CBIO1-9
1/444
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
445 Terms
cancer
a group of diseases where cells divide and grow uncontrollably, forming a malignant tumour and invading other parts of the body
malignant cell
cell has the properties of a cancer cell
benign tumour
tumours that are not cancerous or malignant but can still cause problems due to their growth, but they do not spread to other parts of the body
neoplasia
the presence or formation of new, abnormal growth of tissue
metastasis
movement of cancer cells from their site of origin to a new location
somatic mutation
occurs in any cell of the body after conception, can only be passed on to daughter cells
germline mutation
occurs in germ cells and is therefore passed into all cells in the body of the offspring as well as to their offspring
apoptosis
programmed cell death to eliminate faulty cells
angiogenesis
blood vessel formation
biopsy
sample of cells or tissue removed for examination to determine presence of cancer and/or stage of disease
carcinogen
substance which increases risk of cancer upon exposure
Describe how the development of cancer is a multi-step process
Incidence of many cancer types increases with age, suggesting that it occurs as a result of accumulative damage
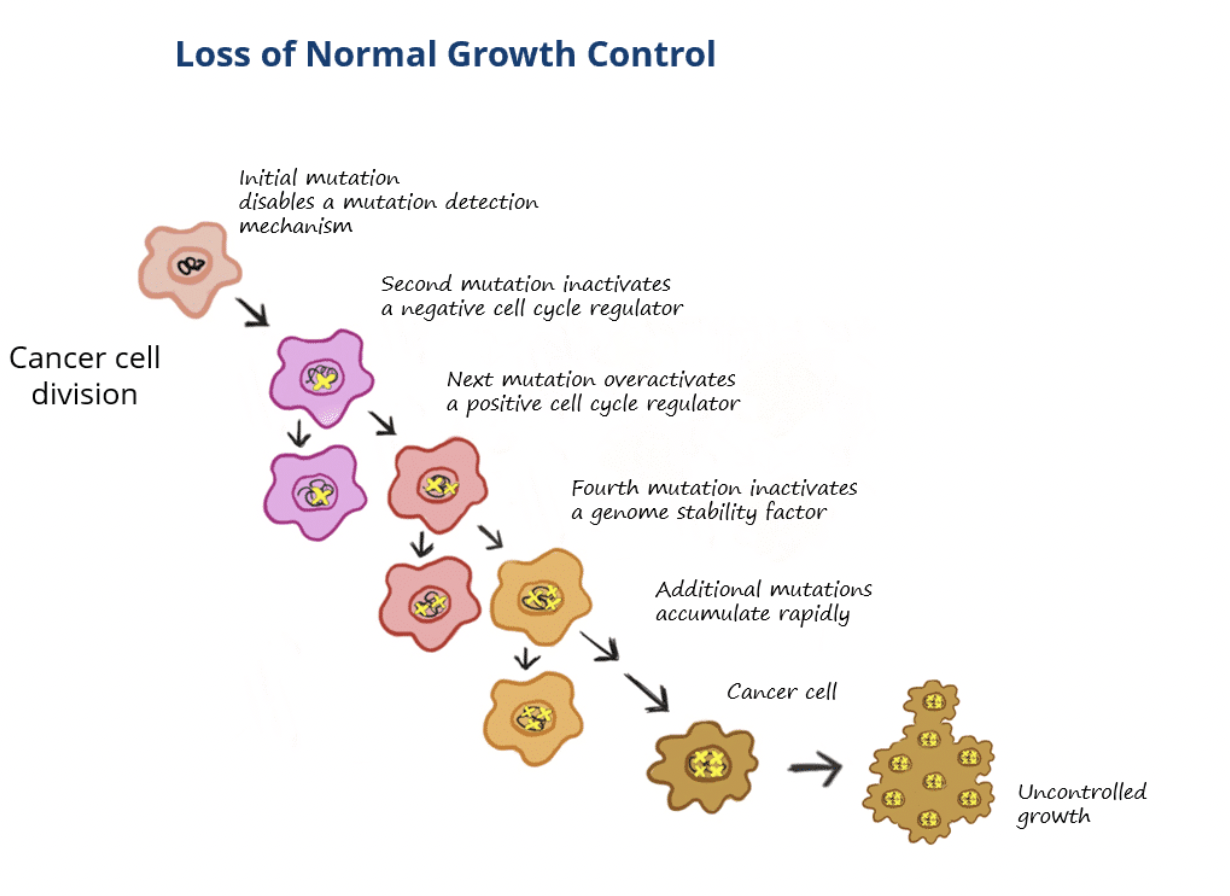
Usually 3-7 ‘events’ must occur for normal cells to transform into cancerous cells
These events are genetic alterations
The type and combination of mutations widely vary between tumour types and between the tumours themselves
Pathological and genetic/genomic analyses of various organ sites reveals that genetic alterations represent the intermediate steps from normal cell function into premalignant stages to cancer
If a mutation occurs then proteins in the cell can detect the defects, which may get repaired or the cell may undergo apoptosis instead of continuing to divide as cell division is a stringently controlled event
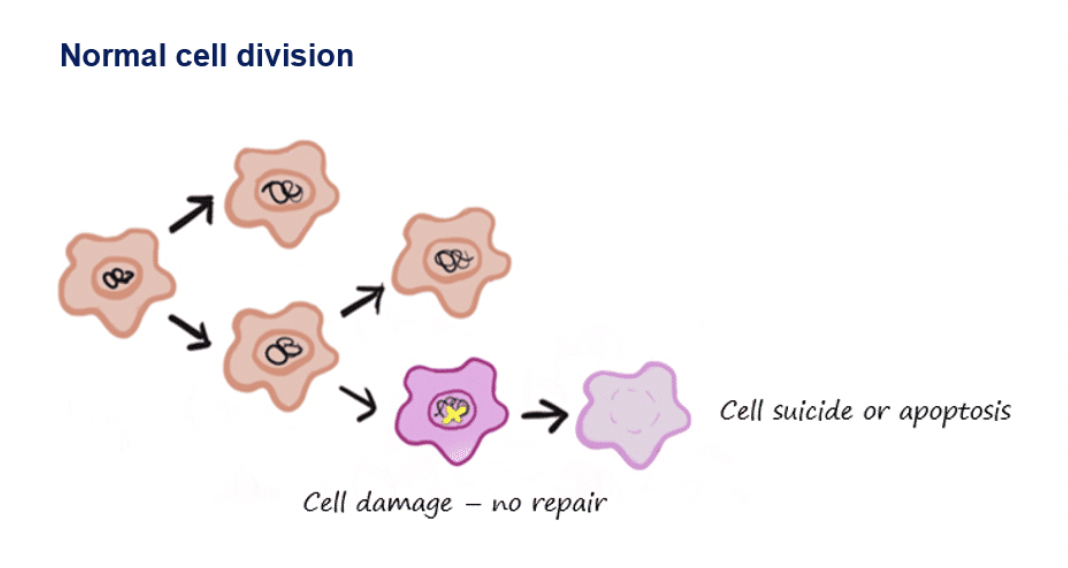
In cancer cells, multiple mutations are needed to overcome this protective mechanism, for example:
The first mutation may occur in a gene encoding a protein that detects mutations which disables protection
This in itself does not cause cancer but if a second mutation arises that causes the cell to grow fast, eg by inactivating a cell cycle regulator, then these cells will be selected for
carcinoma
Most common type of cancer (80-90% of diagnoses)
Arises from epithelial cells that cover external and internal body surfaces
Lung, breast, prostate and colon are the most common carcinomas in the West
These are examples of adenocarcinomas which develop within an organ or gland
Another common suptype is squamous cell carcinoma which is a type of skin cancer arising in the top layer of the skin or epithelial covering of certain organs
sarcoma
Much more rare
Arise in supporting tissues of the body, eg. bone, cartilage, fat, connective tissue, muscle
lymphoma
One of the most common cancers in children and young adults
Arise in white blood cells (lymphocytes)
Two main types are Hodgkin’s lymphoma and non-Hodgkin’s lymphoma (more common)
Non-Hodgkin’s is divided into B-cell and T-cell lymphoma
myeloma
Arises in the immune system in antibody-producing plasma cells
leukaemia
Cancers of the blood, not solid tumours
Arise in immature blood cells that grow in bone marrow and accumulate in large numbers in the bloodstream
Acute leukaemia progresses very quickly while chronic leukaemia is less aggressive and may not cause symptoms for many years
Lymphocytic leukaemia affects lymphocytes
Myelogenous leukaemia affects myeloid cells which normally develop into various mature blood cells, eg. RBCs, WBCs and platelets
Where can other types of cancers occur?
Other types of cancers can occur in different body parts, including cancers of the CNS (brain and spinal cord - most common in the brain is a glioma) and germ cell tumours (arising in pluripotent cells in gonads)
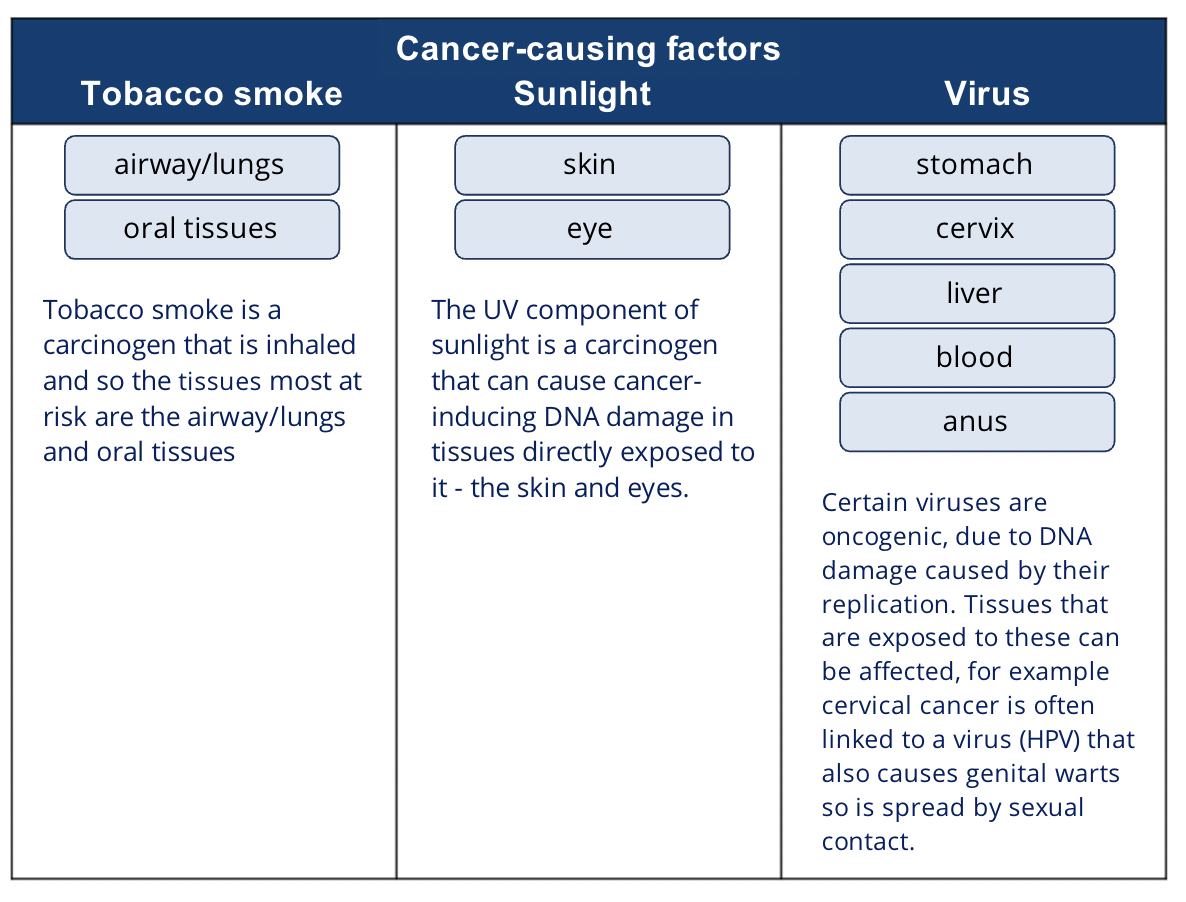
What organs are affected by the cancer causing factors tobacco smoke, sunlight and viruses?
How can ageing affect the risk of cancer?
