lec 12 - proteomics & bioinformatic tools for protein studies (glytsou)
1/52
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
53 Terms
what is bioinformatics
bioinformatics = scientific subdiscipline that involves using computer technology to collect, store, analyze and disseminate biological data and information such as DNA and AA sequences or annotations about those sequences
scientists and clinicians use databases that organize and index such biological information to increase our understanding of health and disease and, in certain cases, as part of medical care
bioinformatics is an interdisciplinary field
connected to:
biology
genetics
chemistry
medicine
pharmacy
engineering
statistics
mathematics
CS
applications of bioinformatics
identifying new drug targets
understanding disease mechanism
designing new drugs
predicting interactions between a compound and enzyme
predicting drug responses/safety
streamlining clinical trials
shortening timeline and reducing the cost of drug development
reducing the risk of side effects
fostering the growth of personalized medicine
what is the relevance of bioinformatics to pharmacology?
bioinformatics play a role in
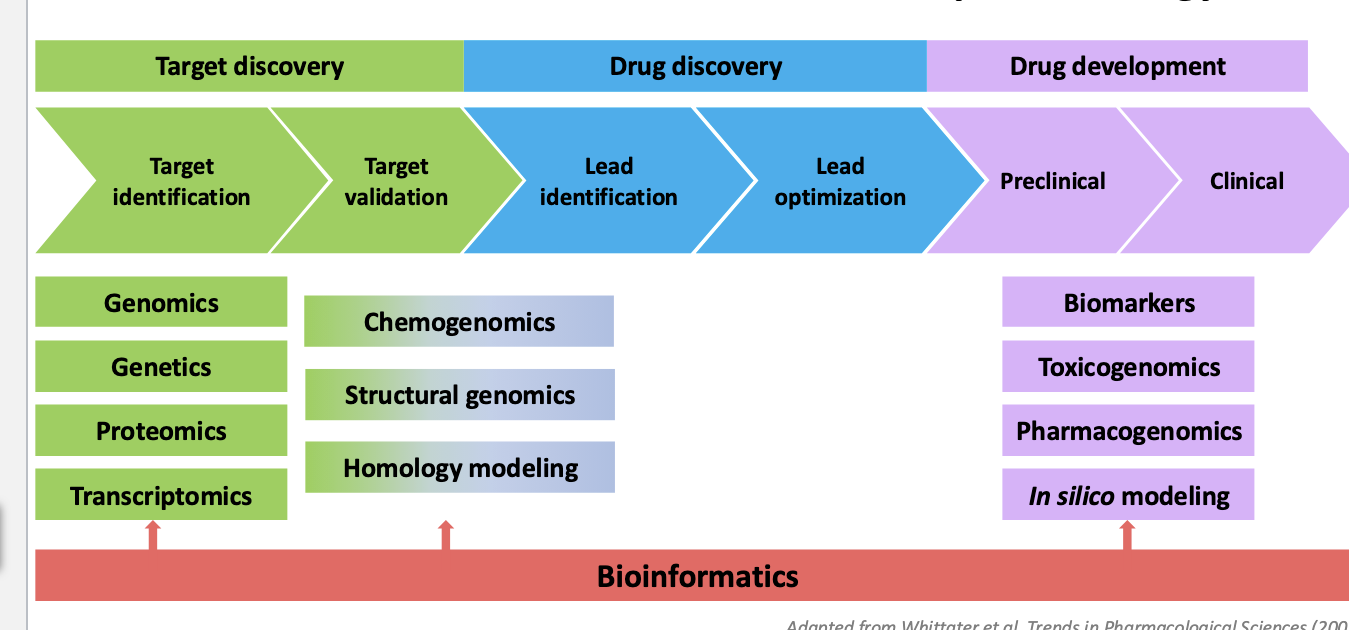
target discovery → transcriptomics and homology modeling
drug development → in silico modeling
human genetics evidence supports 2/3 of the 2021 FDA approved drugs
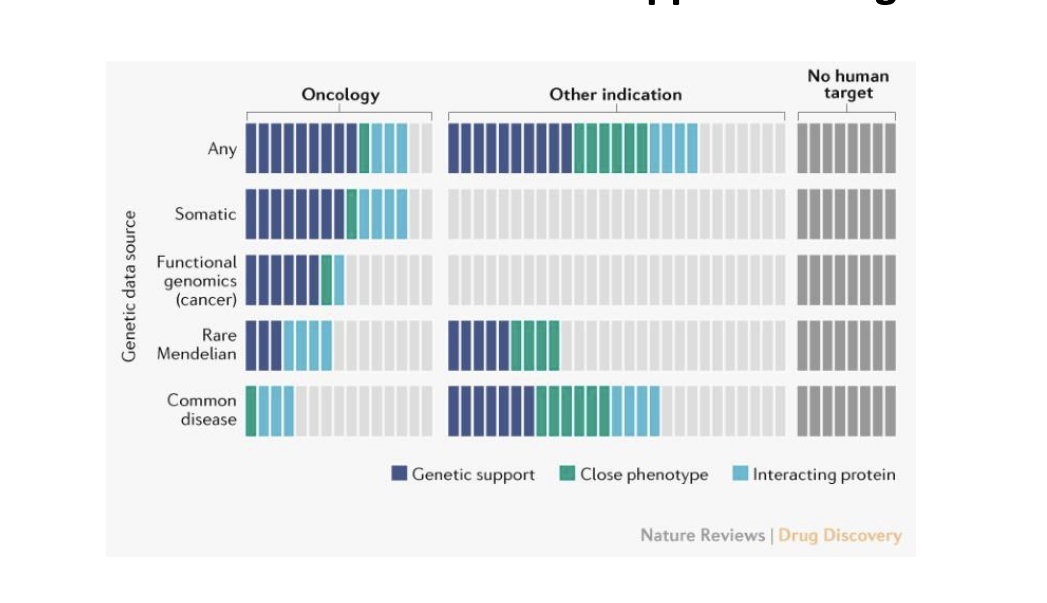
example of developing a therapeutic hypothesis based on genetic evidence
kerendia (finerenone) - bayer
mineralocorticoid receptor antagonist
selective
nonsteroidal
used to treat chronic kidney disease (CKD) from type 2 diabetes
microalbuminuria = increased levels of urinary albumin-to-creatinine ratio (UACR) → early indication of CKD
genome-wide association studies (GWAS) and functional genomic analyses → intronic variant in NR3C2 associated with microalbuminuria
NR3C2 = gene that encodes the mineralocorticoid receptor (MR)
kerendia = mineralcorticoid receptor antagonist → blocks MR
reduces the risk of CDK and slows CKD from getting worse
since NR3C2 (MR) is genetically linked to kidney damage via albuminuria → blocking MR helps
bioinformatics techniques and tools
sequence analysis
structural bioinformatics
high-throughput techniques
biomedical image-based analysis
network and systems biology
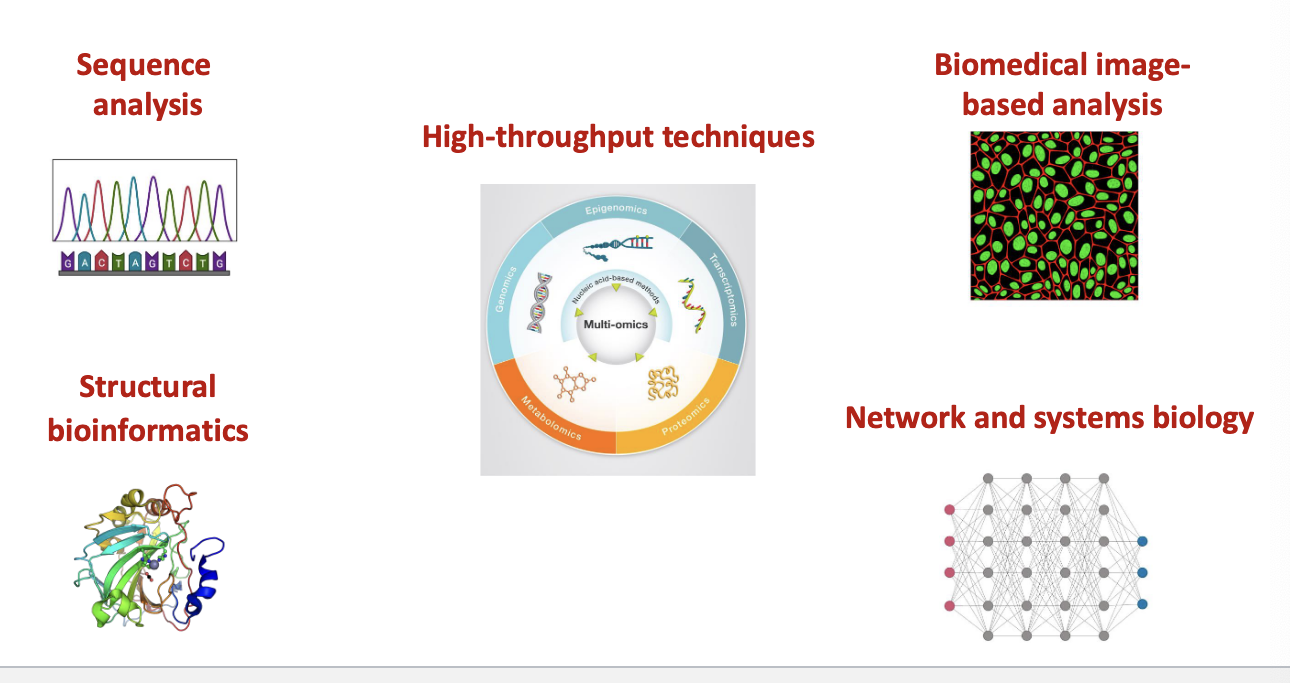
bioinformatics tools
databases
sequences storage databases
archival database: genbank, protein data bank (PBD)
curated database; knowledge base
