of cobalamin which body needs
B12 absorption and metabolism process
cobalamin binds to haptocorrin (HC)- like protein in stomach
Binds to intrinsic factor in small intestine (duodenum)
Cobalamin is released into the duodenum by pancreatic proteases
passes through jejunum into ileum
Binds to specific IF receptor on microvilli of ileal mucosal cells
IF-B12 taken in by mucosal cells
Cobalamin released from IF into blood
IF degraded
B12 foods
meat, fish, dairy, eggs
B12 absorption inhibition
Lack of intrinsic factor, atrophic gastritis, ileal disease, and certain medications (e.g., proton pump inhibitors).
What are the major mechanisms that inhibit absorption of B12?
Diseases that prevent binding of IF–cobalamin complex in the ileum (malabsorption)
Crohn's disease, Tropical sprue, Celiac disease, and surgical resection of the ileum
Certain medications
Conditions that lead to a buildup of bacteria in the bowel (bacteria take up vitamin)
Can’t separate B12 from food (gastric acidity)
Can’t separate B12 from haptocorrin (protein R) – acid or pancreatic issues
Main issues of B12 defiecency
Impaired DNA synthesis- magaloblastic anemia, impaired methylen-THF, etc
Defective fatty acid metabolism- leads to neurological problems due to demyelination of nerve fibers
Pernicious anemia
most common cause of B12 deficiency: absence of IF meaning cobalamin can’t be absorbed
leads to autoimmune diseases, immune destruction, etc
phases of B12 absorption
Intragastric events B12 released from food proteins)
Duodenal and jejunal events ( R protein degraded and B12 binds to IF
Ileal events (B12-IF attaches to receptor and brought to enterocyte and enter blood)
Enterohepatic circulation
Lab tests distinguishing B12 from folate deficiency:
Elevated MMA (B12 deficiency) vs normal MMA (folate deficiency); elevated homocysteine in both.
Bone marrow findings in B12 deficiency
Hypercellular marrow, megaloblastic erythroid precursors, and nuclear-cytoplasmic asynchrony.
Two major biochemical reactions involving B12:
Conversion of homocysteine to methionine.
Conversion of methylmalonyl-CoA to succinyl-CoA.
Reaction using both B12 and folate:
Homocysteine to methionine (methionine synthase reaction).
Pathophysiology comparison of B12 and folate deficency
B12 deficiency: DNA synthesis defect and neurological damage (fatty acid metabolism issue).
Folate deficiency: DNA synthesis defect only.
Additional lab tests for PA (pernicious anemia):
Intrinsic factor antibodies, anti-parietal cell antibodies, and gastrin levels.
Other causes of megaloblastic anemia:
Medications (e.g., methotrexate), myelodysplastic syndrome, and inherited disorders.
Normoblastic macrocytic anemias:
Alcoholism, liver disease, hypothyroidism, and reticulocytosis.
Alcohol-induced macrocytic anemia (4 causes):
Folate deficiency, liver disease, toxic effects on marrow, and poor diet.
Liver disease-induced macrocytic anemia (4 causes):
Altered lipid metabolism, folate deficiency, splenic sequestration, and reticulocytosis.
Poikilocyte associated with liver disease:
target cells, spur cells, and leptocytes (thin cell)
Describe the CBC/diff, retic, BM, and additional testing that would be performed for liver disease.
Anemia mild (~12 g/dL)
Macrocytic (MCV not > 115 fL), normocytic or microcytic
Reticulocytes may be ↑, but RPI < 2
BM either normocellular or hypocellular
Precursors qualitatively normal
Liver function tests abnormal
↑ serum bilirubin, ↑ hepatic enzymes (ALT, AST, AP)
Thrombocytopenia, abnormal platelet function
Algorithm for classifying anemias by morphology:
Microcytic, normocytic, macrocytic.