Most cancer types become more common with age
The changes in the cell that make it cancerous take a long time to develop
The longer we live, the more time there is for genetic mistakes to happen in cells
How can genetic predisposition affect the risk of cancer?
Sometimes a person is already born with cancer-causing mutations (i.e. a germline mutation)
This increases the statistical likelihood of developing cancer
How can the immune system affect the risk of cancer?
Compomised immune system increases the likelihood of developing some cancers (eg. lymphomas and cancers caused by viruses)
This could be due to existing medical syndrome affecting immunity, immunosuppressant medications (eg. for organ transplant patients or in autoimmune diseases), or diseases such as HIV/AIDS
How can body weight and diet affect the risk of cancer?
Estimated that 1/3 of UK cancer cases are linked to smoking, alcohol, diet or being overweight
Diets with high red and processed meat, saturated fats and dairy are associated with higher cancer risk
Alcohol can directly promote DNA damage or affect the absorption of vitamins and other important nutrient, and affect the breakdown of hormones
How can the environment affect the risk of cancer?
Apart from environmental carcinogens, natural radiation from sunlight can cause cancer through direct DNA damage
UVA and UVB are both linked to melanoma development
How can viral infection affect the risk of cancer?
Certain viruses can cause genetic changes in cells that make them more likely to become cancerous
Examples of cancers and viruses that are linked (though not all people with that cancer had that virus and vice versa):
Cervical cancer and other cancers of the genital and anal area linked with human papilloma virus (HPV)
HPV also may lead to oropharyngeal cancer and non-melanoma skin cancers
Primary liver cancer linked with Hepatitis B and C viruses
Lymphomas and Epstein-Barr virus
T cell leukaemia in adults and human T cell leukaemia virus
How can bacterial infection affect the risk of cancer?
Helicobacter pylori infection of stomach causes inflammation of stomach lining which increases risk of stomach cancer
H. pylori can be treated with combination of antibiotics
Further research shows that certain types of bacteria in the digestive system produce substances that increase risk of eg. bowel cancer or stomach lymphomas
Bacterial infections can often be cured with antibiotics which can reduce risk of these cancers
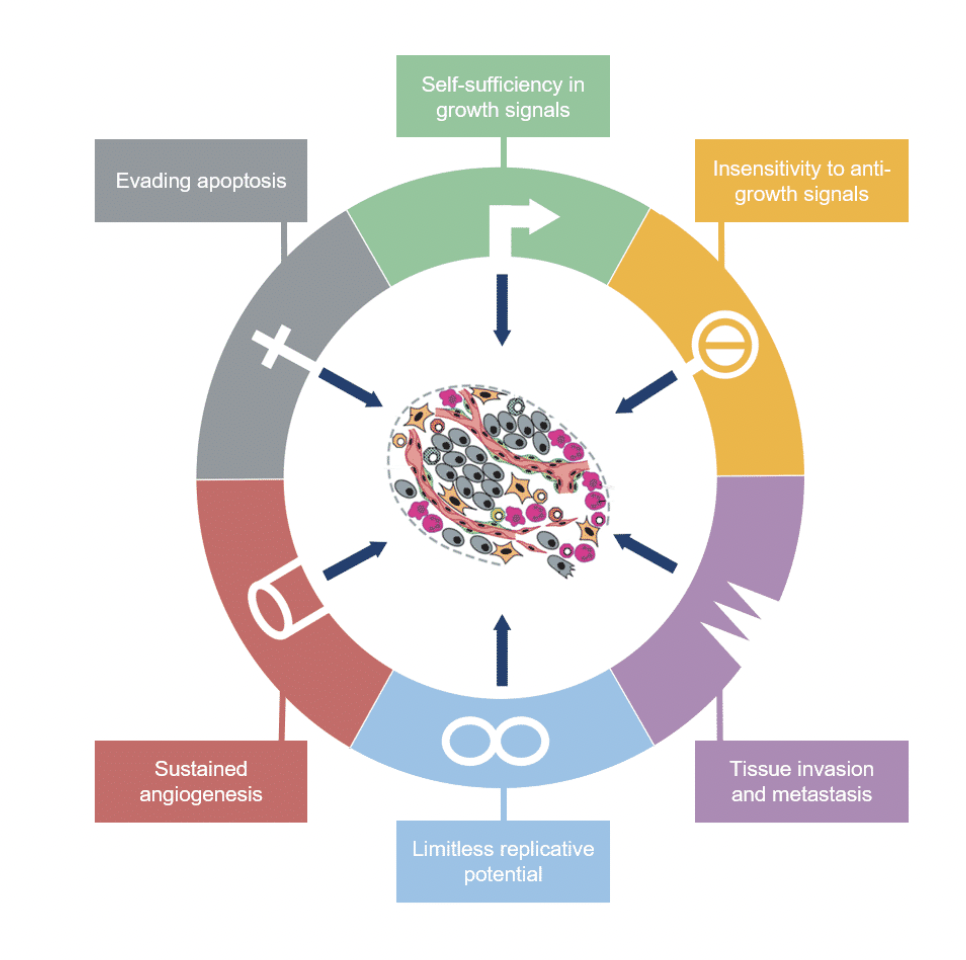
What are the six hallmarks of cancer proposed by Hanahan and Weinberg?
cancer stem cells
cancer stem cells in some cases are believed to be the progenitor cells from which the tumour originates
pericytes
integral components of blood vessel walls which wrap around endothelial layer and regulate capillary blood flow
cancer-associated fibroblasts (stromal cells)
found within the vicinity or microenvironment of a tumour can promote tumorigenesis eg. by secreting paracrine factors
Outline self-sufficiency in growth signals as a hallmark of cancer
Normal cells need external signals in the form of growth factors to grow and divide
Growth signals are transmitted through receptors in the cell membrane and when signals are absent the cells become quiescent and cease to grow
Cancer cells can grow and divide without external signals using one or more mechanisms:
Alteration of extracellular growth signals
Alteration of response to growth factors
Alteration of intracellular response
How can cancer cells grow and divide without external signals by altering extracellular growth signals?
Some cancer cells acquire the ability to make their own growth factors which creates a positive feedback loop
Eg. glioblastomas produce their own platelet-derived growth factors (PGDF) and sarcomas produce their own transforming growth factor-α (TGF-α)
How can cancer cells grow and divide without external signals by altering their response to growth factors?
Some cancers overexpress receptors which make them hyper-responsive to levels of growth factors that would not normally trigger growth
Eg. the epidermal growth factor receptor (EGF-R/erbB-1) is overexpressed in stomach, brain and breast cancers while HER2/neu receptor (erbB-2) is often overexpressed in stomach and breast cancers
How can cancer cells grow and divide without external signals by altering their intracellular response?
In some cancer cells the downstream cytoplasmic components that receive and process growth factor signals can signal without stimulation from upstream regulators
Eg. SoS-Ras-Raf-MAPK cascade - in 25% of human tumours, mutant Ras proteins trigger the downstream cascade in the absence of external growth signals
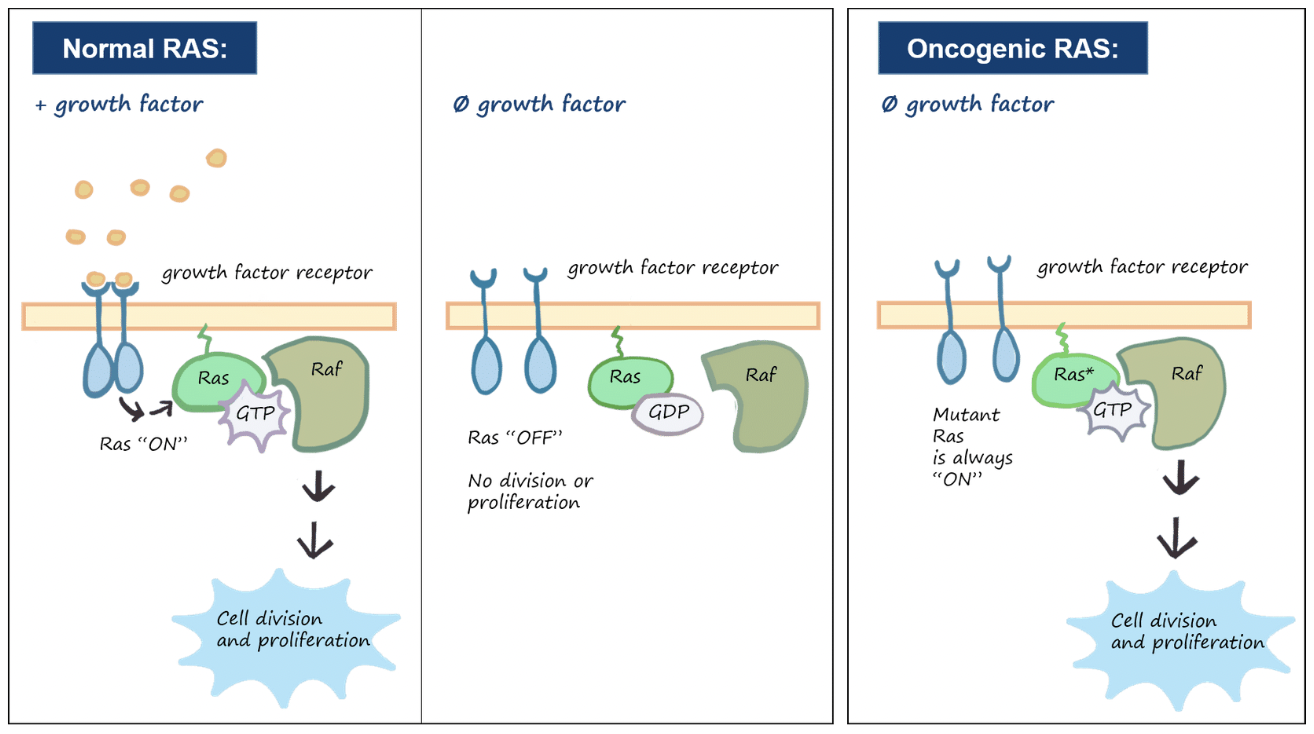
In some cases tumour cells can co-opt neibouring cells to produce growth factors that enable the cancer cells to divide
Outline insensitivity to anti-growth signals as a hallmark of cancer
Growth inhibitors in the surrounding environment, ECM and on neighbouring cell surfaces help regulate growth of normal cells
Growth inhibitors often act via transmembrane receptors and the transmitted signal acts on the cell cycle click to interrupt mitotic division
Cancer cells are resistant to growth-preventing signals from neighbouring cells
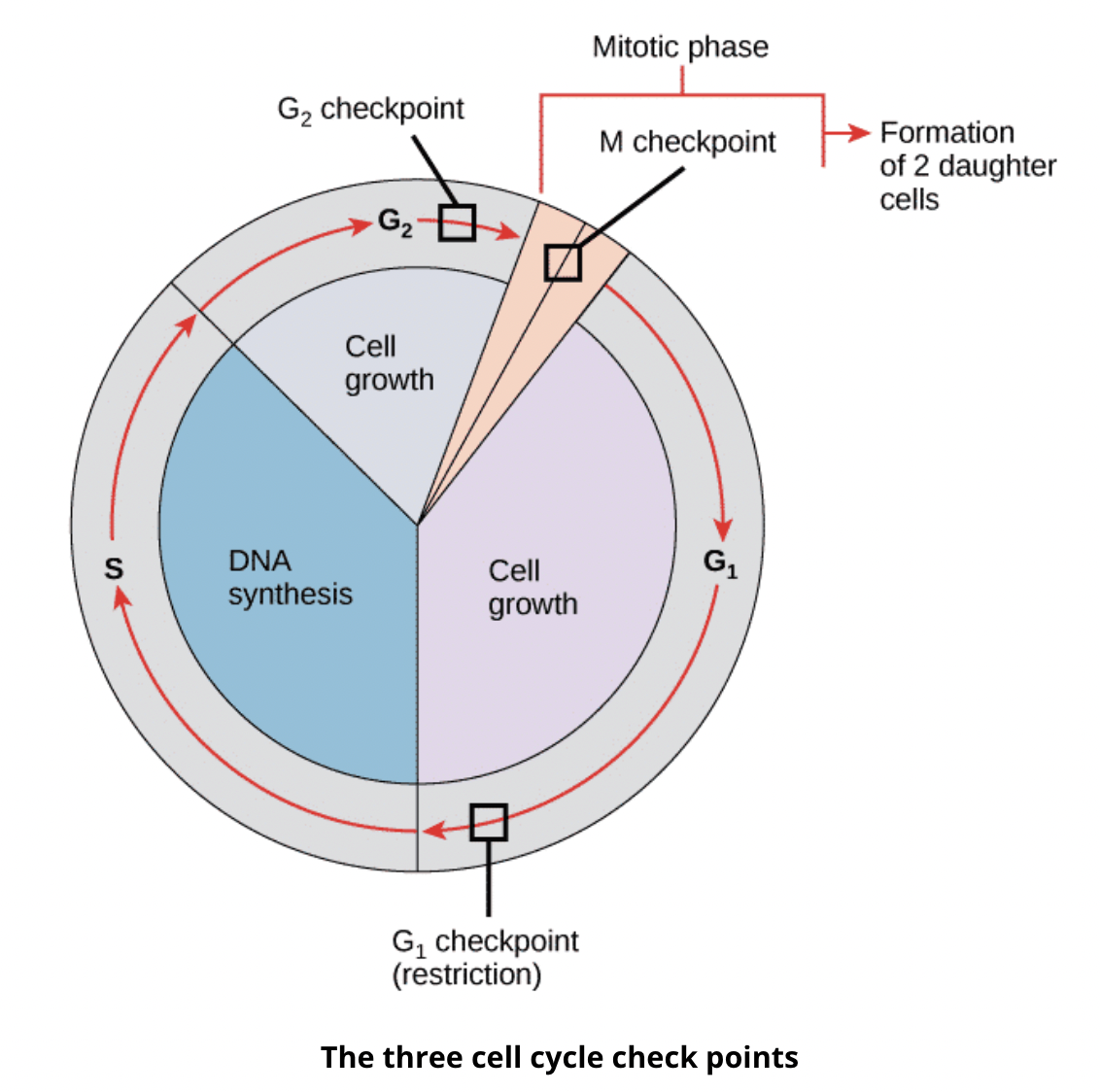
How can damage to the retinoblastoma protein lead to tumourigenesis?