interpro → protein families, motifs and domains
uniprot → sequence and functional information on proteins
computational models
mathematical models to describe biological system
software tools
sequence alignment tools
BLAST
clustalW
function analysis tools
GEO
pathway tools
image analysis software
clinical tools (ORACLE)
machine learning applications
protein structure predictions
drug response predictions
uniprot knowledgebase: hub for protein information
freely accessible database of protein sequence and functional information
contains description of:
function of proteins
interactions
polymorphisms
secondary structure
quaternary structure
etc

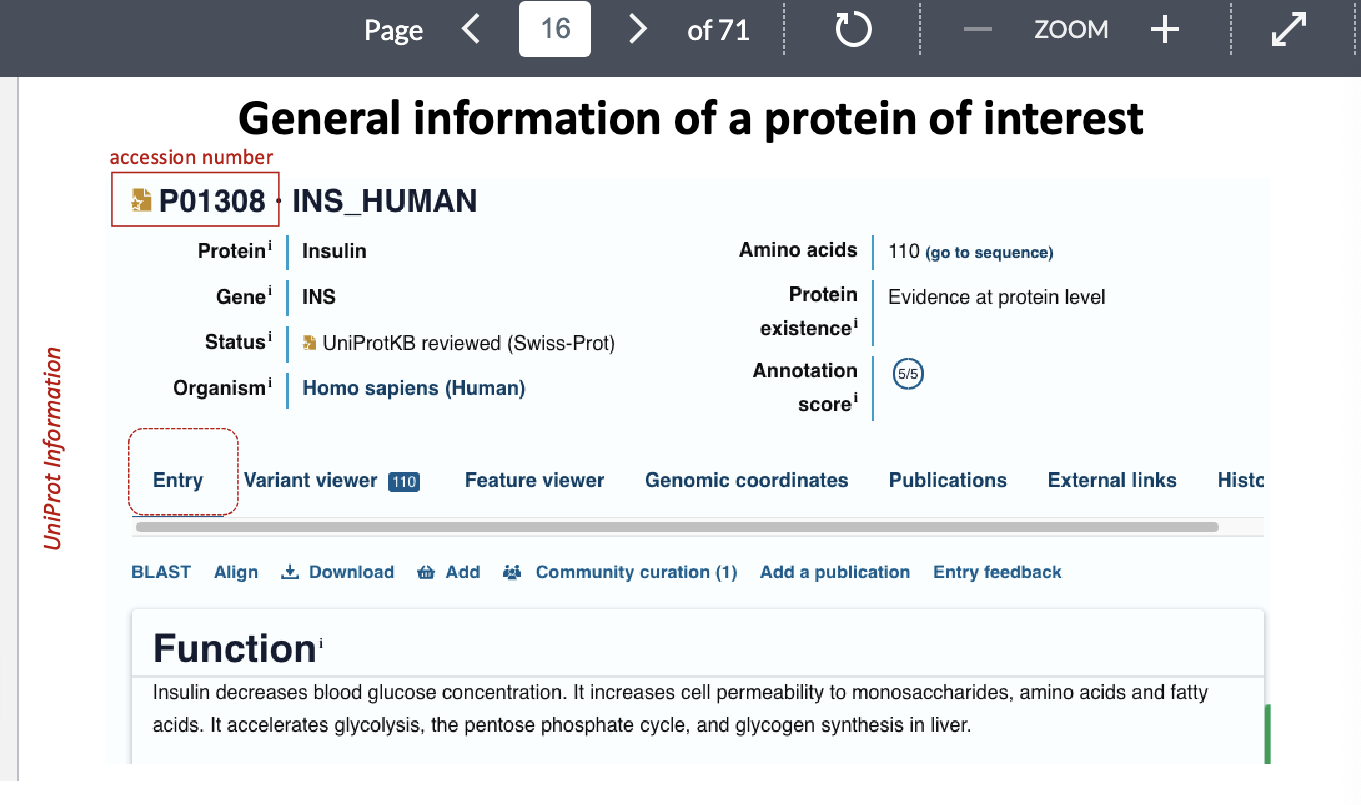
uniprot info: general information of a protein of interest
accession numnber = unique ID for protein
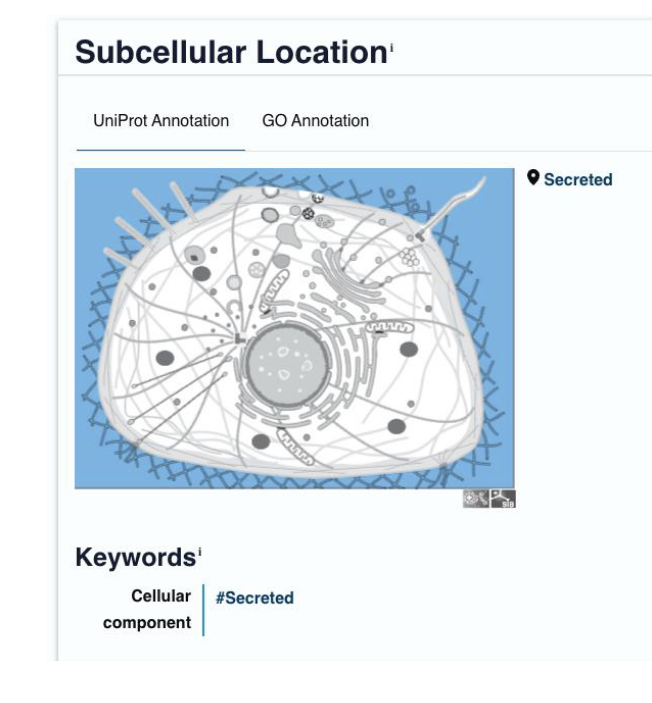
uniprot info: subcellular location
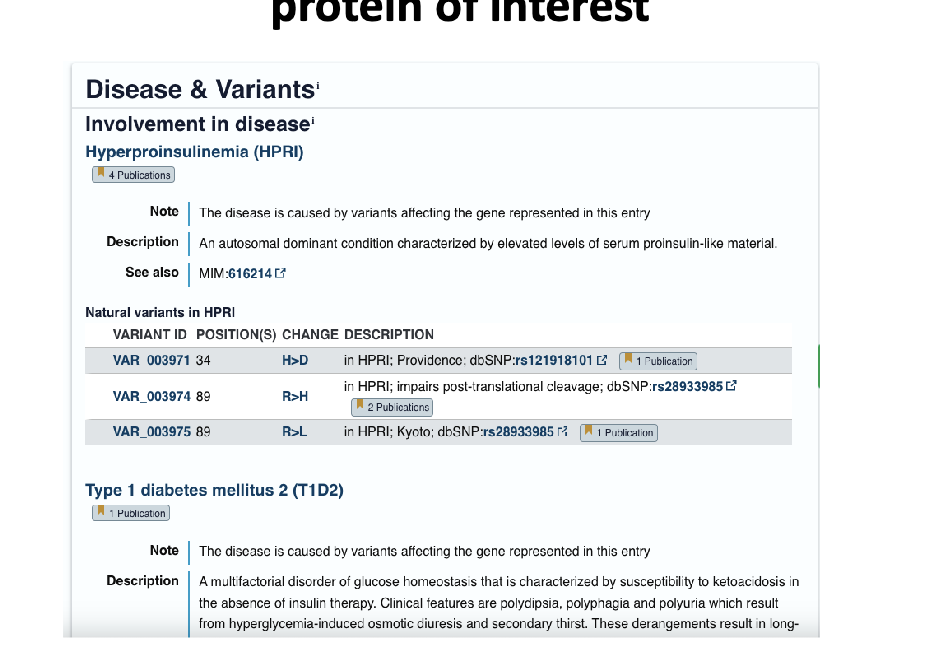
uniprot info: diseases caused by variants/mutations in the gene
uniprot info: pharmaceutical (the use of protein as pharmaceutical drug)

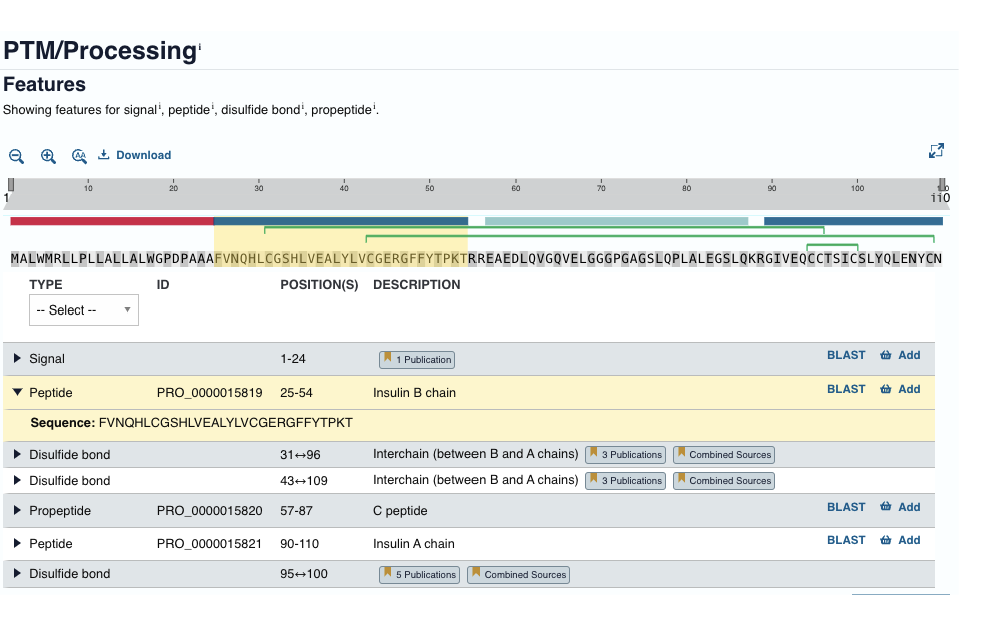
uniprot info: post-translational modifications/processing
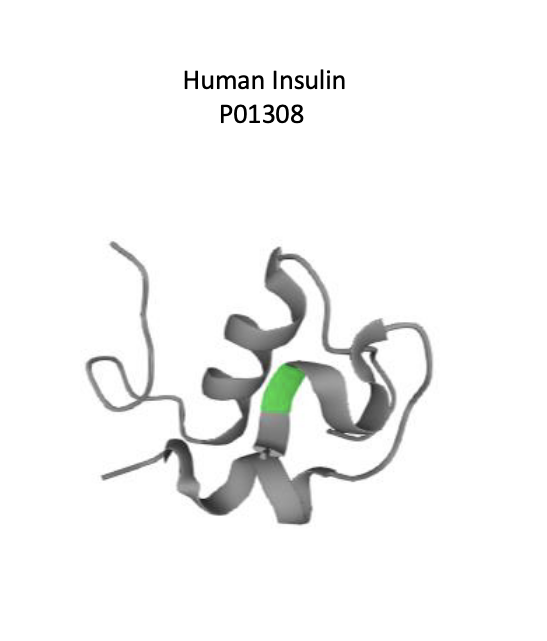
uniprot info: protein structure
2024 nobel prize in chemistry: protein structure design and prediction
david baker → for computational protein design
demis hassabis and john M/ jumper → for protein structure prediction
the importance of determining the protein structure
medicine
drug design
predict the function of a mutated protein
biochem
predict the MOA and develop tools to manipulate it
biotech
design of new enzymes
protein structure can be determined experimentally
by 2024, >227,000 proteins are known in atomic detail
techniques used to determine the protein conformation (3D shape or spatial arrangement)
x-ray crystallography
use diffracted x-rays from a protein crystal to generate an electron density map which indicates the atomic positions of protein
ONLY for proteins that can readily crystallize
nuclear magnetic resonance (NMR) spectroscopy
reveals the structure and dynamics of proteins in solution by identifying protons in close proximity to one another
cryo-electron microscopy
rapidly developing method that can elucidate the structure of large multimeric complexes and increasingly higher resolutions
protein structure prediction
protein structure = primary, secondary, tertiary, quaternary
one of the most important goals of computational biology
extremely challenging
approaches to predict 3D structures
Ab initio predictions