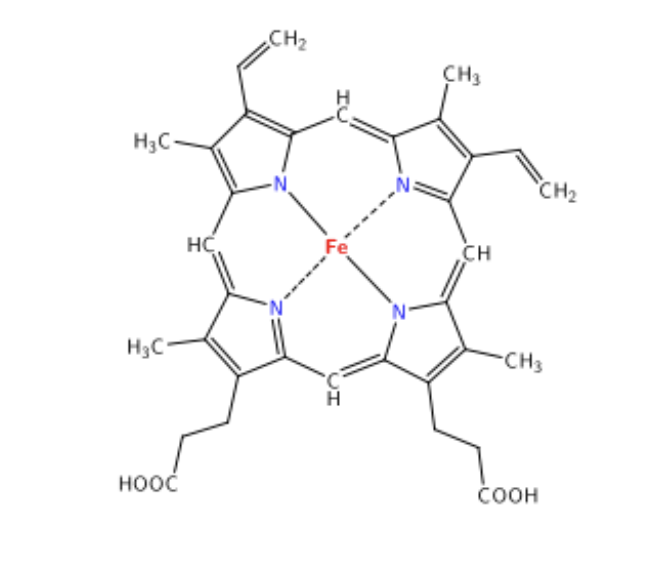
what is the structure of heme?
Iron-chelated porphyrin ring
non- amino acid portion of the protein made of flat tetrapyrrole ring with ferrous iron in the center
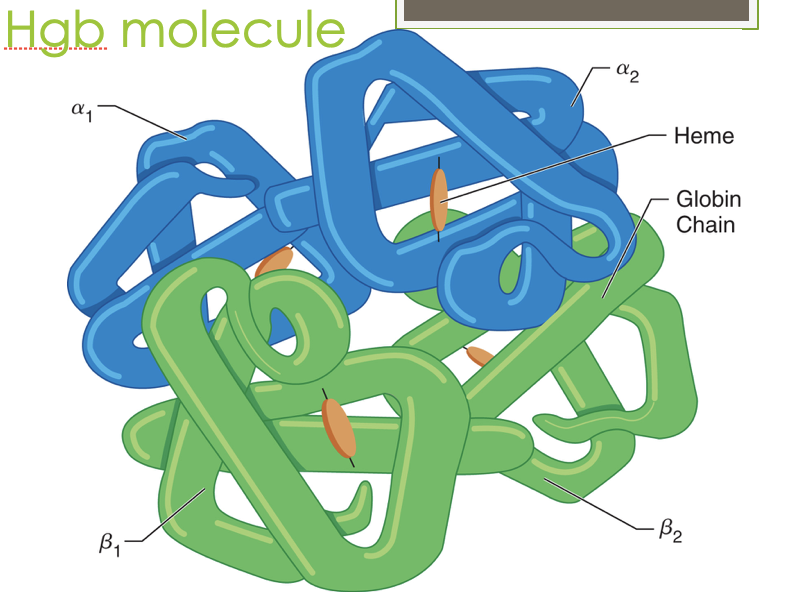
What is the structure of hemoglobin? How many chains/what types?