The transition from G1 to S is a checkpoint, where if there is a reason for cells to not divide such as DNA damage, checkpoint proteins like retinoblastoma protein (Rb) activates the checkpoint to cause cell cycle arrest until the damage is repaired
If the damage can not be repaired then the cell will apoptose
If Rb itself is damaged, the damaged cell would continue to divide
Describe the extrinsic apoptotic pathway
T lymphocytes express Fas ligand (Fas L) on their cell surface
Fas L binds to Fas receptors on the surface of the target cell
This triggers a cascade of intracellular events resulting in apoptosis
Sequence of events is mediated by Fas associated death domain (FADD)
In the final step of the pathway, caspases activate each other in a self-amplifying caspase cascade, initiating the proteolytic breakdown of cellular materials causing apoptosis
Describe the intrinsic apoptotic pathway?
Regulated by maintaining a balance of anti-apoptotic proteins (eg. Bcl-2 and Bcl-x) and pro-apoptotic proteins (eg. Bax and Bak) in the mitochondrial membrane
Anti-apoptotic proteins bind with pro-apoptotic proteins in healthy cells to inhibit each other’s actions
If a cell is damaged or no longer receives survival signals then Bcl-2 and Bcl-x get blocked, allowing Bax and Bak to form channels in the mitochondrial membrane
This allows mitochondrial substances such as cytochrome C to leak into the cytoplasm
Leaked cytochrome C binds to APAF-1 proteins to create a compound that activates the caspase cascade, resulting in apoptosis
What is p53 and why is it important for cancer cells?
p53 is a transcription factor governing anti-proliferative cell programmes
p53 is an important tumour suppressant which blocks angiogenesis, induces cell cycle arrest and DNA damage repair, or identifies irreparable DNA damage and signals for the cell to apoptose
Defective p53 or absence of it allows the cell to avoid apoptosis and replicate defects and accumulate DNA damage
Outline limitless replicative potential as a hallmark of cancer
Even when uncoupled from growth-promoting/inhibitory signals, most cells do not divide uncontrollably as they have an inbuilt limit (Hayflick limit) which limits their multiplication to 60-70 doublings, after which they become senescent
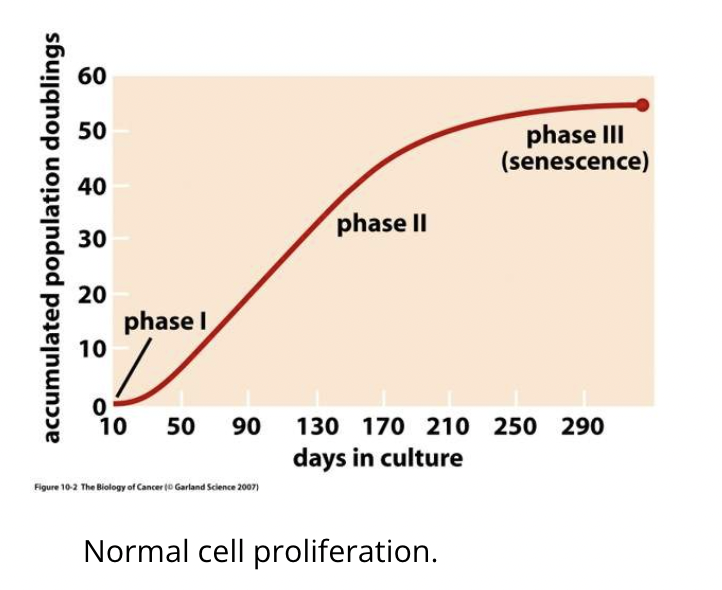
Senescence is initiated due to telomere shortening (by 50-100bp at each division due to inability of polymerase to replicate right to the ends)
The limit is reached when telomeres are eroded and no longer protect the chromosome
Telomerases exist to maintain telomere length and many cancer cells upregulate its expression to avoid progressive telomere shortening and avoid triggering senescence
Outline sustained angiogenesis as a hallmark of cancer
Angiogenesis is the formation of new blood vessels by branching from existing ones
This is essential for cells to receive enough oxygen and nutrients and to carry away wates
Almost all cells in a tissue must be within 0.1mm of a capillary blood vessel
Without dedicated blood supply tumour growth is limited to the size of a pinhead, but cancer cells acquire the ability to initiate angiogenesis
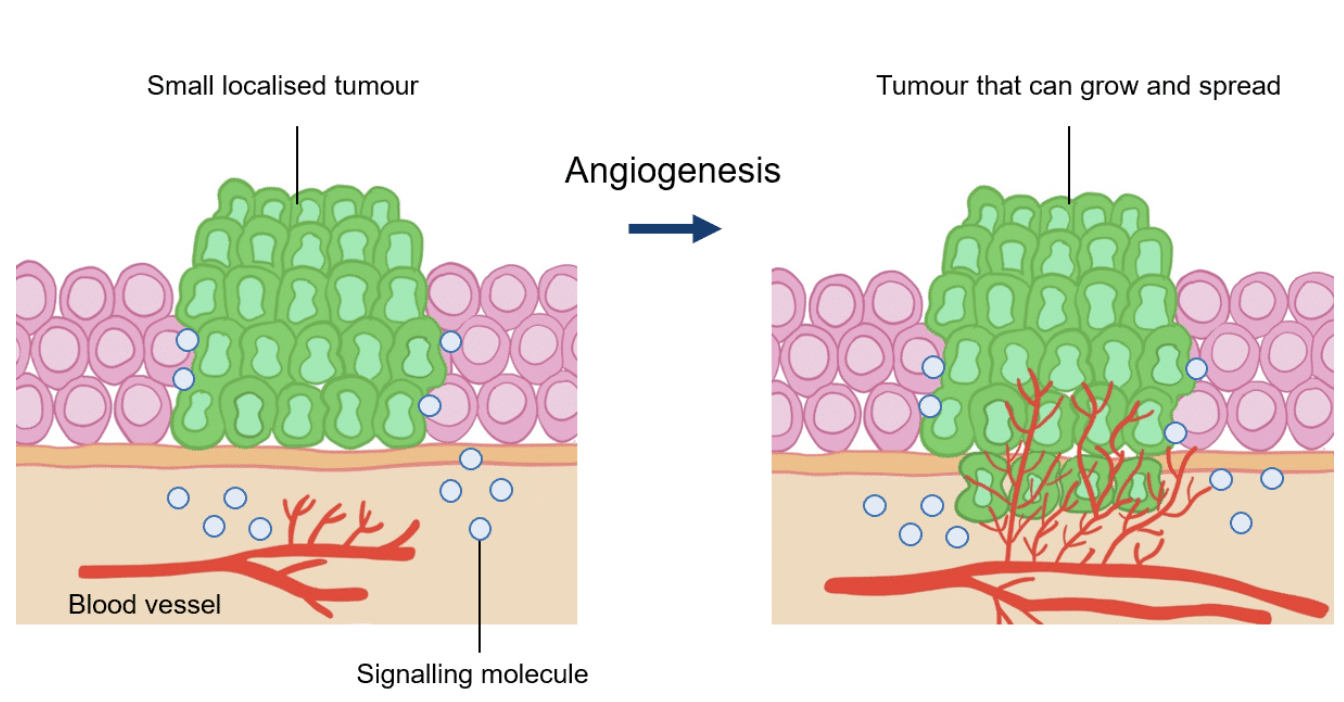
Tumours can release growth factors such as vascular endothelial growth factor (VEGF) which binds to surface receptors of endothelial cells to trigger the formation of new blood vessels
Endogenous inhibitors of angiogenesis, eg thrombospondins, which can be downregulated in çancer cells
What is the angiogenic switch?
The angiogenic switch is a discrete step in tumour development which may occur at different stages of tumour progression, based on the nature of the tumour and its microenvironment
It refers to the point where the balance between pro- and anti-angiogenic factors shifts to favour pro-angiogenesis
Avascular tumours reach a steady state where there is a balance between proliferation and apoptosis, resulting in no net growth
Oxygen and nutrient supply is required via the angiogenic switch for growth in cell mass
What are the steps of the angiogenic switch?