without prior knowledge
calculations that attempt to minimize the free energy of a structure
knowledge based methods
an unknown primary structure is examined for compatibility with known protein structures/fragments
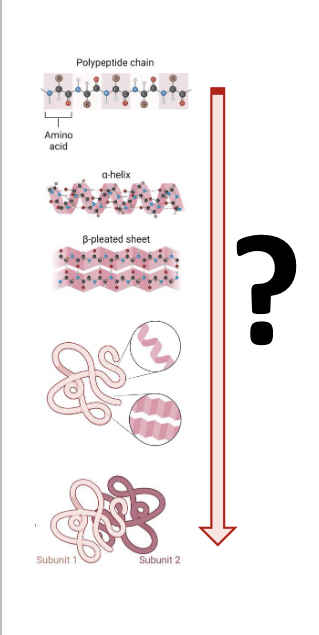
prediction and analysis of 3D structures of biomolecules
computational structural prediction methods
homology modeling
to predict the structure of an unknown protein from existing homologous proteins
protein-ligand docking and virtual screening
to predict and analyze the binding interactions between small molecules (ligands) and proteins
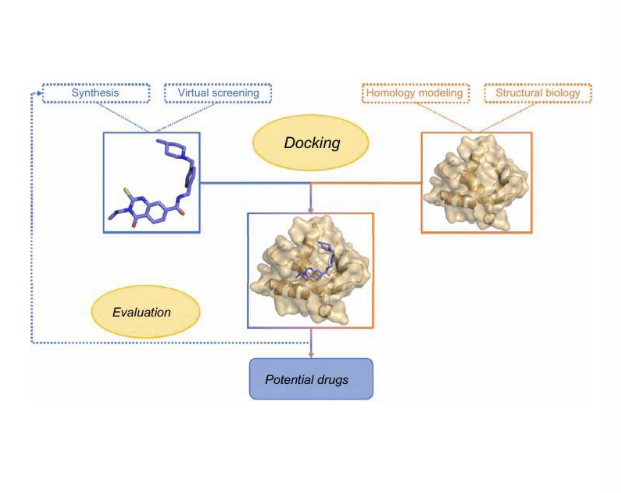
rational drug design
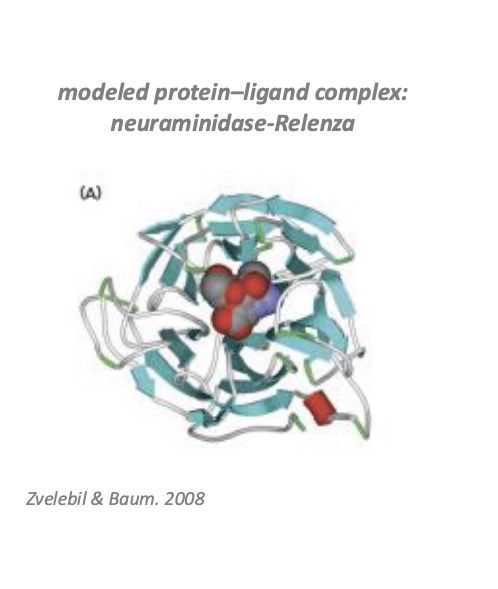
relenza (zanamivir)
treatment of illness due to influenza A and B virus in adults and peds pts aged 7 years of age and older who have been symptomatic for no more than 2 days
structure based design
selecting molecules that were likely to bind to the conserved regions of the enzyme neuraminidase
neuraminidase = enzyme produced by the flu virus to release newly formed virus from infected cells
alphafold
a significant step forward in protein folding prediction
since 1994, protein structure prediction challenge “critical assessment of structure prediction” (CASP)
alphafold structures = vastly more accurate than competing methods
alphafold = freely available, AI program that can predict the shape of a protein, almost instantly, down to atomic accuracy
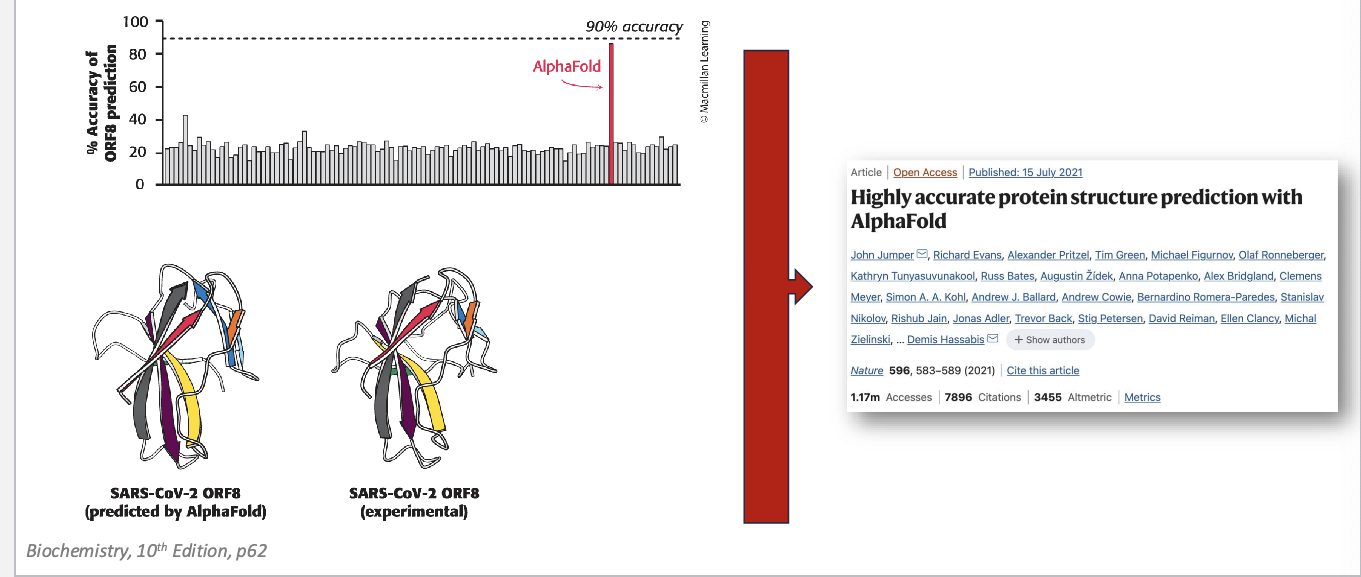
deepmind software…
that can predict the 3D shape of proteins is already changing biology
alphafold mania = deepmind software
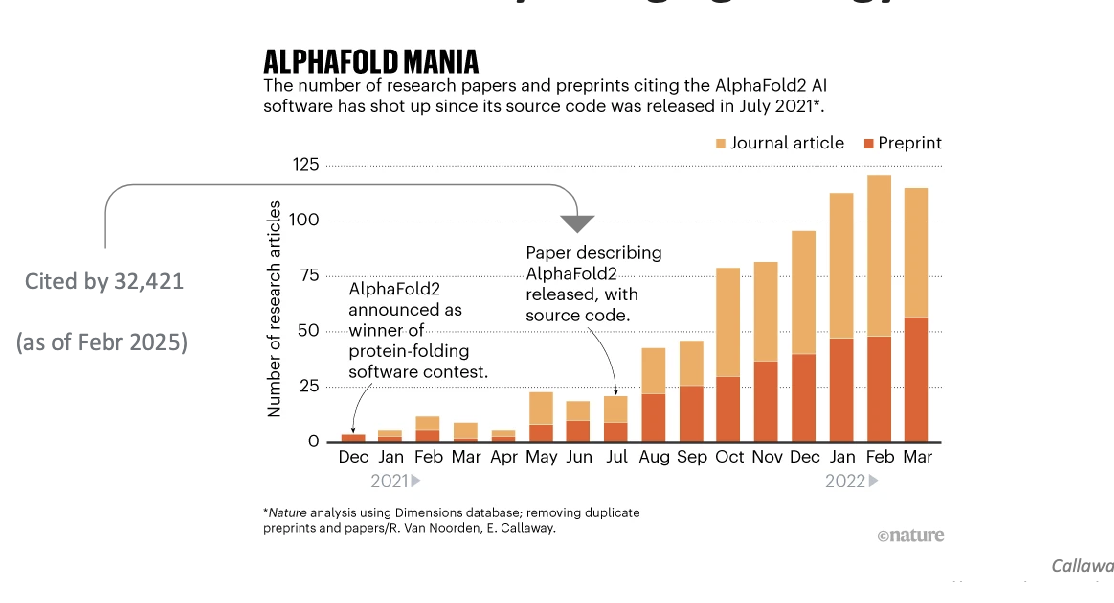
alphafold limitations and future directions
alphafold predictions are valuable hypotheses and accelerate but do NOT replace experimental structure determination
limitations
CANNOT predict the consequences of new mutations in proteins since there are NO evolutionarily-related sequences to examine
CANNOT deal with proteins that can adopt different structures in different states/environments
CANNOT predict protein structures bound to ligands
isomorphic labs = deepmind’s drug discovery spin off
predict the structure of proteins when they are bound to drugs and other interacting molecules
protein data bank (PDB)
RCSB protein data bank
structures are published and can be accessed for visualization and analysis
high-throughput techniques
multi-omic approach for proteins = proteomics
molecular read-out → protein
results → abundance of peptides, peptide modifications, and interactions between peptides