Four globin chains (2 alpha, 2 beta) with a heme nestled in a hydrophobic crevice to protect the Fe
an allosteric protein that is affected structurally and functionally by the molecules around
Heme synthesis steps in mitochondria
Step 1: glycine and succinyl CoA to form 5-aminolevulinic acid (ALA)
*Rate limiting step that needs enough Fe2+ and vitamin B6
Last step: inserting Fe into protophyrophyrin IX that was created in cytoplasm; after this the heme will leave mitochondria and attach to globin in cytoplasm
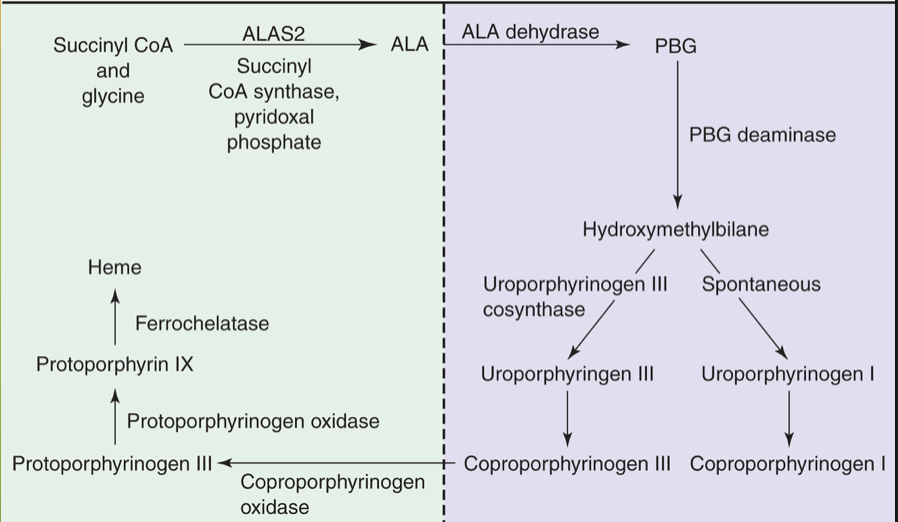
Cytoplasmic steps of heme production
Intermediate steps involving porphobilinogen to protoporphyrinogen IX- creating the ring itself
What are the regulating mechanisms on the production of heme
Negative feedback by heme.
Regulation of Hb synthesis
Concentration of iron,Heme feedback inhibition,rate limiting step, regulation of globin chain synthesis, Activity and concentration of ALAS2, activity of PBGD
T state vs R state of hgb
R= relaxed, low O2 affinity (stabilized salt bridges so unable to bind to O2)
2,3 DPG bound to salt bridges to stabilize it
T= tense state, high O2 affinity (broken salt bridges so able to bind to O2)
2,3 DPG unbound to allow O2 to bind freely
Oxygen disassociation curve left shift
Increased O₂ affinity (alkalosis, low 2,3-DPG).
Oxygen disassociation curve right shift