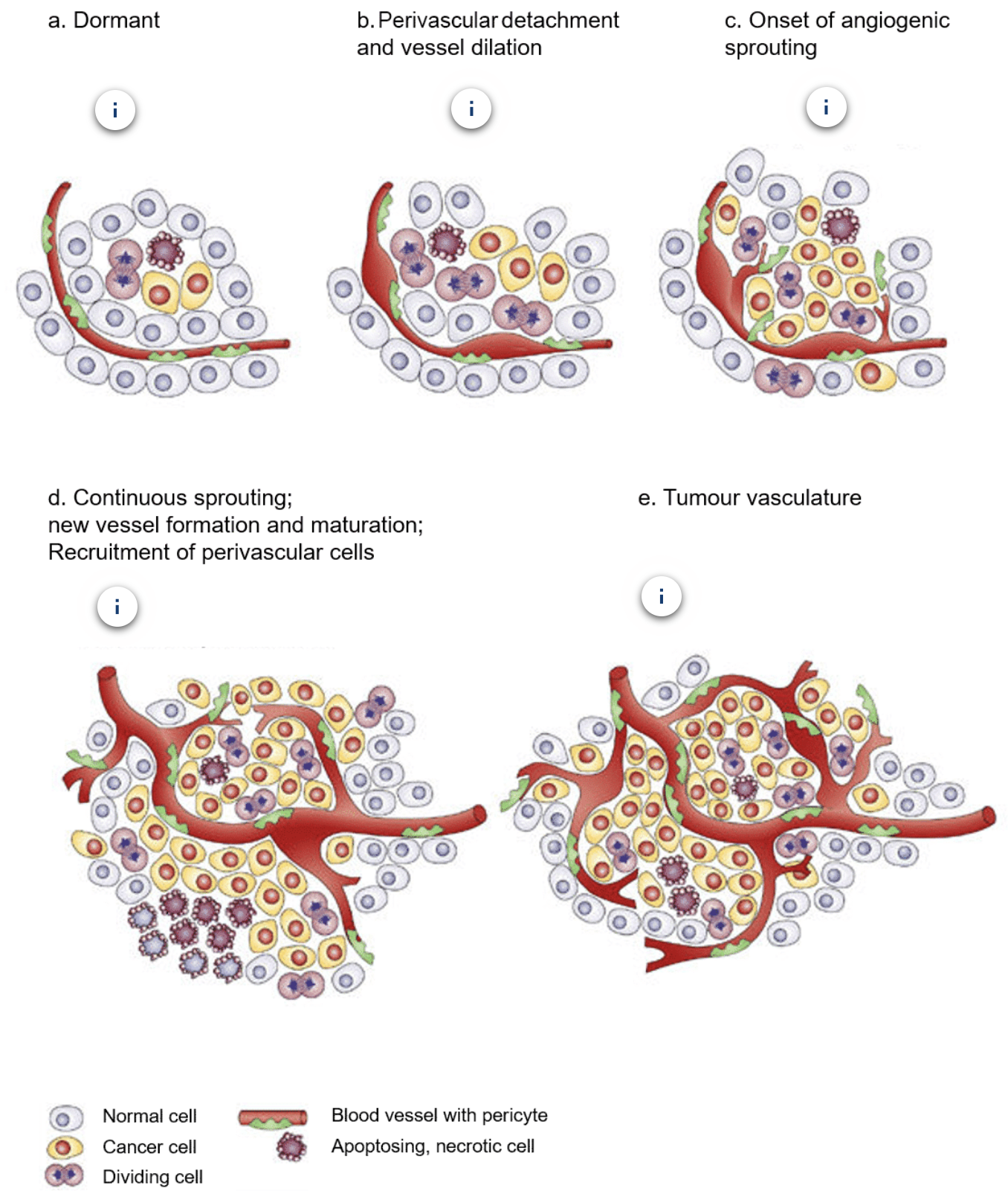
a. Most tumours start growing as avascular nodules until they reach a steady state of proliferating and apoptosing cells
b. Angiogenic switch occurs to facilitate exponential tumour growth, beginning with perivascular detachment and vessel dilation
c. Perivascular detachment and vessel dilation is followed by angiogenic sprouting
d. New vessels form and mature and perivascular cells get recruited
e. Blood vessel formation continues as long as the tumour grows and the blood vessels specifically feed hypoxic and necrotic areas of the tumour to provide essential nutrients + oxygen
Outline tissue invasion and metastasis as a hallmark of cancer
Primary tumour masses spawn ‘pioneer cells’ which invade adjacent tissues and can travel to distant sites through the circulatory sites, establishing metastases
Newly formed metastatic tumours are amalgams (mixture/combination) of cancer cells and normal supporting cells conscripted from the tissue
To break away from the tumour the cells must overcome cell-cell adhesion mediated by molecules such as E-cadherin
Migrating cancer cells encounter new environments and different ECM proteins and may alter integrin expression to accomodate these
What is the role of E-cadherin expression cancer?
E-cadherin forms bridges between cells by contacting intracellular proteins called catenins, strengthening links between cells by forming adherens junctions
E-cadherin is often downregulated in cancer
Upon colonisation of new sites the downregulation of E-cadherin is somewhat reversed to reform contacts with neighbouring cells
What is the role of integrin expression cancer?
Integrins facilitate cell-ECM adhesion with substrate preferences
Migrating cancer cells preferentially express those integrins which facilitate tissue invasion by binding to a wider range of substrates
Some integrins facilitate matrix degradation by certain proteases which is needed for invasion into stroma, across blood vessel walls and through epithelial layers
What are the new emerging hallmarks of cancer according to a review in 2011?
Deregulating cellular metabolism
Avoiding immune destruction
Genome instability and mutation
Tumour promoting inflammation
What are the new emerging hallmarks of cancer according to a review in 2022?
Unlocking phenotypic plasticity
Non-mutational epigenetic reprogramming
Senescent cells
Polymorphic microbiome
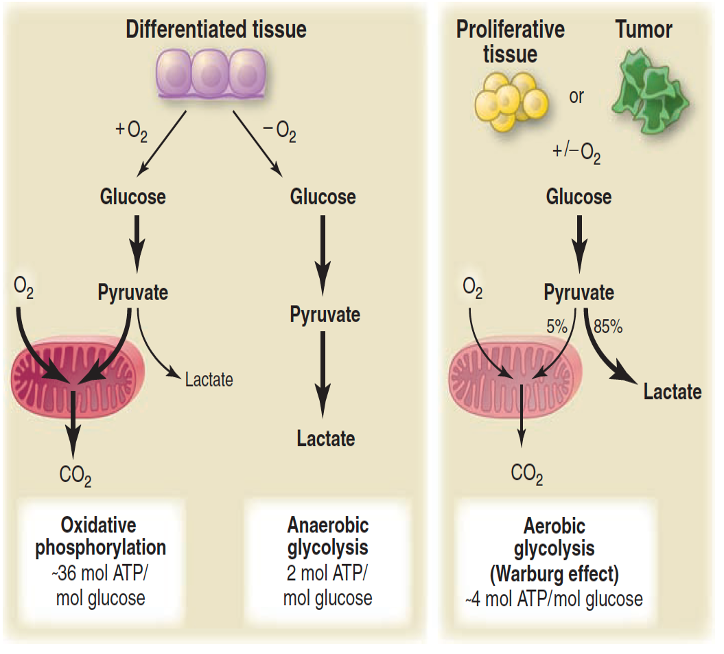
Warburg effect
Refers to the fact that cancer cells, prefers fermentation as a source of energy rather than the more efficient mitochondrial pathway of oxidative phosphorylation
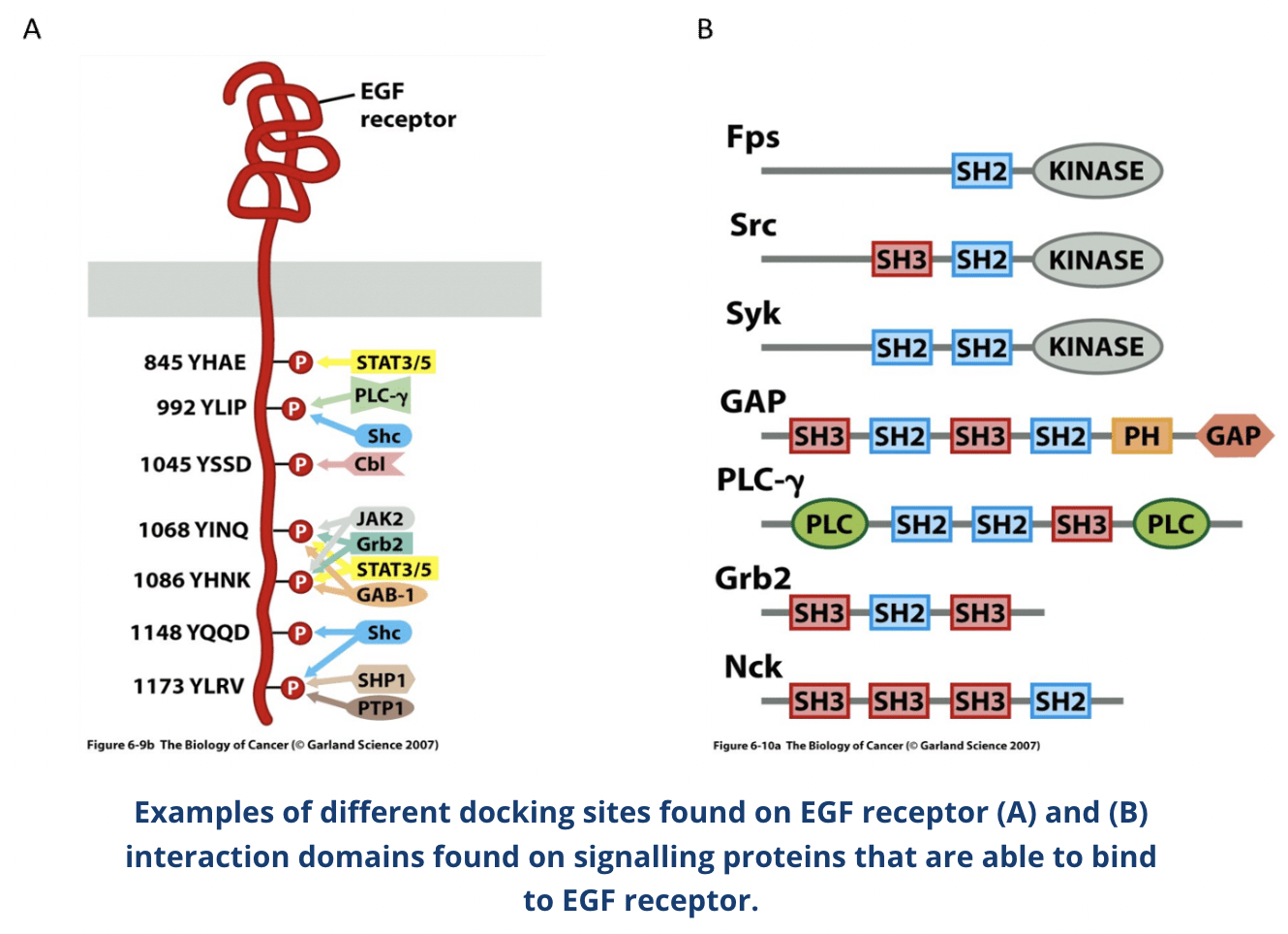
Describe the EGFR subfamily
Epidermal growth factor receptor (EGFR, aka ErbB-1 and HER1) is the first discovered, most studied and best characterised member of the RTK family
ErbB family consists of four related receptors: EGFR (aka ErbB-1 and HER1), ErbB-2 (HER2), ErbB-3 (HER3), and ErbB-4 (HER4)
ErbB receptors are activated by growth factors of the epidermal growth factor (EGF) family and have disulphide bonds that determine binding specificity
Contain structural motifs such as immunoglobulin (Ig)-like domains, heparin-binding sites and glycosylation sites
ErbB receptors bind to each other in different combinations, for example ErbB2 does not bind to ligands and instead acts as a co-receptor for other members of the ErbB subfamily
EGFR is found in various organs and abnormal expression or activation of it is associated with various cancers
How do RTKs work?
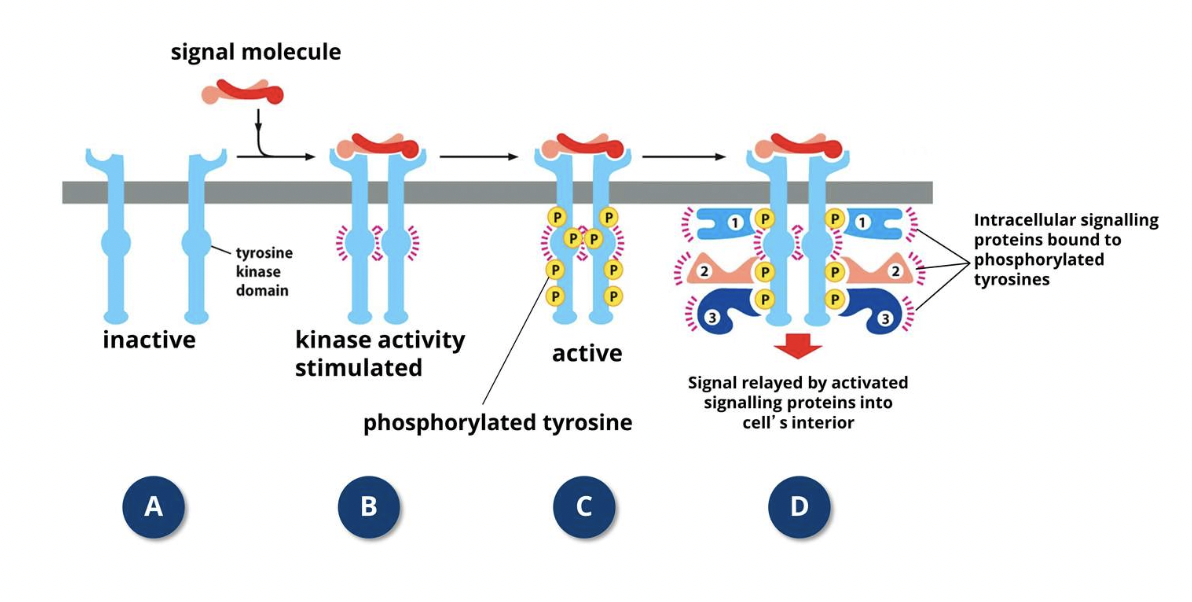
RTKs respond to growth factors and other proteins from the surrounding environment which cause adjacent RTKs to form cross-linked dimers
Cross-linking triggers tyrosine kinase activity of RTKs via cross-phosphorylation
After cross-phosphorylation, the cytoplasmic tails of RTKs behave as docking sites for specific intracellular cytoplasmic signalling proteins
These proteins have a SH2 (SH= Src homology) domain that binds to specific sequences of the RTKs which contain a phosphorylated tyrosine
More than one SH2-containing protein can bind to an activated RTK at a given time because RTK tails have multiple phosphorylated tyrosines, leading to simultaneous activation of different signalling pathways
Some signalling proteins (eg. PLC-γ) bind directly to the activated RTK whereas others bind via adaptor proteins such as Shc and Grb2
Shc (SH2-containing sequence (SHC)-transforming protein 1) and Grb2 (growth factor receptor-bound protein 2) are adaptor proteins involved in signal transduction and cell communication
Many signalling proteins contain SH3 domains in addition to SH2
SH3 binds to proline-rich sequences on other proteins and recruit them to to the activated RTK to propagate the growth factor signal and effect alterations in gene transcription
Signals get transferred from the membrane to the nucleus due to crosstalk occurring between different intermediates in the cell signalling pathways
What is the role of adaptor proteins in the signalling pathway of RTKs?