technology → mass spectrometry, western blotting, and ELISA
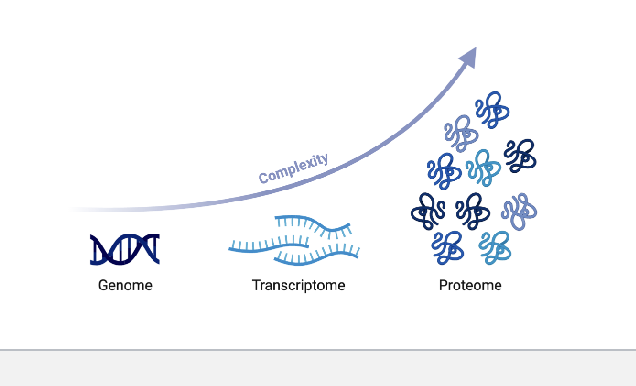
proteomics → large-scale study of proteins
proteome = the whole set of proteins expressed in a cell at a particular time
proteomics = the investigation of the proteome
explore the complete catalogue of proteins expressed in a cell type at a given time point
investigate how this inventory changes when the conditions are altered
unlike the genome, the proteome is NOT a fixed characteristic of the cell
a transcribed gene may be differentially translated or NOT translated and different proteins have different degradation rate so transcriptomic data is often NOT a good predictor of protein abundance
protein methods
protein purification
isolate one specific protein from a complex mixture
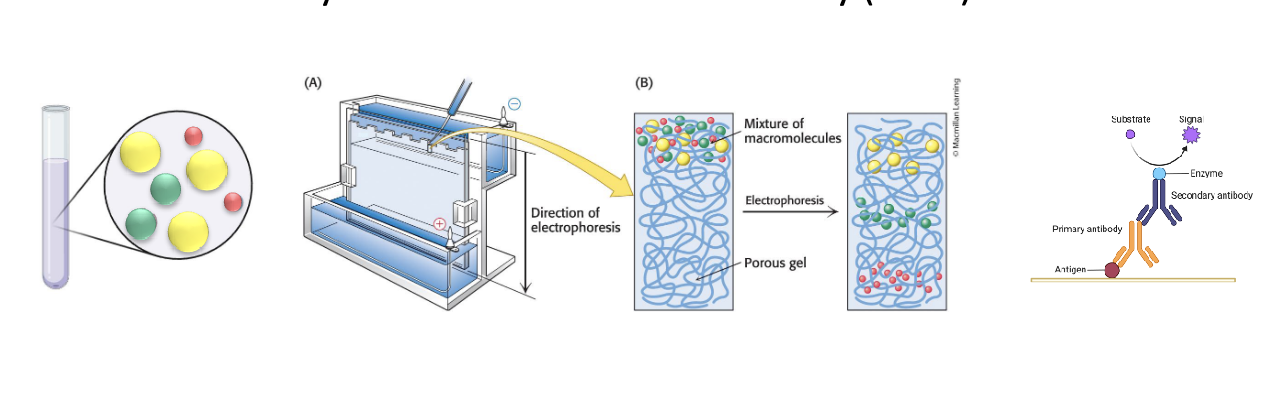
polyacrylamide gel electrophoresis and western blotting
electrophoresis → separate proteins based on size
western blotting → after electrophoresis, proteins are transferred to membrane; antibodies are used to detect specific protein
enzyme-linked immunosorbent assay (ELISA)
quantifies a specific protein
capture antibody fixed on plate with an enzyme that produces a color change or signal when target protein is present
limitations of protein methods discussed before
limited in # of proteins studied per condition/assay
limited to the detection of proteins for which an antibody is available
mass spectrometry…
is a powerful technique for ID of peptides and proteins
used to investigate:
when and where proteins expressed
rate of protein production, degradation, and steady-state abundance
how proteins are modified (for example, post-translational modifications such as phosphorylation
the movement of proteins between subcellular compartments
how proteins interact with one another
mass spec allows the highly precise and sensitive detection of the mass of an analyte
mass spectrometers:
convert the analyte into gas-phase ions
matrix-assisted laser desorption/ionization (MALDI) = technique used to produce ions using a laser energy-absorbing matrix
electrospray ionization (ESI) = techinque used to produce ions using an electrospray in which a high voltage is applied to liquid to create aerosol
apply electrostatic potentials to measure the mass-to-charge ratio (m/z)
consists of 3 components
ion source
mass analyzer
detector
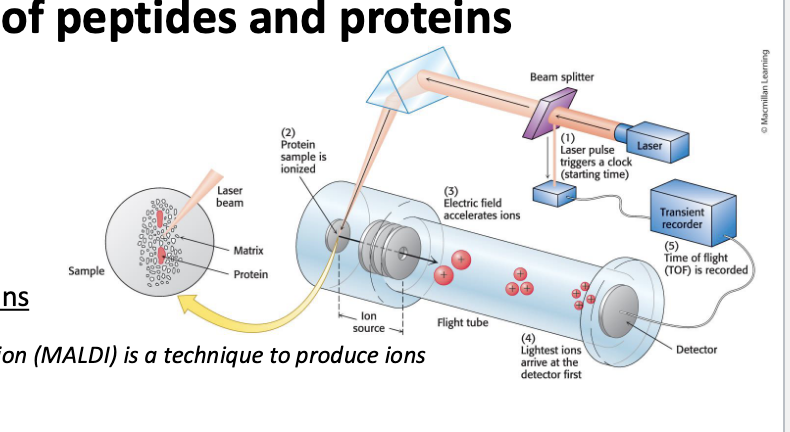
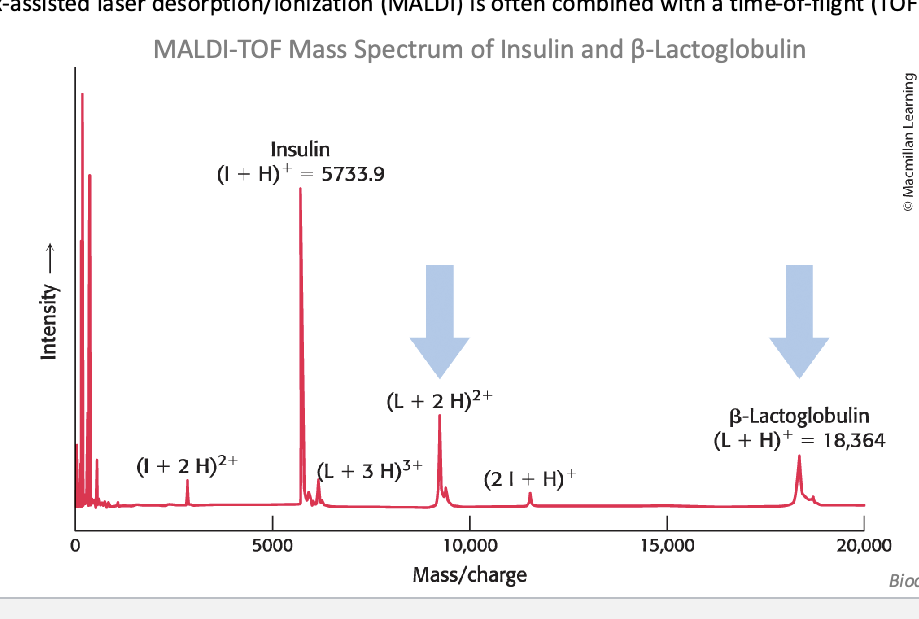
mass spec can detect…