Decreased O₂ affinity (acidosis, high 2,3-DPG).
Ways CO2 is transported throughout body
Dissolution in the plasma -7%
Formation of carbonic acid -70%
Binding to the N-terminal amino acids of Hb (carbaminohemoglobin) – 23%
Methemoglobin
Nonfunctional Hgb with ferric Fe3+ rather than Fe2+ meaning it can’t bind to O2
remaining normal HGB has increased O2 affinity
Reversible
<1.5% normally formed per day
Hereditary form: deficiency/ abnormality in NADH MetHb reductase causing levels to reach 10-20%
Inherited form: Exposure to toxic substances can oxidize large amounts of Hgb causing stabilization to be acquired by substituting amino acid in globin chain
*chocolate brown color
Sulfhemoglobin
Sulfur atom binds to periphery of porphyrin ring causing decreased O2 affinity and irreversible oxygenation due to drugs or sulfur exposure
*green blood
Carboxyhemoglobin
Hgb that has been exposed to carbon monoxide, affinity for CO 200X higher than O2 affinity; caused by smoking, city living
incapable of transporting oxygen since CO bound where O2 goes
Curve shifted left since remaining normal Hbg has increased affinity but doesn’t release
*Cherry red color to blood and skin
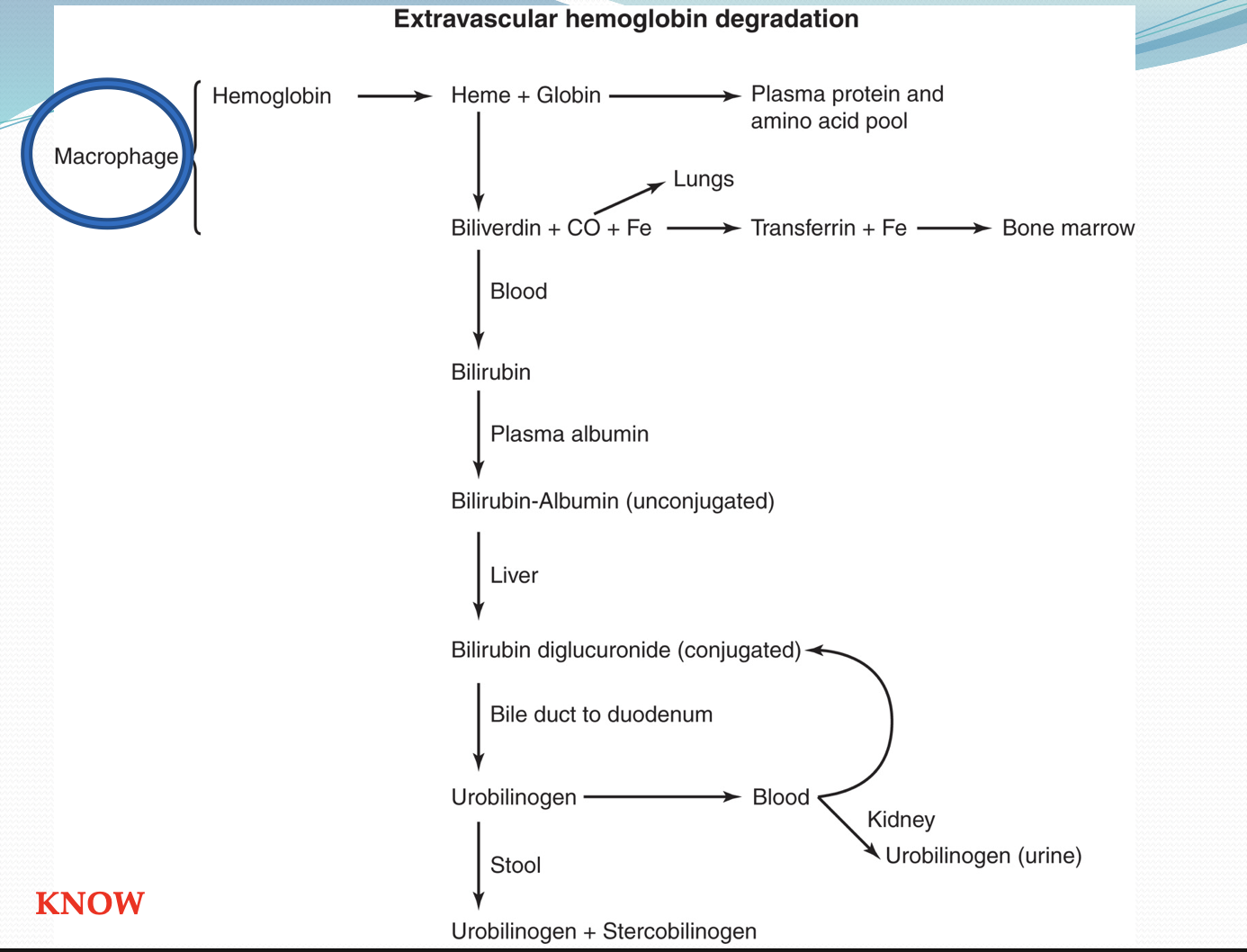
Extravascular Hemolysis
Takes place in macrophages of spleen; most efficient, recycling amino acids and iron
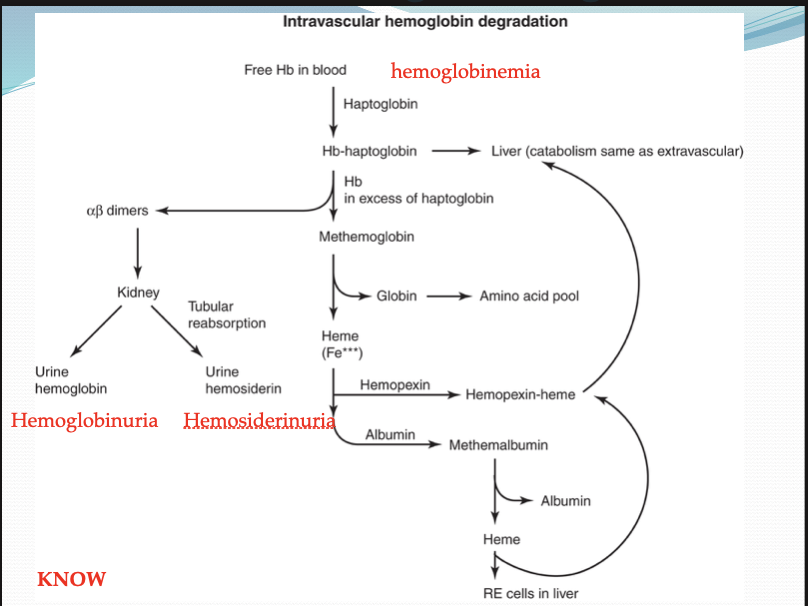