SH2 and SH3 domain-containing proteins (eg. Grb2) are known as adaptor proteins
Adaptor proteins link activated receptors to other parts of the signalling pathway but they themselves lack intrinsic signalling abilities
Grb2 SH2 domain binds to phosphotyrosine docking site of the RTK and the two SH3 domains of Grb2 recruit Sos, a guanine exchange factor which converts GFP to GTP on Ras
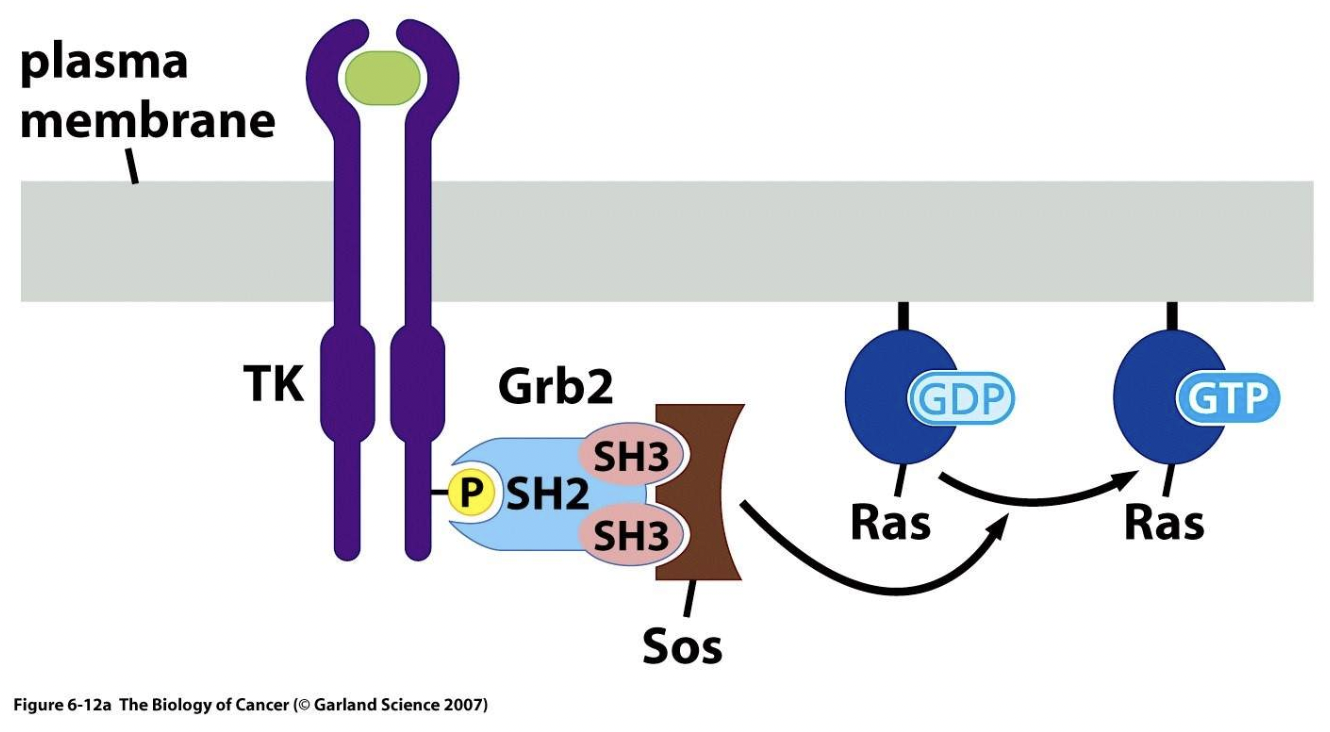
What is Ras and how is its activity modulated?
Ras is a key signalling molecule in pathways switched on by RTK activation
It is a membrane-bound GTP/GDP-binding protein which acts as a molecular switch, transforming signals from the cell membrane to the nucleus
An SH2 domain in Grb2 binds to a specific phosphotyrosine residue in the activated RTK
Grb2 also contains two SH3 domains which bind and activate Sos, acting as an adaptor protein for the EGF receptor
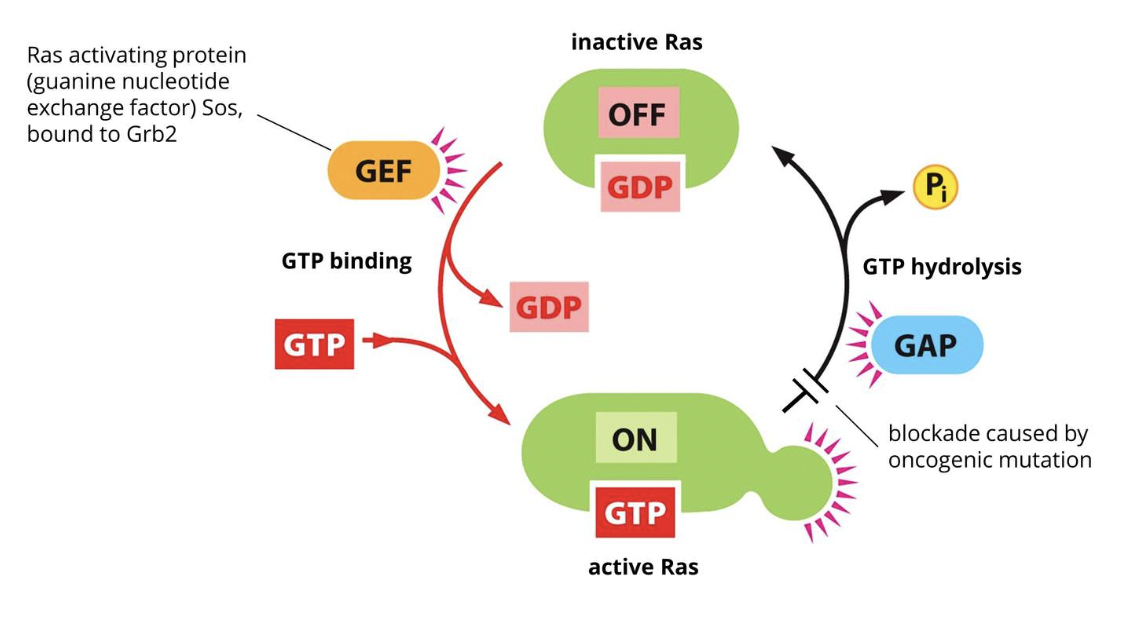
Sos is a guanine exchange factor (GEF) which converts the inactive GDP-bound Ras (which is located in the membrane) to the active GTP-bound form
A GEF aids in the separation of GDP from Ras
GTP then spontaneously binds to Ras and GEF detaches which converts Ras into the active Ras-GTP form
Hydrolysis of bound GTP restores Ras-GTP to the inactive Ras-GDP form
This is promoted by GTP-ase activating protein (GAP)
How can cancer-causing mutations affect signalling pathways?
Genetic changes seen in cancer cells can be linked with signalling pathways that drive processes underlying tumorigenesis and cancer progression, causing aberrant signalling that can escape normal control mechanisms
Oncogenic mutations can cause affected genes to be overexpressed (eg via gene amplification) or make defective proteins (by point mutations or chromosomal rearrangements)
Examples of these proteins include chose involved in signalling pathways, such as growth factor RTKs (including EGFR) and specific proteins affected - small GTPases like Ras, serine/threonine kinases Raf and Akt, the cytoplasmic tyrosine kinases Src and Abl, lipid kinases (phosphoinositide 3-kinases, PI3Ks), nuclear receptors (eg. estrogen receptor)
Other pathways can also be affected, as well as downstream targets of signalling pathways such as transcription factors like c-Myc and cell cycle effectors like cyclins
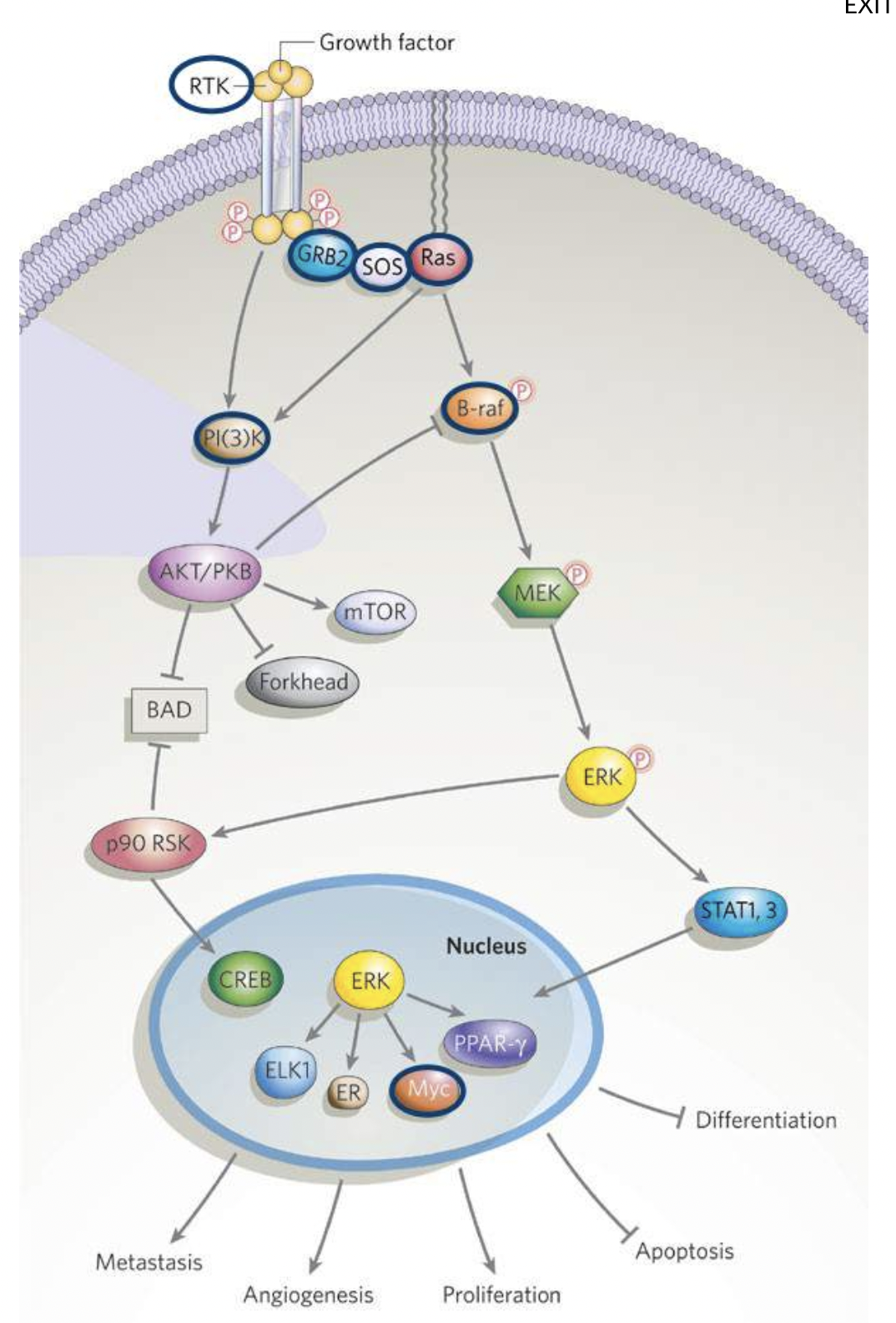
Deletions and loss-of-function mutations can suppress negative regulators that act as tumour suppressors
One of the most commonly mutated genes in cancer is the tumour suppressor p53 which controls cell proliferation and stress response, including apoptosis and DNA damage repair
How is RTK activity regulated?