molecular masses with a high degree of sensitivity and accuracy
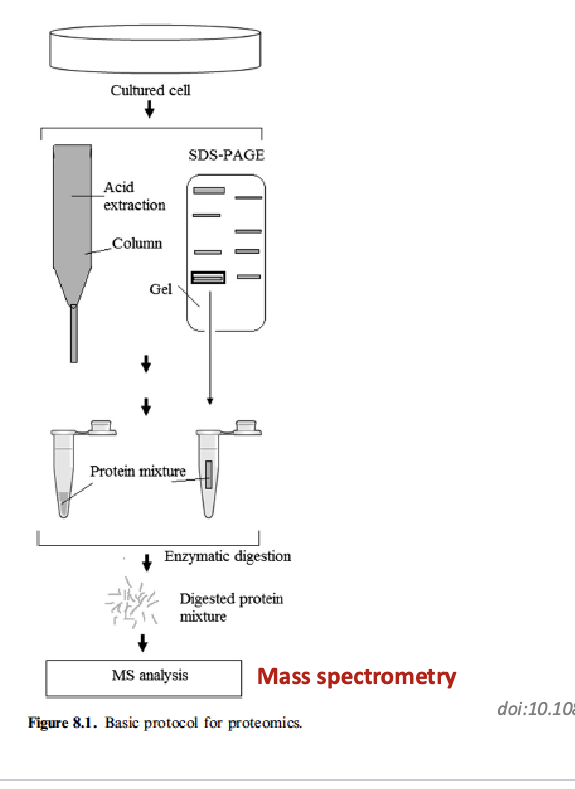
basic protocol for proteomics
start with cultured cells → proteins are extracted using acid extraction or column purification (helps isolate protein from other cellular materials) or proteins can be run through SDS-PAGE which also extracts proteins → enzymatic digestion: proteins are digested into peptides using enzymes → digested protein mixture is put for MS analysis → Ms identifies and quantifies peptides based on m/z ratio
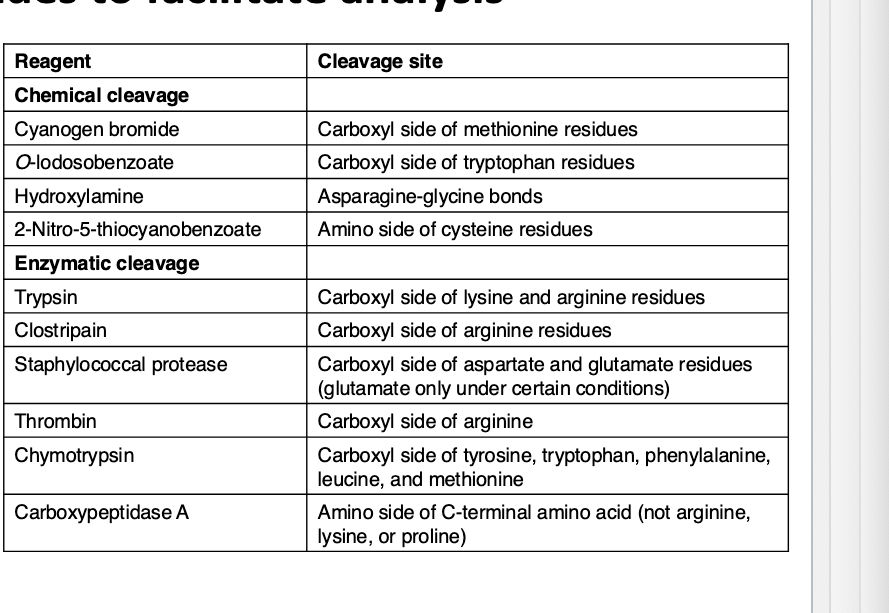
peptides can be specifically cleaved into small peptides to facilitate analysis
sequencing of long peptides by MS yields complex spectrums that are difficult to interpret
to sequence an entire protein
protein is chemically or enzymatically cleaves to yield peptides
peptides are ionized and their m/z
fragment ion spectra are then assigned peptide sequences based on database comparison and protein sequences are predicted
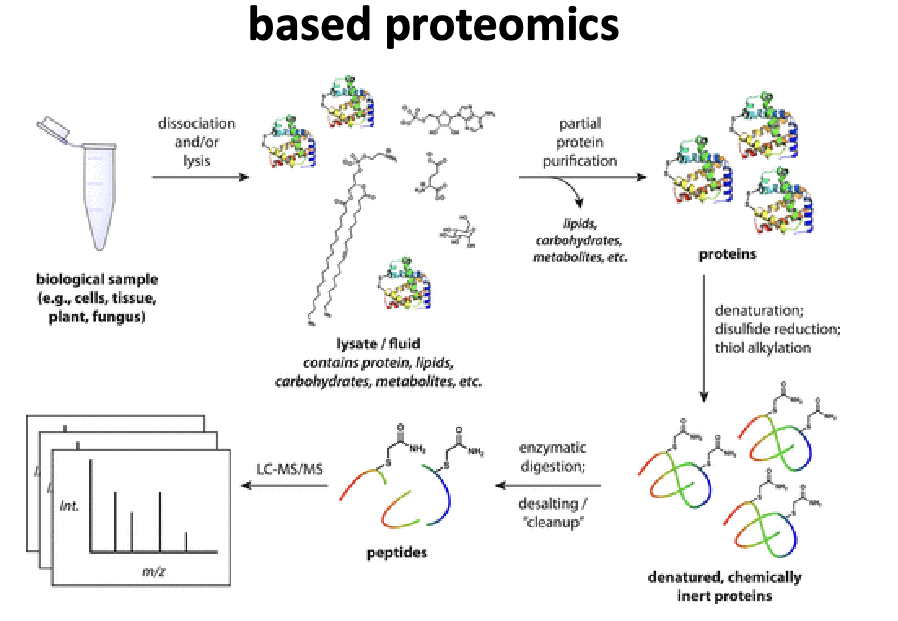
generic sample prep workflow mass spec based proteomics
biological sample → dissociation and/or lysis → get lysate/fluid that contains proteins, lipids, carbs, metabolites, etc. → partial protein purification: lipids, carbs, metabolites all leave → proteins → denaturation; disulfide reduction; thiol alkylation → denature, chemically inert proteins → enzymatic digestion; desalting/cleanup → peptides → LC-MS/MS
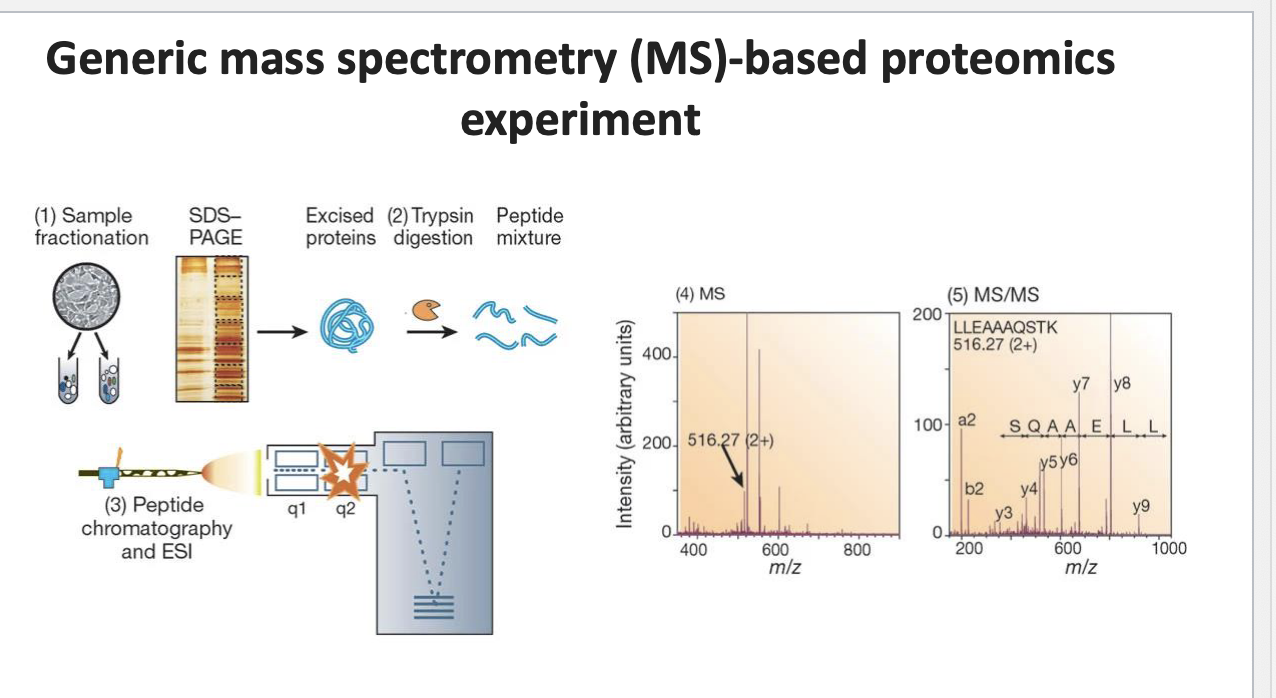
generic MS-based proteomics experiment
sample fractionation: biological sample fractionated to isolate different proteins → run thru SDS-PAGE which sorts them by size → proteins excised from gel → trypsin digestion → peptide mixture → peptides separated using chromatograph and then ionized by ESI for MS analysis
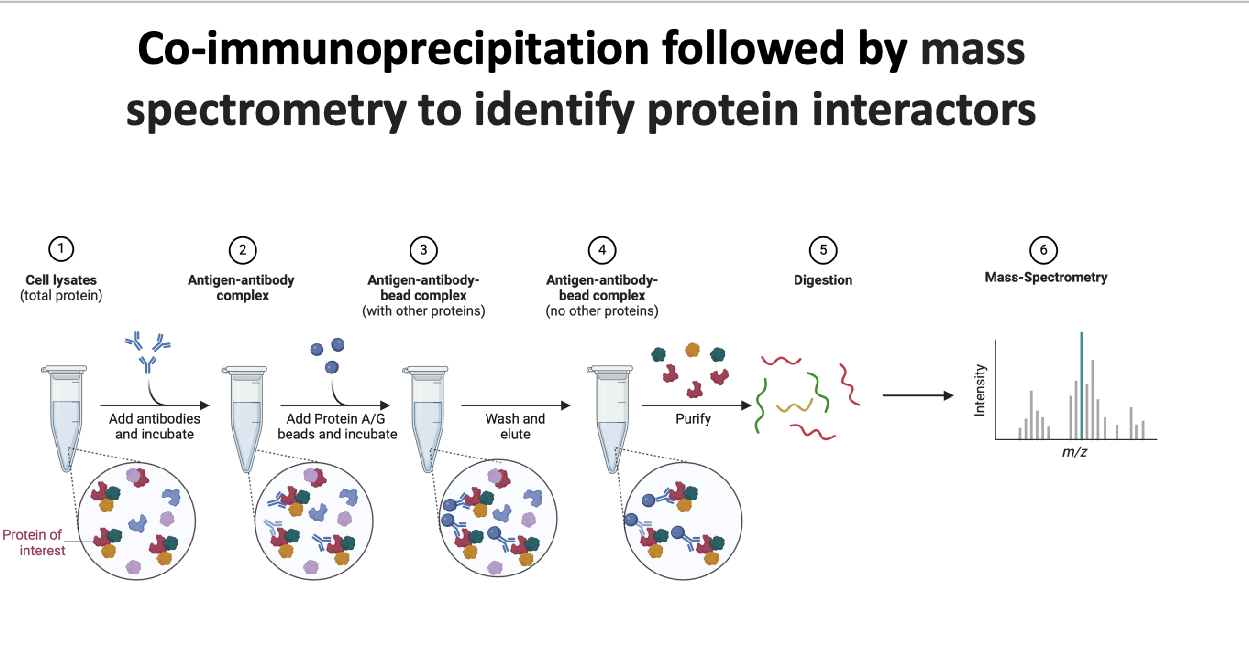
co-immunoprecipitation followed by MS to identify protein interactors
cell lysates (total protein) → add antibodies that specifically binds to target protein (aka antigen) and incubate → antigen-antibody complex → add protein A/G beads, which binds to antibodies, and incubate → antigen-antibody-bead complex (with other proteins)→ wash and elute to remove unbound proteins → antigen-antibody-bead complex w/NO other proteins → purify → digestion → peptides → MS
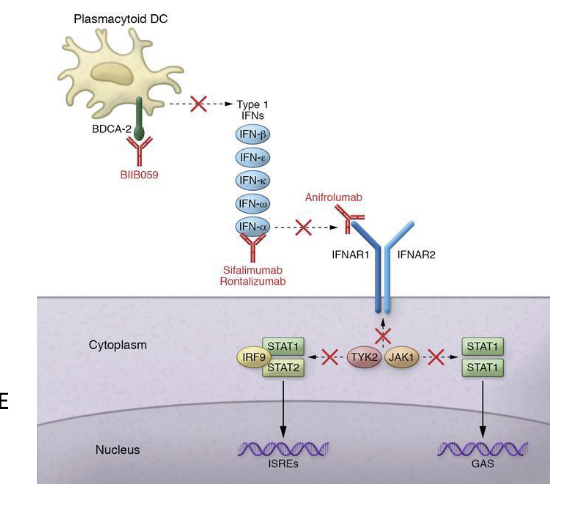
example of developing a therapeutic hypothesis based on protein interactions: saphnelo (anniforlumab)
saphnelo (anniforlumab) - astrazeneca
mAb used for treatment of systemic lupus erythematosus (SLE)