Intravascular hemolysis
Caused by mechanical trauma, compliment fixation, fibrin formation, etc in vascular system, causing schistocytes
Hgb dissociate into alpha-beta dimers and bind to haptoglobin, too big for kidney to filter out so its carried to liver, hepatocytes break down similarly to extravascular
haptoglobin and hemopexin decrease and free HGB, methemoglobin, urine sedament, schistocytes, and spherocytes all increase in urine
Haptoglobin
Acute phase reactant, prevents loss of free Hgb through kidney
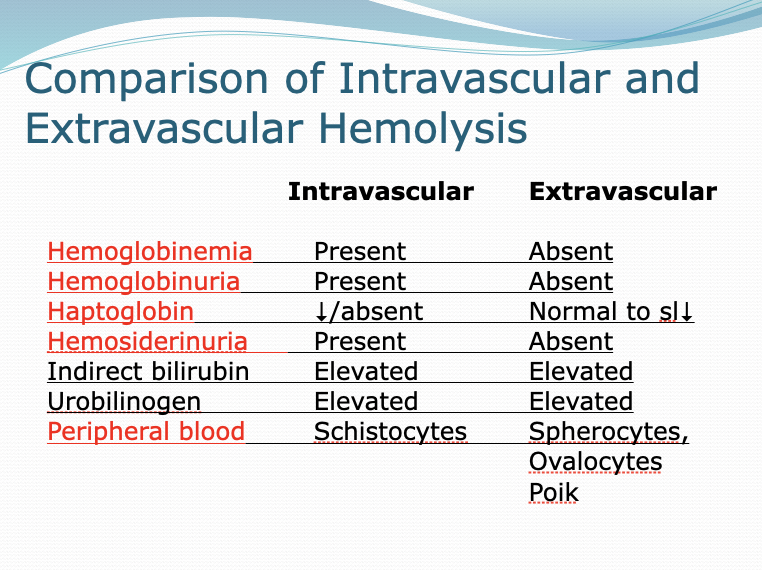
Intravascular vs extravascular hemolysis
Intravascular: Hemoglobinemia, hemoglobinuria, hemosiderinuria all present and Haptoglobin decreased or absent
Schistocytes
Extravascular: Hemoglobinemia, hemoglobinuria, hemosiderinuria all absent and Haptoglobin normal
spherocytes and ovalocytes
intrinsic vs extrinsic
intravascular vs extravascular
inside vs outside RBC
inside veins vs in spleen or tissue
What is HGB broken down to and what is recycled?
Recycled components: Iron (stored as ferritin or transferred to transferrin)
Degraded components: Heme (converted to bilirubin)
defect that causes thalassemia
Mutations in one or more globin gene causing decreased or absent synthesis of globin chains
What regions are more commonly associated with thalassemia and how does it relate to malarial protection?