RTK activity is regulated in various ways such as:
Low availability of growth factors and ligands in the extracellular environment
Internalisation and degradation of activated growth factors
Tyrosine phosphatases (PTPases) which dephosphorylate phosphotyrosine
How can mutations affect RTKs in cancer cells?
In cancer, genetic mutations can result in RTK activation and clustering in the absence of growth factor/ligands
Activating mutations have been found in the kinase domain of the EGFR as well as the extracellular, transmembrane and juxtamembrane domains
Juxtamembrane domain - intracellular region adjacent to the transmembrane domain which is not part of the kinase domain
Chromosomal rearrangements can cause fusion of RTK genes (eg. ALK, FGFR, PDGFR) with genes that make proteins containing an oligomerization domain, resulting in oncogenic fusion proteins whose kinase activity is permanently switched on
Gives cancer cells a growth advantage
What signalling pathways are activated by Ras and which are most relevant in cancer?
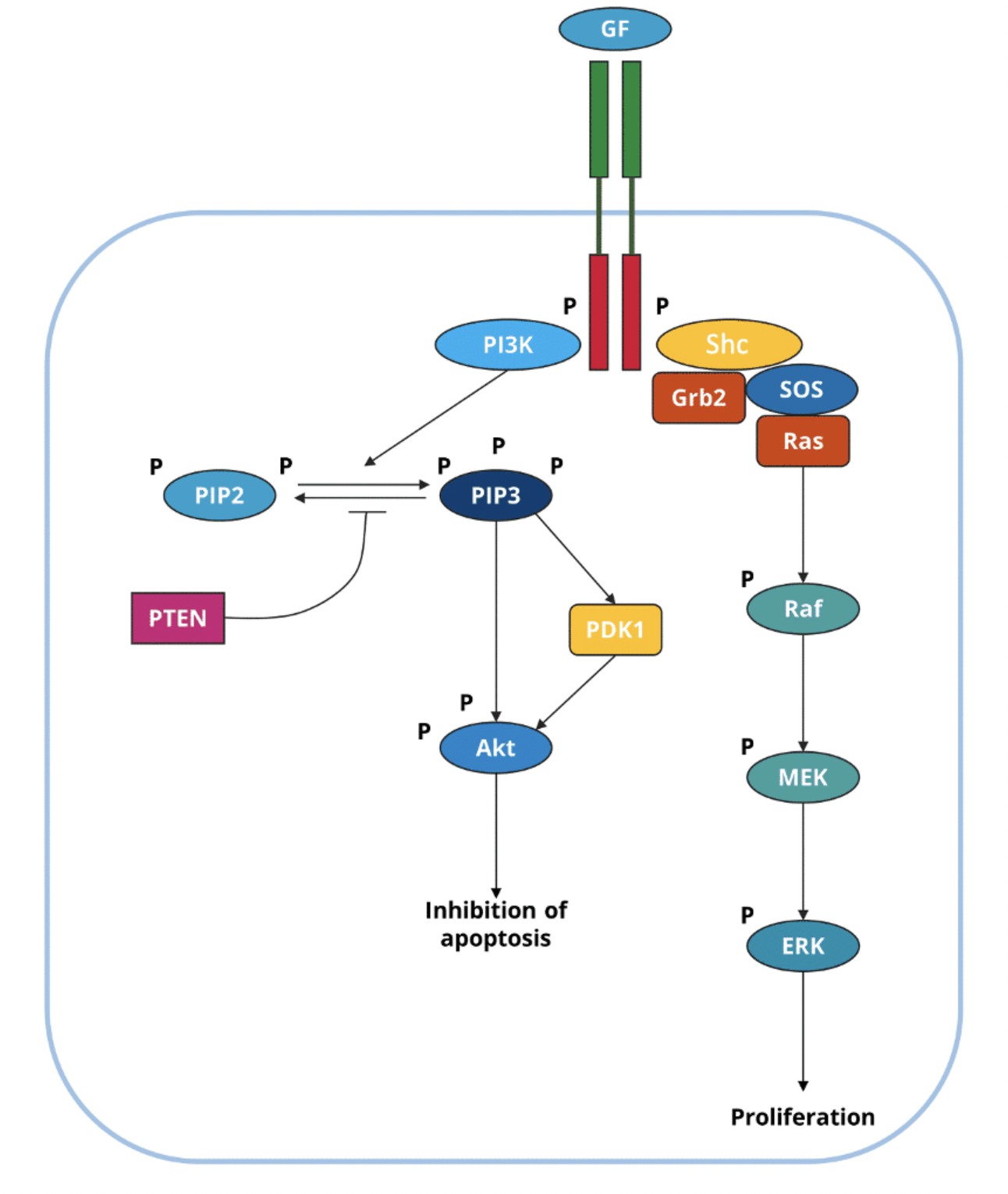
GTP-bound Ras can activate MAP kinase (MAPK), phosphoinositide 3-kinase (PI3K) an Ral-GEF signalling pathways
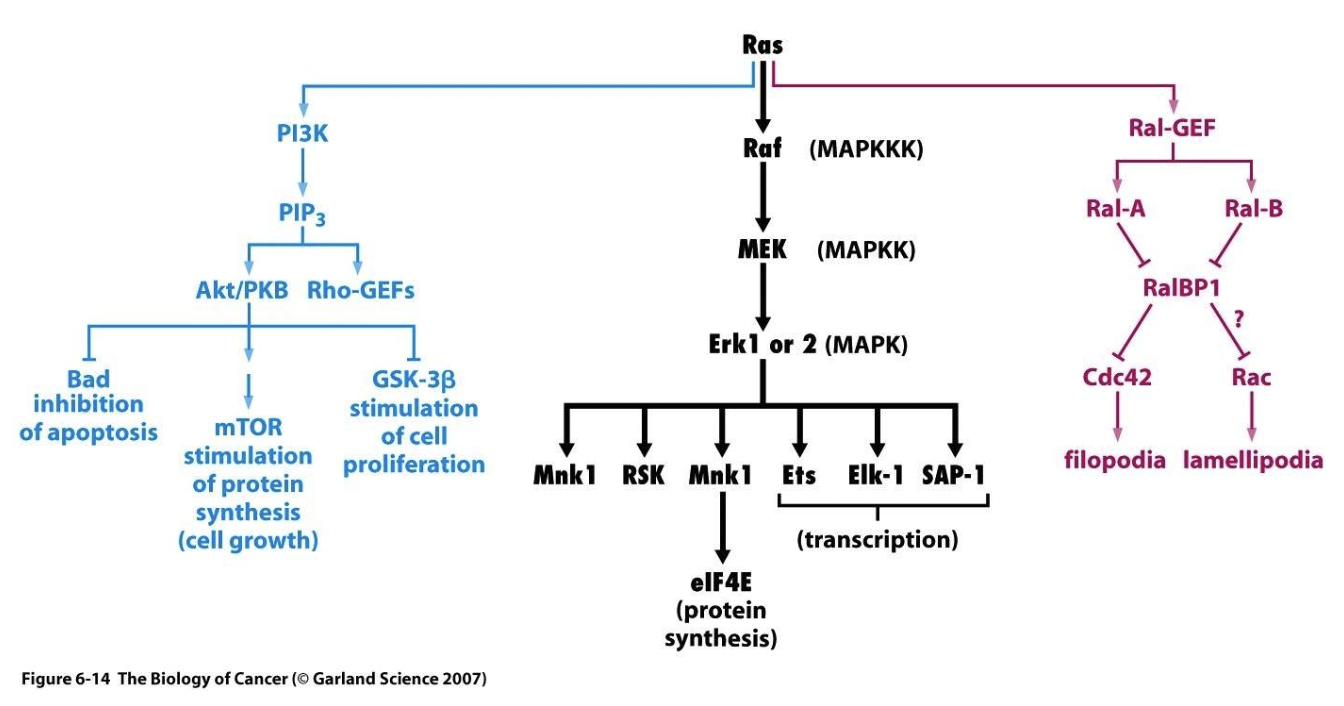
MAPK and PI3K pathways are most relevant to cancer
What is the relevance of the MAPK pathway to cancer?
The Mitogen-activate protein (MAP) kinase cascade is among the most common intracellular signalling pathways triggered by RTKs, involving three serine-threonine kinases
Starts with Ras activation
Active GTP-Ras activates the serine-threonine kinase Raf (MAPKKK)
Raf phosphorylates MEK (MAPKK)
MEK phosphorylates Erk1 or 2 (MAPK)
All three kinases in these pathway phosphorylate several different factors, hence the initial signal is amplified at each step
The final enzyme in the pathway phosphorylates one or more transcription regulators which alters gene transcription
Many growth factors, including nerve growth factor (NGF) and platelet-derived growth factor (PDGF) signal through MAPK pathways
What is the relevance of the PI3K pathway to cancer?
In the PI3K pathway Ras activates PI3K by binding to its Ras-binding domain (RBD) which stimulates its catalytic domain, but PI3K can also be activated directly through RTKs such as PDGFRβ which has a PI3K docking site
Activated PI3K phosphorylates PIP2 to produce the secondary messenger PIP3 (both phospholipids residing in the cell membrane)
PIP3 binds directly with PH domains, eg. phosphoinositide-dependent kinase 1 (PDK1) and Akt (also known as protein kinase B or PKB)
PIP3 brings PDK1 and Akt to the cell membrane where PDK1 activates Akts by phosphorylating it
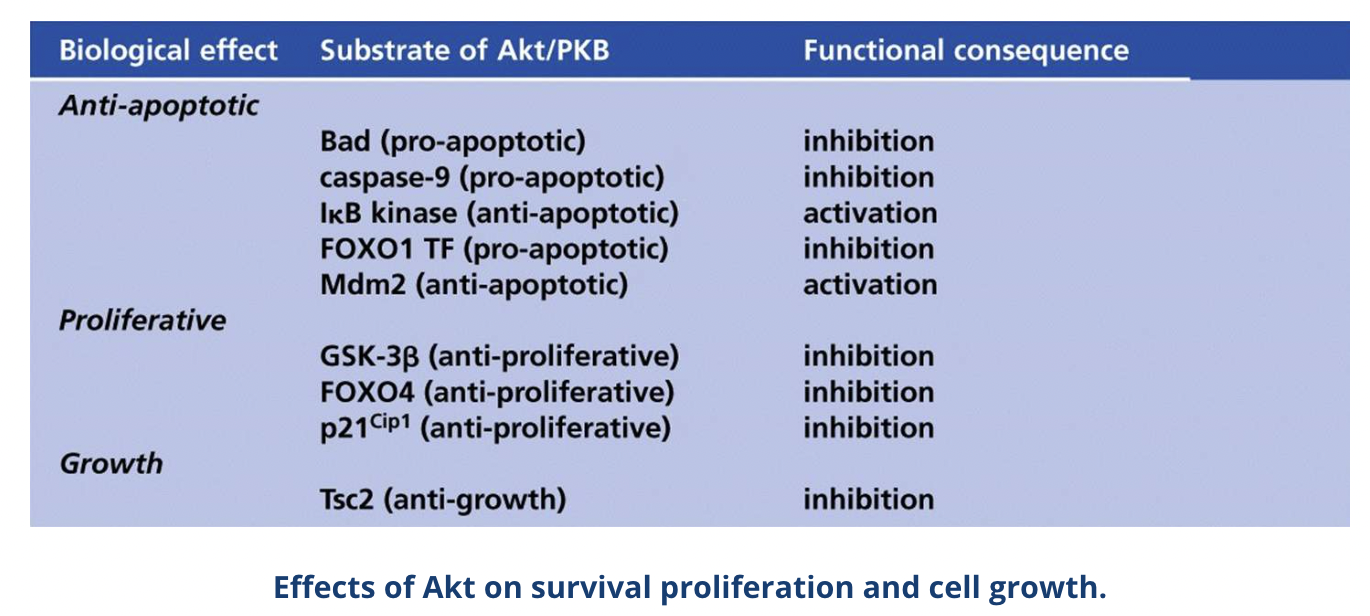
Activated Akt inhibits the proapoptotic Bcl-2 family members BAD and BAX, promoting cell survival
Akt also phosphorylates various other proteins which promotes cell survival, proliferation and growth
What is the role of PTEN in the PI3K pathway and cancer?