blocks the signaling of IFNAR1 → prevents downstream JAK-STAT signaling → reduces immune overactivation in lupus
binds the type I interferon receptor (IFNAR1)
blocks the activity of type I interferon (INF)
NO direct genetic evidence linking SLE and IFNAR1
missense variants in TYK2 associated with SLE
TYK2 = kinase that physically interacts with IFNAR1
quantitative proteomics
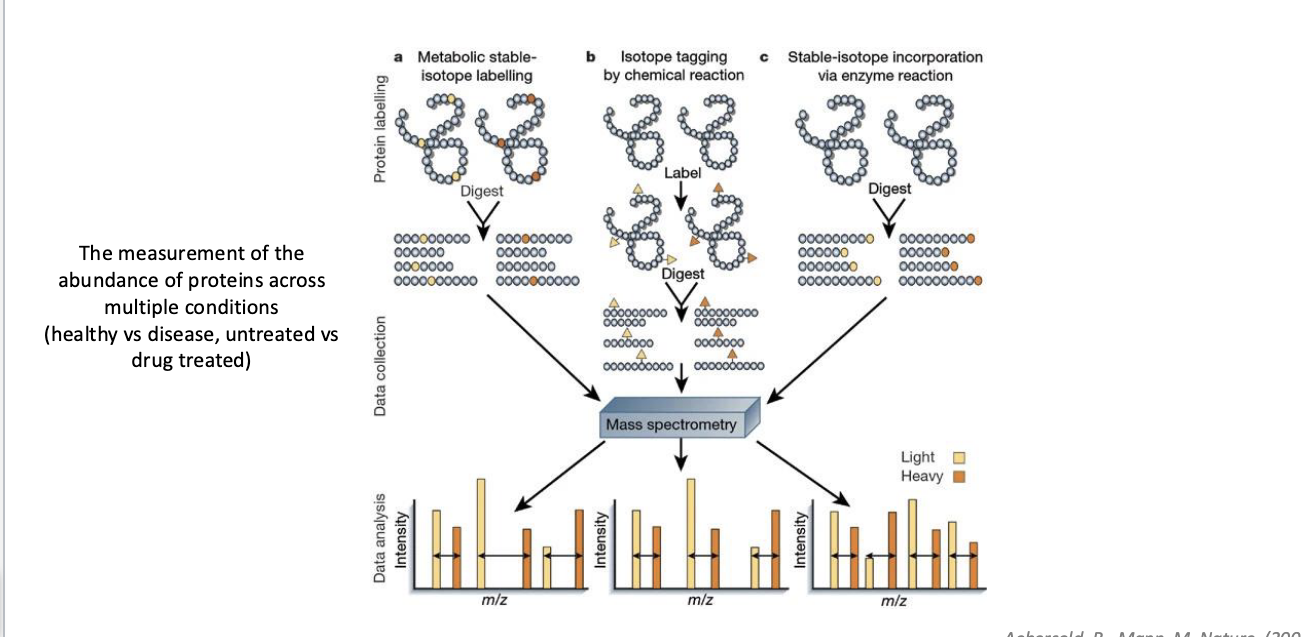
the measurement of the abundance of proteins across multiple conditions
healthy vs disease
untreated vs drug treated
3 approaches to quantify
metabolic stable isotope labeling
cells are grown in media containing “heavy” AA → proteins made in these cells incoporate the label during synthesis → labeled and unlabeled samples are mixed, digested, analyzed by MS
isotope tagging by chem rxn
proteins from different conditions digested first → peptides chemically labeled with isotopic tags
stable-isotope incorporation via enzyme rxn
stable isotopes is introducing during enzymatic digestion → label is added to peptides post-digestion
quantitative proteomics: stable isotope labeling by AA in cell culture (SILAC)
cells are either grown in
light isotope-containing media
heavy isotope-containing media + treatment
after you harvest + lyse cells → quantitate extracted protein → mix lysates → SDS-PAGE → excise bands → trypsin digestion → LCMS/MS: calculate ratio of heavy:light peptides; indicates relative protein abundance between conditions
applications of proteomics-based tech
detection of various diagnostic markers
candidates for vaccine production
understanding pathogenicity mechanisms
alteration of expression patterns in response to different signals/medications
interpretation of functional protein pathways in different diseases
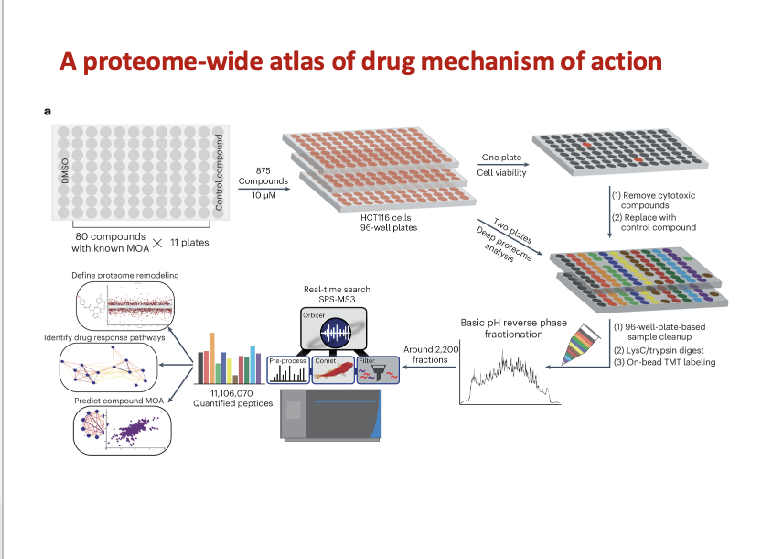
proteomics reveals small molecules’ secrets
high throughput quantitative proteomics
875 small molecule compounds
comprehensive profiling method to characterize the MOA of small molecules
each compound altered the expression of 15 proteins
revealed potential new targetes for commonly used small molecules
elucidated compound MOA and drug repurposing
revealed off-target effects which can increase efficiency and safety profiling in drug discovery
high-throughput techniques: genomics and transcriptomics
genomics
molecular read out → genes (DNA)
results → genetic variants; gene presence or absence; genome structure
technology → sequencing, exome sequencing
transcriptomics
molecular read out → RNA and/or cDNA
