3. genomics and transcriptomicsn
1/13
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
14 Terms
What are the fields of genomics and transcriptomics and how do they differ
Genomics
study of whole organisms genetic material
Technologies like whole genome sequencing, whole exome sequencing, amplicon sequencing, and single cell genomics
Transcriptomics
Complete set of RNA transcripts produced BY GENOME
Involves whole transcriptome sequencing, targeting gene expression analysis, single cell transcriptomics, and spatial transcriptomics
While genomics provide DNA, transcriptomics show real time expression and regulation (RNA) of genes
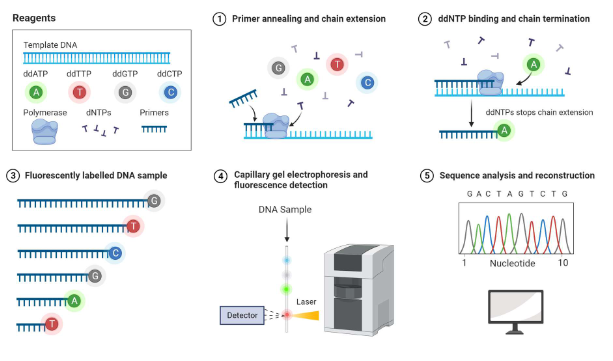
What is sanger sequencing
Selectively incorporates chain-terminating dideoxynucleotides (ddNTPs) during DNA replication
Relies on the condensation reaction where DNA polymerase extends the primer by adding deoxynucleotides
Used ddNTPs which lack the 3’-OH group, causing extension stopping and allowing fragments to be size separated to determine sequence
How does the chain termination method in Sanger sequencing work
Reaction begins with a primer annealed to a DNA template
DNA polymerase extends the chain by adding standard deoxynucleotides
When an dideoxynucleotiden(lacking the 3’OH group) is added, it prevents the addition of further nucleotides, terminating the chain
Result fragments of various lengths are then separated (usually by capillary electrophoresis) to read the DNA sequence
What is illumina sequencing (next-generation sequencing)
Involved library preparation where DNA/RNA fragments are lighted with adapters
Uses cluster generation on a flow cell, allowing massively parallel sequencing
The sequence-by-synthesis process records the incorporation of nucleotides in real time
a bioinformatics pipeline converts the raw data (typically stored in FASTQ files) into interpretable sequences.
Outline the main stages of Illumina sequencing and explain why each step is important.
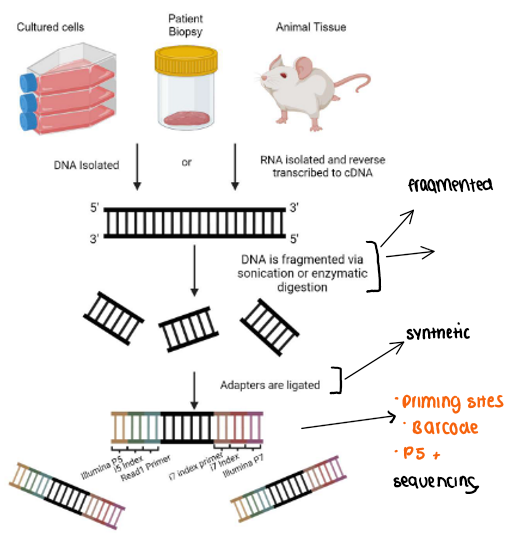
DNA or RNA isolation from human or animal tissues
library preparation
fragmentation of DNA/RNA samples
fragmented DNA is end-repaired to ensure all fragments have blunt ends
An adenosine (A) overhang is added to the 3’ ends to allow sufficient ligation of adaptors
cluster generation
illumina adaptors are added to complementary oligonucleotides
birdge amplication creates clusters of clonal DNA, which improves signal detection
priming sites
barcode (index) sequencing for multiplexing multiple samples
P5 + P7 flow sites for immunobilization during sequencing
detection via imaging
Bioinformatic analysis
raw data are output as FASTQ files containing sequence reads and quality scores
Define bioinformatics
leverages computational tools to manage, analyze, and interpret biological data such as DNA and RNA sequences.
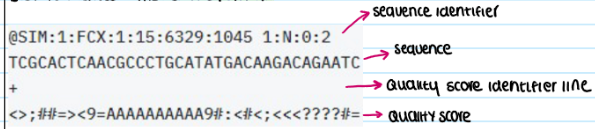
define the role of FastQ file formats in sequencing data analsysi
A standard text-based format that stores both nucleotide sequences and their corresponding quality scores.
hat are the key steps in a bioinformatics pipeline for transcriptomics data, and what tools are used at each stage?