The tumour suppressor phosphatase and tensin homologue (PTEN) inhibits this pathway by dephosphorylation PIP3 to PIP2
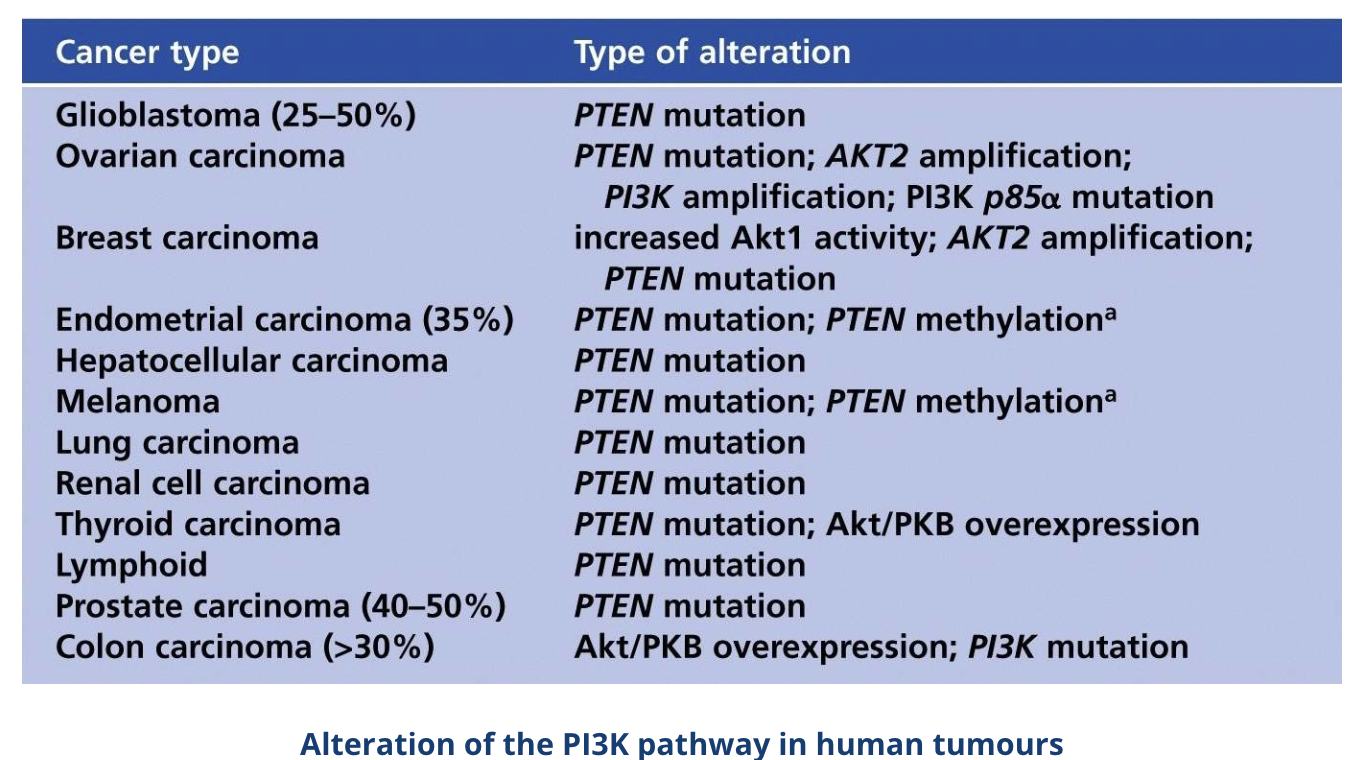
Alterations of the PI3K pathway such as inactivating PTEN mutations are found in ~30-40% of cancers
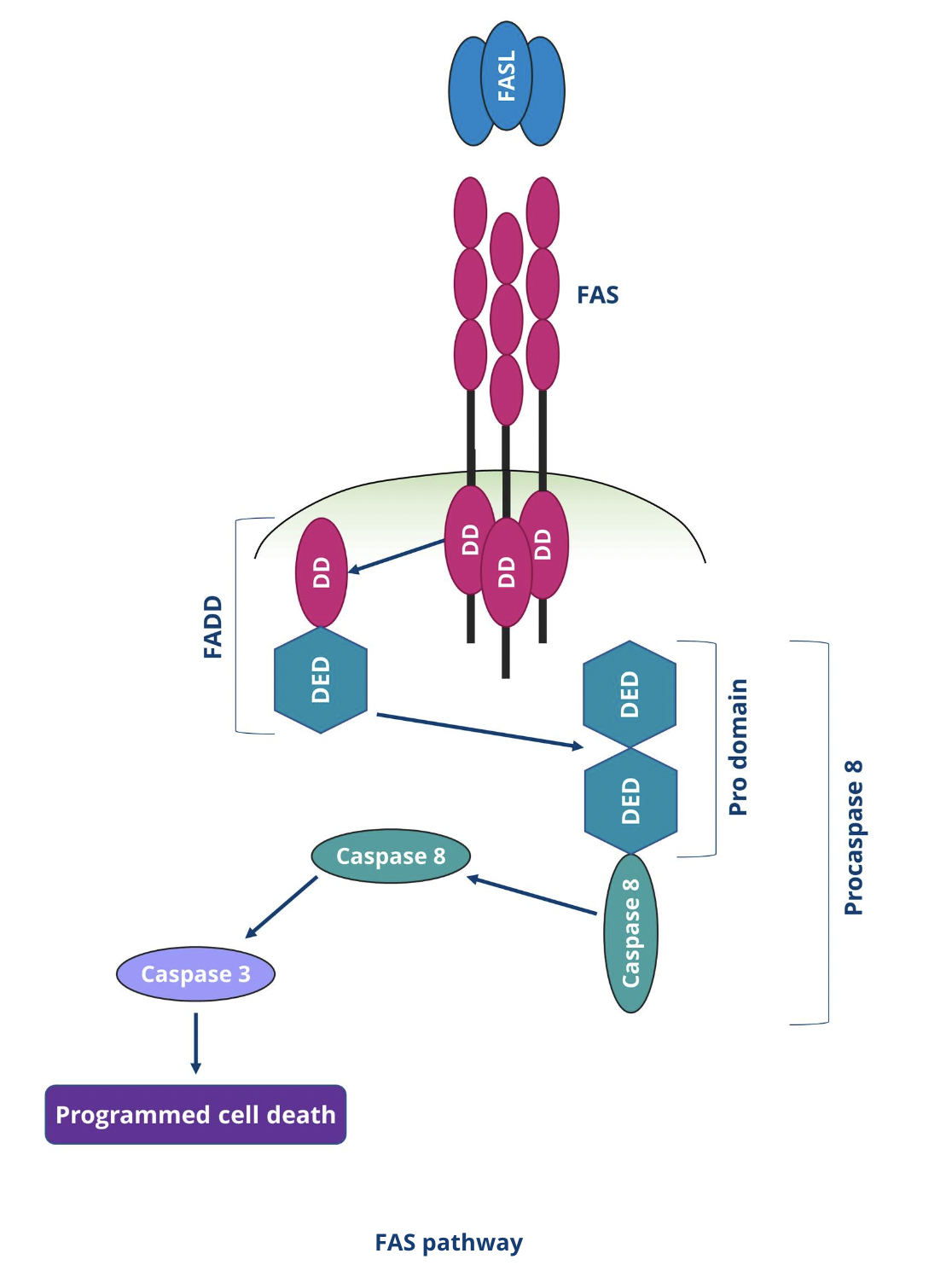
How is apoptosis initiated by the extrinsic pathway?
Extrinsic apoptosis is initiated by the extracellular microenvironment and driven by death receptors

Surface receptor interaction with death ligands leads to the assembly of the death-inducing signalling complex (DISC) which is a dynamic multiprotein complex at the intracellular tail of the death receptor
Death receptors contain cytoplasmic death domains which help transmit death signals from the cell surface to intracellular signalling pathways by recruiting proteins with the appropriate death domain
During FasL and FAS binding, Fas-associated proteins with death domains (FADD) are recruited and during TNF and TNFR binding, a TNFR1-associated death domain protein (TRADD) is recruited with FADD
Adaptor proteins recruit procaspase 8 to form the death effector domain (DED)
Through dimerisation and proteolytic cleavage, procaspase 8 is converted to its active form caspase 8 which initiates apoptosis by cleaving and activating executioner caspase-3
How is apoptosis initiated by the intrinsic pathway?
Intrinsic apoptosis (mitochondrial pathway) is initiated by cellular stressors like DNA damage or withdrawal of growth factors
Apoptotic cells retain their plasma membrane and metabolic activity during the apoptotic process due to the clearance by macrophages and other phagocytic cells via efferocytosis
At the end stage, apoptotic cells have a complete breakdown of the plasma membrane and acquire an necrotic morphotype (secondary necrosis)
Intrinsic apoptosis is irreversible and regulated by pro- and anti-apoptotic members of the Bcl2 family of apoptosis regulator proteins
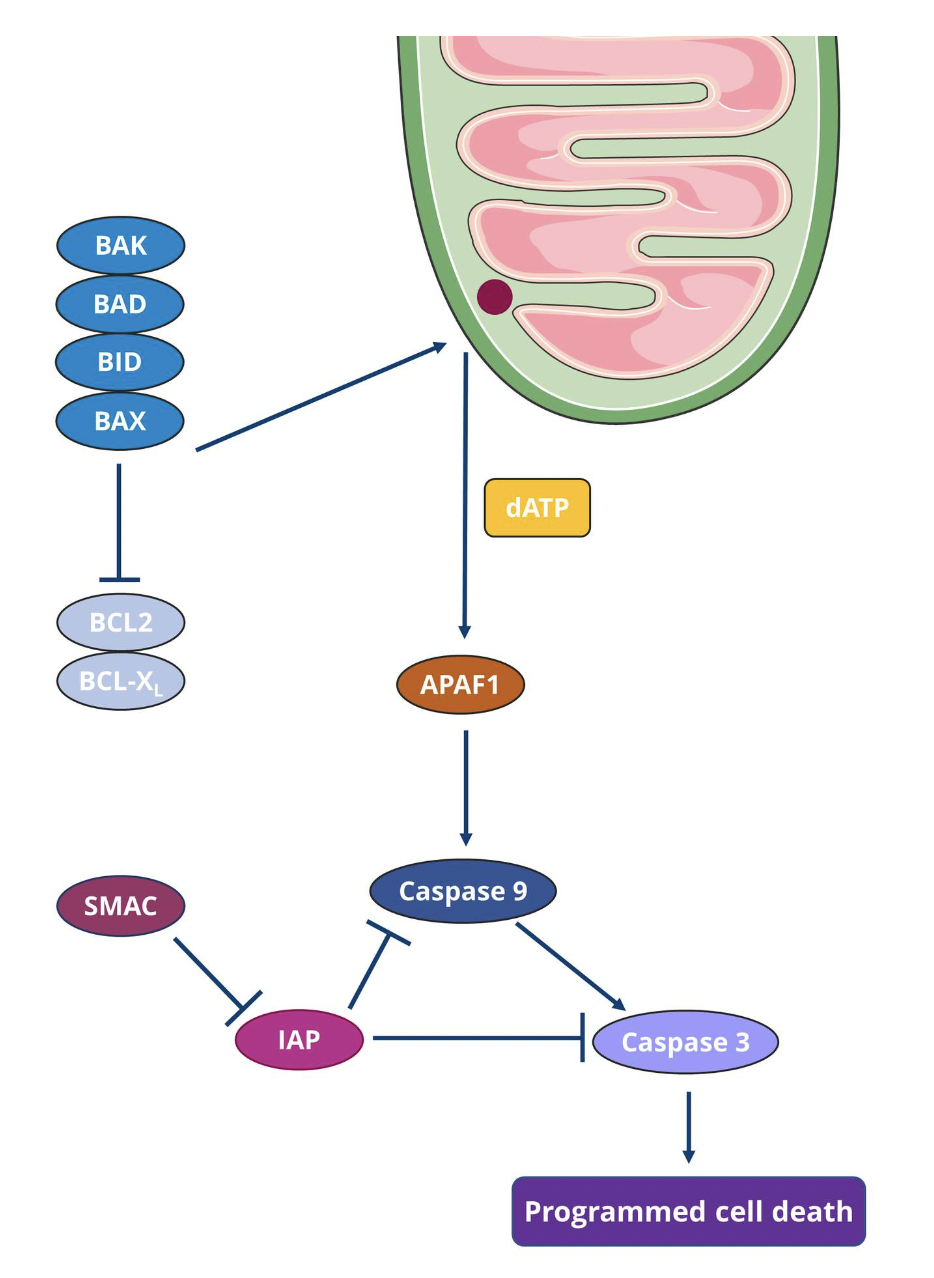
The first step is widespread mitochondrial outer membrane permeabilisation (MOMP) which is mediated by BAX and/or BAK
BAX continuously travels between the outer mitochondrial membrane and the cytosol in an inactive form whereas BAK is located in the outer mitochondrial membrane
When apoptosis is triggered, BAX ceases to translocate, and BAX and BAK get activated by pro-apoptotic proteins such as BID and BAD
Apoptogenic factors are released, including cytochrome C and SMAC
Cytochrome C binds to APAF1 and pro-caspase 9 to form the supramolecular complex apoptosome
The apoptosome activates caspase 9, which then catalyses the proteolytic activation of the executioner caspase 3
SMAC associated with the inhibitor of apoptosis (IAP) protein family to regulate apoptosis
Activation of executioner caspases triggers morphological and biochemical changes in the cell, including DNA fragmentation, phosphatidylserine (PS) exposure and formation of apoptotic bodies
Caspase 3 catalyses the proteolytic inactivation of DNA fragmentation factor subunit alpha (aka. inhibitor of CAD, ICAD) and releases the catalytic activity of caspase-activated DNAse (CAD) to trigger DNA fragmentation
anoikis
Anoikis is a variant of intrinsic apoptosis, caused by loss of integrin-dependent attachment of cells to the ECM
What are some mechanisms of cell death?