results → gene expression; gene presence or absence; splice sites’ RNA editing sites
technology → RT-PCR (reverse transcription-PCR) and RT-qPCR; gene arrays; RNA-sequencing

genomics - the large-scale study of DNA
genome = organism’s complete set of DNA
genomics = the study of all of a person’s genes (the genome)
sequencing = determine the exact order of bases in a strand of DNA
human genome project
the sequence of the human genome has been completed
started in 1990 and completed in 2003
led at the NIH by the national human genome research institute
comprised of approx
3 billion BP of DNA distributed among 24 chromosomes
23,000 genes
methods for sequencing DNA
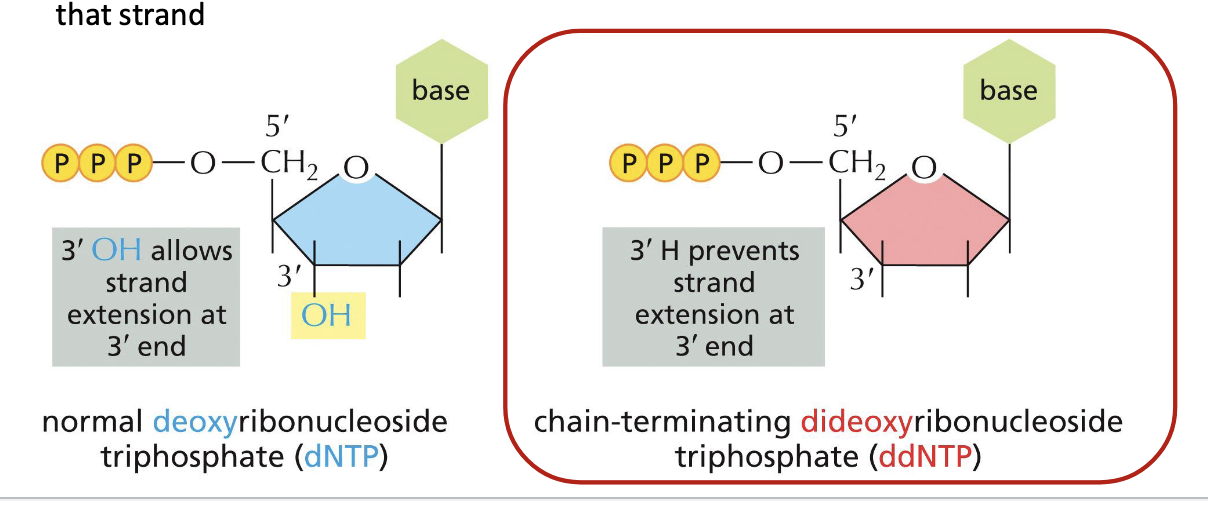
sanger sequencing or dideoxy sequencing
method using DNA polymerase along with special chain-terminating nucleotides called dideoxyribonucleoside triphosphates
when incorporated into a growing DNA strand, they block further elongation
normal deoxyribonucleoside triphosphate (dNTP) → 3’ OH allows strand extension at 3’ end
dideoxyribonucleoside triphosphate (ddNTP) → 3’H prevents strand extension at 3’ end
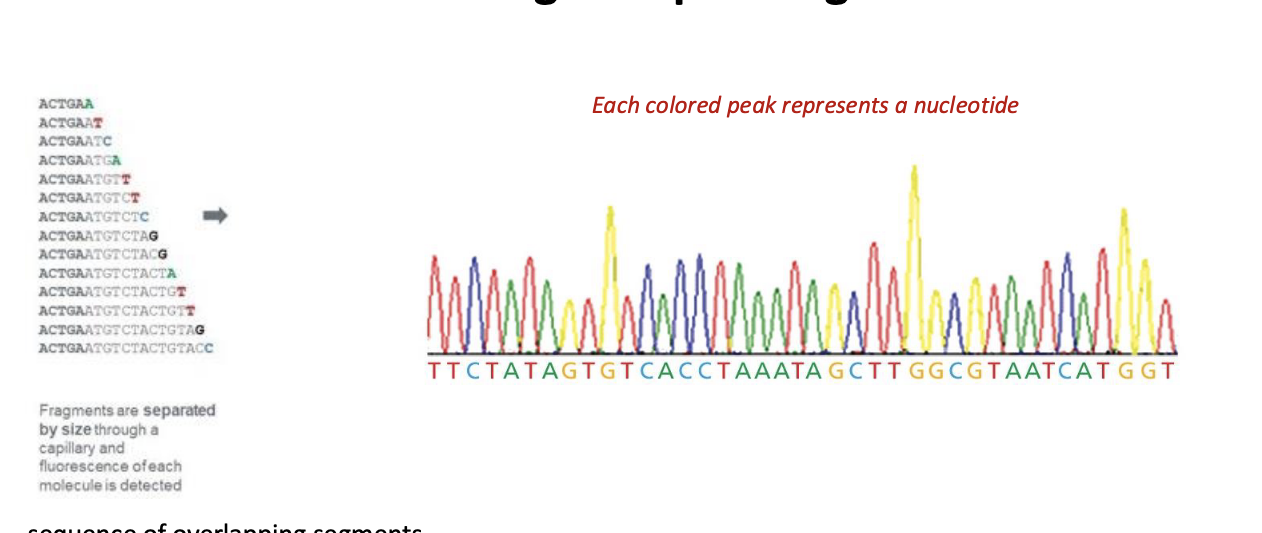
automated sanger sequencing of DNA process
4 different chain terminating nucleotides have been chemically tagged with a different colored fluroescent label
the rxn is loaded onto thin capillary gels which separates the rxn products into series of distinct bands
a detector records the color of each band
a computer translates the info → nucleotide sequence
diagram
single-strand DNA fragment to be sequenced → add primer → add small amounts of labeled chain terminating ddNTPs and add excess amounts of unlabeled dNTPs → mixture of DNA products each containing a chain-terminating ddNTP labeled with a specific fluorescent marker → products loaded onto capillary gel → size separated products are read in sequence

automated sanger sequencing of DNA results
each colored peak = nucleotide
sequence of overlapping segments
longer sequence are assembled from shorter pieces
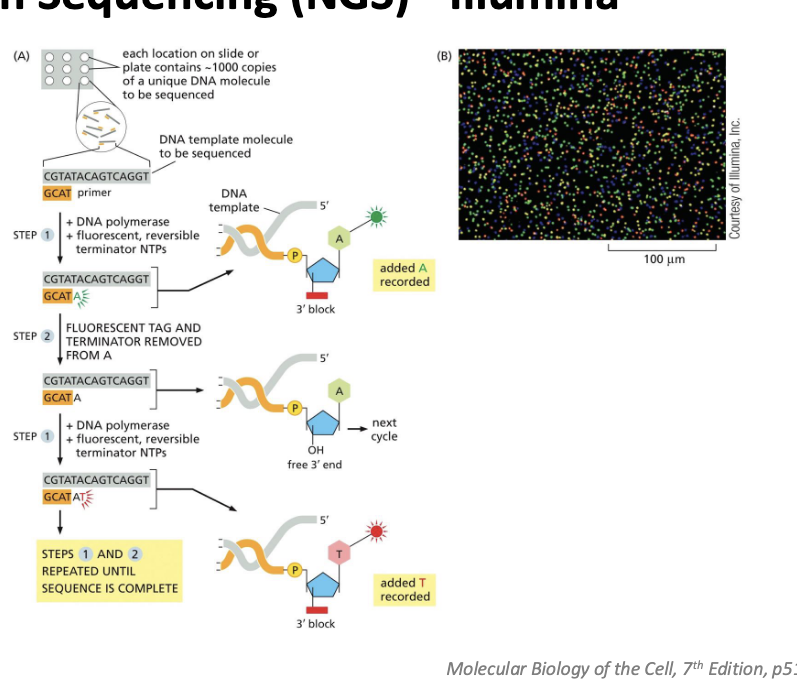
next generation sequencing (NGS) - illumina
allows sequencing of thousands of millions of DNA molecules simultaneously
high speed, reduced cost
a genome or other large DNA sample is broken into millions of short fragments
the sequences are amplified on a solid surface with covalently attached linkers
diagram
each location on slide or plate contains ~1000 copies of a unique DNA molecule to be sequenced
add DNA polymerase, fluorescent, reversible terminator NTPs → A is added and recorded