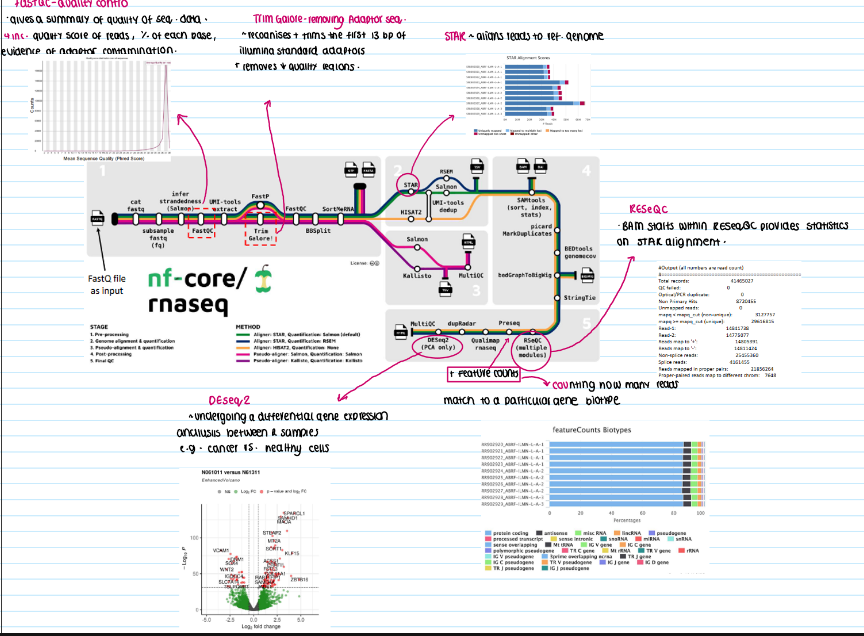
Quality control
Tool: FastQC
purpose: evaluates sequence quality scores (%) of each base and detects adapter contamination
Trimming of adaptor sequencing
Tool: Trim Galore
purpose: removes adapter sequences (e.g. the first 13 bp of illumina adapters) and low-quality regions
Alignment
Tool: Star aligner
purpose: maps reads to a reference genome, providing mapping quality and splice junction information
Alignment quality assessment
tool: RESeQC
purpose: generates detailed statistics on the alignment (e.g. total records, duplicate reads, unmapped reads)
feature counting:
tool: featurecounts
purpose: determines the number of reads mapping to specific genes or features
differential expression analysis
tool: DESeq2
purpose: compares expression levels between different sample groups (e.g. cancerous vs. healthy tissue) to identify altered genes
Why is quality control essential in sequencing,
Ensures the raw sequencing data is accurate and reliable.
Identifies issues such as adapter contamination, low base quality scores, and biases which can adversely affect downstream analysis
what specific metrics does FastQC provide?
Quality Score Distribution: Assesses the overall quality of bases across reads.
Base Composition: Evaluates the percentage of each nucleotide at every position.
Adapter Content: Detects the presence of non-biological adapter sequences in the data.
Read Length Distribution: Verifies consistency in the length of sequencing reads.
Explain the purpose and importance of alignment in the transcriptomics pipeline using STAR.
Purpose of Alignment:
To map each sequencing read to its corresponding location in a reference genome.
Provides the basis for quantifying gene expression and identifying splice junctions.
mportance:
Accurate alignment is critical for ensuring that the subsequent counts (via FeatureCounts) are reliable.
It enables the detection of anomalies such as non-unique mappings (multiple mapping) and helps to assess overall data quality using alignment quality tools like RESeQC.
What is differential gene expression analysis, and how does DESeq2 facilitate this process?
Differential Gene Expression Analysis:
A statistical method used to determine differences in gene expression levels between sample groups (e.g., diseased vs. healthy).
DESeq2:
Performs normalization of count data to account for sequencing depth and technical variation.
Estimates variance and uses a robust statistical model to determine which genes are significantly upregulated or downregulated.
Provides visualizations such as volcano plots
How can the integration of genomics and transcriptomics data advance pharmacogenomics and personalized medicine?
Integration Strategy:
Combining genomic data (e.g., mutations, copy number variations) with transcriptomic data (e.g., gene expression profiles) provides a comprehensive view of an individual's molecular landscape.
Pharmacogenomics Impact:
Enables prediction of drug efficacy based on a patient’s specific omic profile.
Helps in identifying biomarkers for drug response or resistance.
Discuss a case study or literature example that leverages next-generation sequencing for pharmacogenomics or personalized medicine
creation of a living biobank of breast cancer organoids.
Breast cancer organoids are derived from patient tumors and maintained ex vivo.
Next-generation sequencing is used to profile both genomic mutations and transcriptomic expression patterns.
The data helps capture disease heterogeneity, providing insights into which pharmacological agents may be most effective.