More common in Mediterranean, Middle Eastern, Southeast Asian regions
RBC phenotype provides partial protection against malaria
Poilk in thalassemia
target cells and basophilic stippling
How do alpha and non-alpha chains pair? (who do they prefer and what is the
typical ratio)
Two major types of classical thalassemia
α-thalassemia
Impaired α-chains
do not combine
β-thalassemia
Impaired β-chain
Bind with each other to form HbH (β4)
δ-thalassemia
Not clinically significant
Combinations of gene deletions
δ β, γδβ (more severe)
Rare

Alpha Thalassemia
Two α-genes on each of two #16 chromosomes = four α-genes (diploid)
Two pairs, α1 and α2
α2 more severe since more protein encoded
diseases where All four alpha genes deleted
Hydrops fetalis (not compatible w/ life)
α-thal Major
Hgb Barts (gamma tetramer)
diseases where 3 of 4 alpha genes deleted
Hgb H disease (beta tetramer)
diseases where 2 of 4 alpha genes deleted or ¼ genes deleted
2/4 = α-thalassemia minor trait
¼ = silent carrier
Mentzer index
(Mcv/ rbc count)
if <13 thalassemia favored
3 main causes of anemia in thalassemia
decreased Hgb A synthesis, chronic extravascular hemolysis, and ineffective erythropoiesis
CBC in thalassemia
increased RDW (aniso), increased reticulocytes (poly),decreased RBCS, Hgb, and MCV

Generally what is expected to be seen in thalassemia
Hgb H
Excess beta globins bind together to form Hgb H which are unstable and cause hemolytic anemia (visualized with Brilliant cresyl blue stain)
Oxygen disassociation curve shifts left since Hgb H has a high affinity for O2
Hgb detected in thalassemia and what it looks like through electrophoresis
Hemoglobins Detected in Alpha Thalassemia:
Hemoglobin A (HbA)
Hemoglobin A2 (HbA2)
Hemoglobin F (HbF)
Hemoglobin H (HbH)
Hemoglobin Bart's (in severe cases)
Electrophoresis Findings by Alpha Thalassemia Phenotype:
Silent Carrier (-α/ααα): Normal electrophoresis
Alpha Thalassemia Trait (--/αα): Normal or slight HbA2/HbF elevation
Hgb H Disease (--/α-): Presence of HbH band
Hydrops Fetalis (α0α0): Predominantly Hgb Bart's
What chromosome is beta globin gene found? How many are there? How does that impact phenotype?
Found on chromosome 11, only 1 copy of each beta gene so less wiggle room meaning no silent carriers
causes excess alpha globin
Reduced HbA due to lack of beta chains
increased HbA2 and HbF
Beta thalassemia results
excess alpha chains form precipitate inside RBCs decreasing their lifespan causing anemia. BM compensates causing bone changes and brittleness due to ineffective erythropoiesis
facial deformities: prominent cheek bones, flaring teeth, nasal bridge depression
Beta Thalassemia Major
Cooley's anemia
Homozygous (β0/β0, β+/β+) or Double heterozygous(β0/β+)
Symptoms manifest at 6 months: irritability, bone changes, “hairy scull”, heart problems
extreme microcytic, hypochromic, baso stippling, RPI<2
Treatment: transfusions, splenectomy
Causes of increase or decrease in Hgb A2%
Elevated HbA2: Beta thalassemia, megaloblastic anemia, hyperthyroidism.
Decreased HbA2: Iron deficiency anemia, delta-beta thalassemia.
Mutations associated with alpha and beta thalassemia:
Alpha Thalassemia: Deletions in the alpha globin genes on Chromosome 16.
Beta Thalassemia: Point mutations in the beta globin genes on Chromosome 11.
Phenotype Matching:
Alpha thalassemia: Silent carrier, trait, HbH disease, hydrops fetalis.
Beta thalassemia: Minor, Intermedia, Major
Differentiating thalassemia from iron deficiency anemia (CBC/diff, PBSM):
Same except:
Thalassemia
Blood Smear: Target cells, basophilic stippling.
Hemoglobin Electrophoresis: Increased HbA2 and/or HbF.
Iron Deficiency Anemia:
Blood Smear: Microcytosis, hypochromia.
Hemoglobin Electrophoresis: Normal.