fluorescent tag and terminator removed from A → now have free 3’ OH
add DNA polymerase, fluorescent, reversible terminator NTPs again → added T is recorded
RNA-sequencing (RNA-seq)/transcriptomics
RNA-seq = method to detect the presence and quantitation of all the RNA molecules in a cell under specific conditions
isolate RNA from cells or tissue of interest
select for RNA by filtering for sequencing containing polyA tails
synthesize cDNA using reverse transcriptase
cDNA = edited DNA → just the parts of the cell actually used to make proteins
sequence cDNA molecules using an NGS method
use computational algorithms to assemble sequencing data
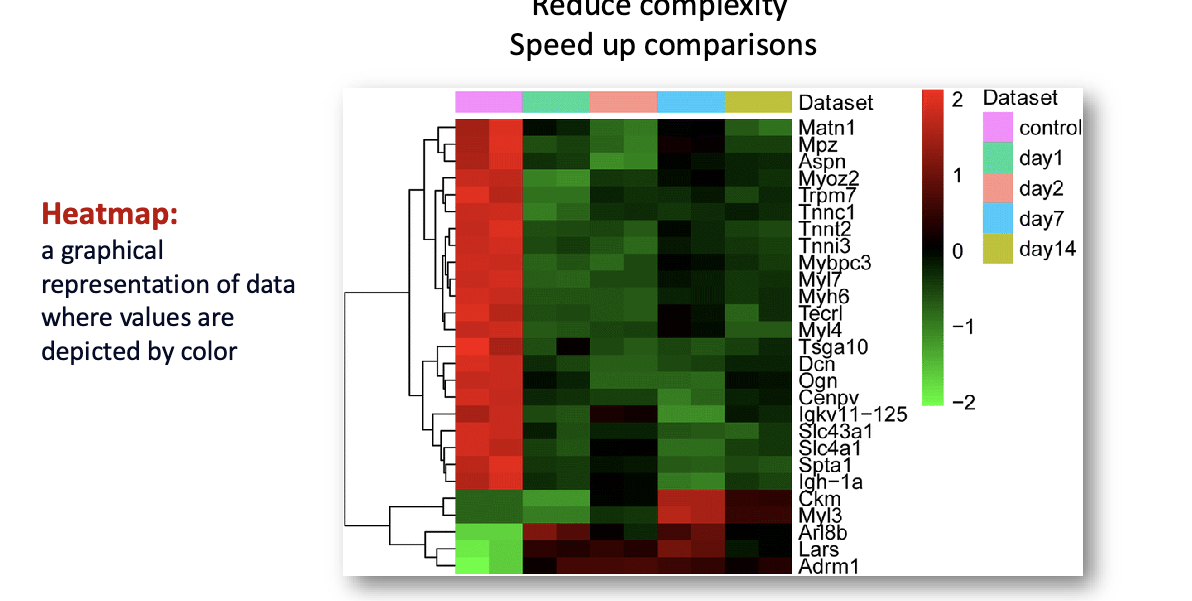
data analysis and visualization: heatmap/clustering
reduce complexity
speed up comparisons
heatmap = graphical representation of data where values are depicted by color
clustering = task of classifying N objects (proteins) into k groups (clusters) in such a way that the objects within a group are similar to each other but the groups are different from each other
clustering methods identify similar and distinct expression patterns
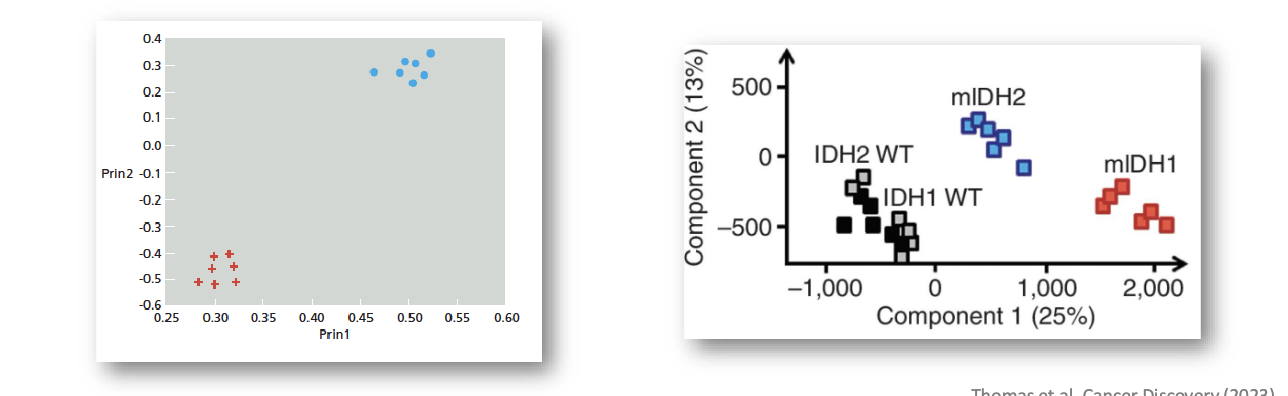
data analysis and visualization: principal component analysis (PCA)
method for combining the properties of an object
can simplify the data to minimize their effect
reduce the data to only 2 or 3 principal components
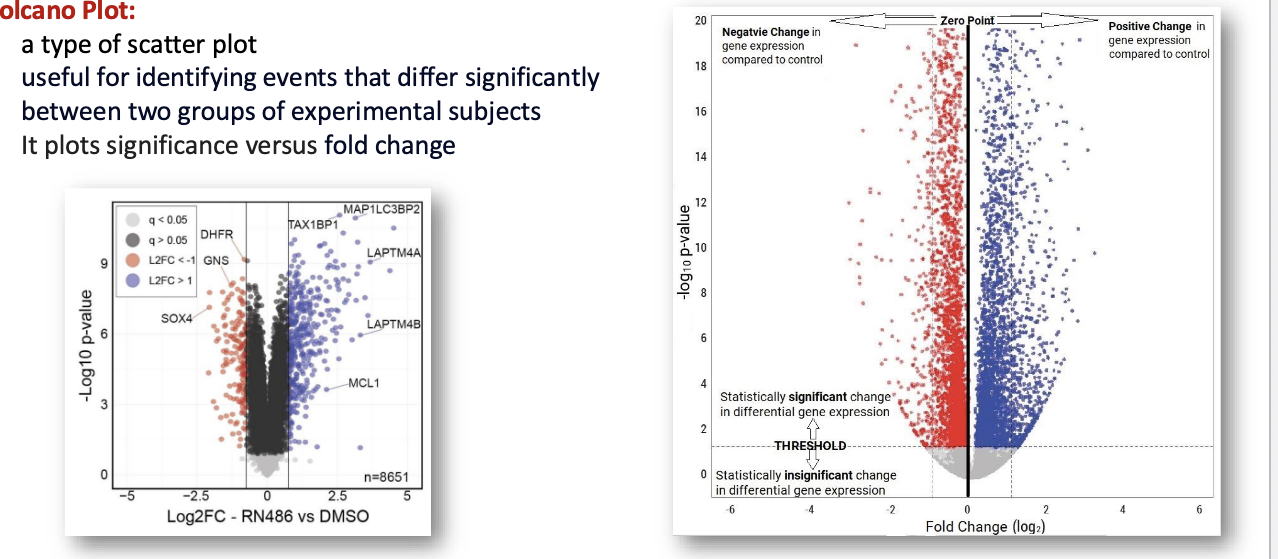
data analysis and visualization: volcano plot
type of scatter plot useful for identifying events that differ significantly between 2 groups of experimental subjects
it plots significance vs fold change
fold change = how much a gene’s expression has changed between 2 groups
lecture summary
bioinformatics = interdisciplinary scientific field tha tprovides sophisicated tools for managing large datasets and computational approaches that acclerate drug discovery and advance medical care
uniprot = central access point for extensive curated protein info
structural bioinformatics = helps predict 3D structures of biomolecules
proteomics = large scale study of proteins
mass spec for proteomics allows the detection of peptides and proteins in sample
quantitative proteomics methods = allows accurate measurement of protein abundance across multiple samples
methods for sequencing DNA: sanger and next generation sequencing
RNA sequencing = used to quantify gene expression in a sample
heatmaps, PCA analysis, volcano plots = statistical methods for data analysis and visualization