Apoptosis: genetically programmed, caspase-dependent
Necrosis - not genetically determined, caspase-independent
Mitotic catastrophe - failure of cells to undergo mitosis, oweing to chromosomal damage
Senescence - cells that are metabolically active but unable to divide
Autophagy - genetically programmed self-digestion, caspase- and p53-independent
List some oncogenes
Akt, Cdk4, bRaf, Ras, Her2, Bcl-2, PI3K
List some tumour suppressor genes
TP53, APC, pTEN, BRCA1, Bax, RB-1
Describe how cells undergo apoptosis via the p53-dependent mechanism
Cells undergoing apoptosis via the p53-dependent mechanism mainly follow the mitochondrial pathway, but p53 can also activate death receptor mediated apoptosis
The p53 pathway acts through cytosolic and mitochondrial p53
In the cytosolic pathway:
Nuclear p53 activates proapoptotic genes including members of the Bcl-2 family such as Bax, NOXA and PUMA
Cytosolic p53 is released from its inactive complex with Bcl-XL and it induces Bax oligomerization and mitochondrial translocation
In the mitochondria:
p53 promotes Bax and Bak activation while blocking anti-apoptotic Bcl-XL
p53 also complexes with cyclophilin D, which regulates mitochondrial permeability, resulting in the disruption of the mitochondrial membrane which triggers the release of cytochrome c and other apoptogenic factors, thereby activating caspase 9
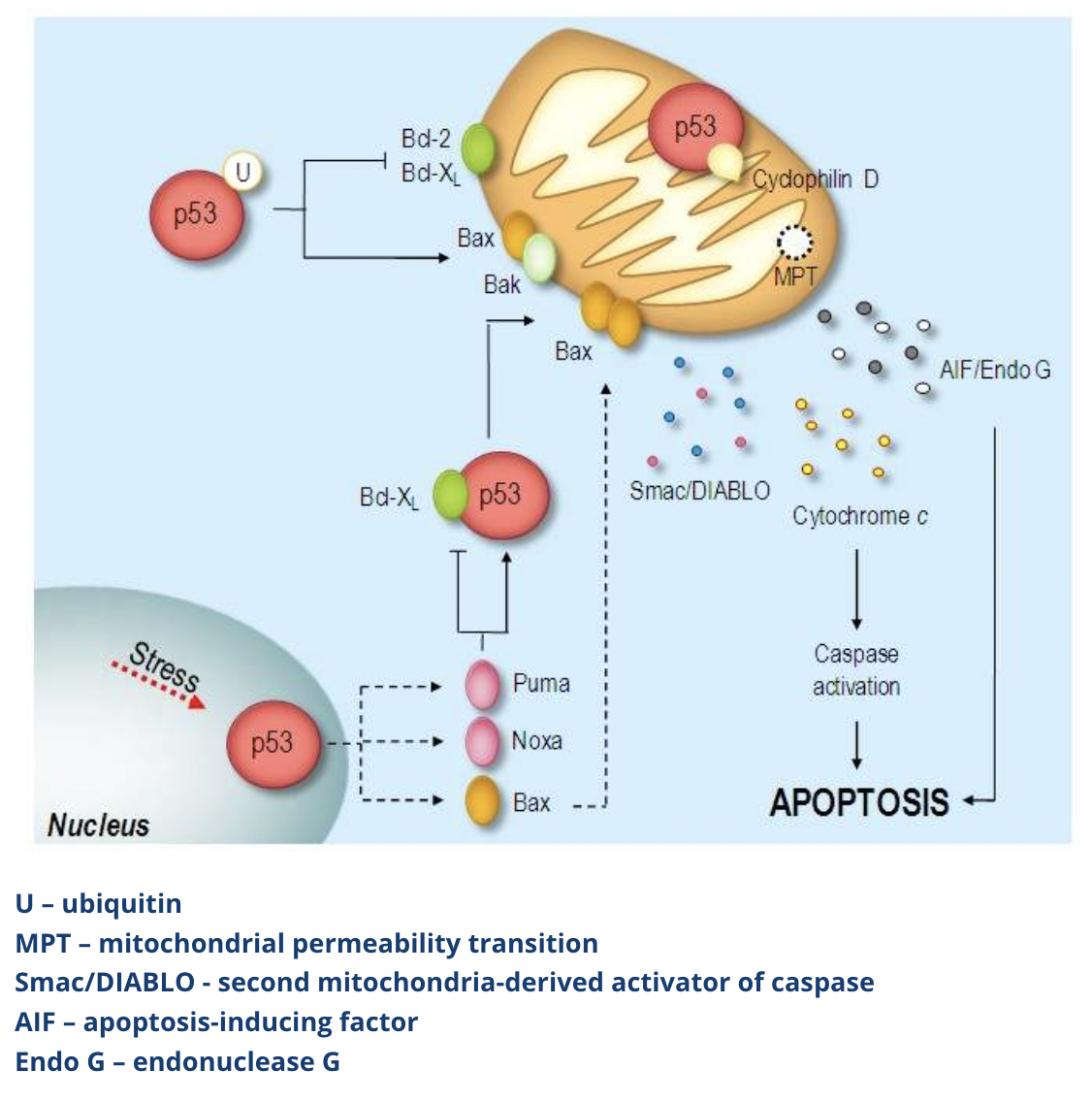
p53 overexpression can also activate death receptor 5 (DR5) which promotes cell death via death receptor mediated apoptosis by activating caspase 8
How can mutant p53 cause cancer?
p53 is encoded by TP53 which is the most frequently mutated gene in human cancers (>50% of human cancers have TP53 loss of function genes)
Mutant p53 can act as an oncogene by inhibiting WT p53 in a dominant-negative manner
Cancer-derived mutant p53 can activate growth-promoting and oncogenic genes like c-MYC, multiple drug resistance gene 1 (MDR1), EGFR and telomerase reverse transcriptase (TERT)
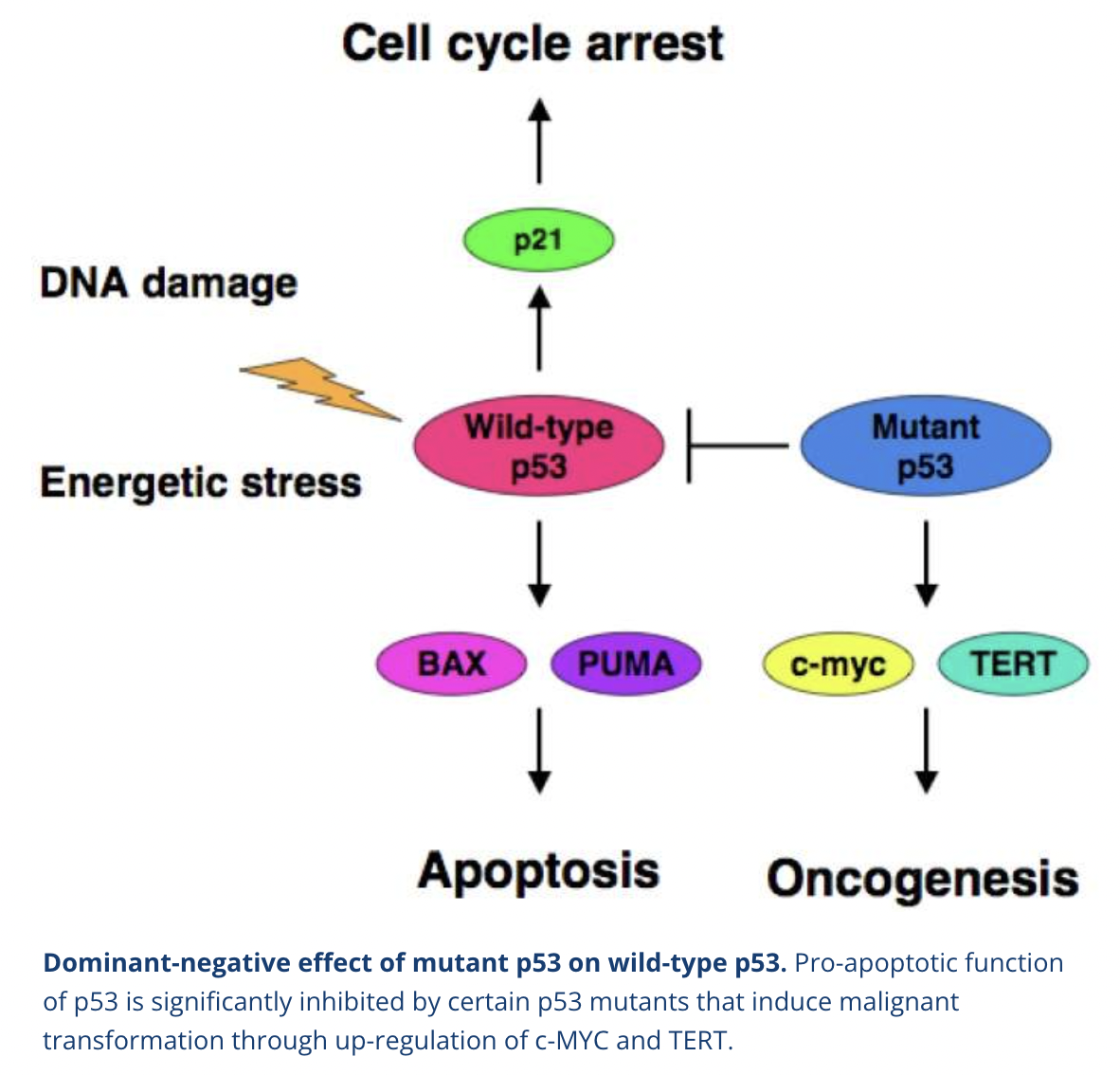
What are the characteristics of p53 mutant cells?
Low sensitivity to DNA damaged-induced apoptosis
Chromosomal damage
Low hypoxia protection
Anoikis protection
What are some other anti-apoptotic alterations that can occur in cancer other than p53?
P14ARF inactivation
MDM2 overexpression
BAX mutation
BCL2 overexpression
NFkB activation
PTEN inactivation
IGF-1/2 overexpression
FLIP overexpression
FAP-1 overexpression
Why is anoikis evasion relevant to cancer cells?
Cancer cells can undergo epithelial to mesenchymal transition (EMT) to travel to other organs and form metastases
In EMT, cells lose connection between each other and the ECM, thereby facilitating their migration as a single cell or cell cluster
Loss of attachment to the ECM causes anoikis in normal cells, but cancer cells can evade this death process
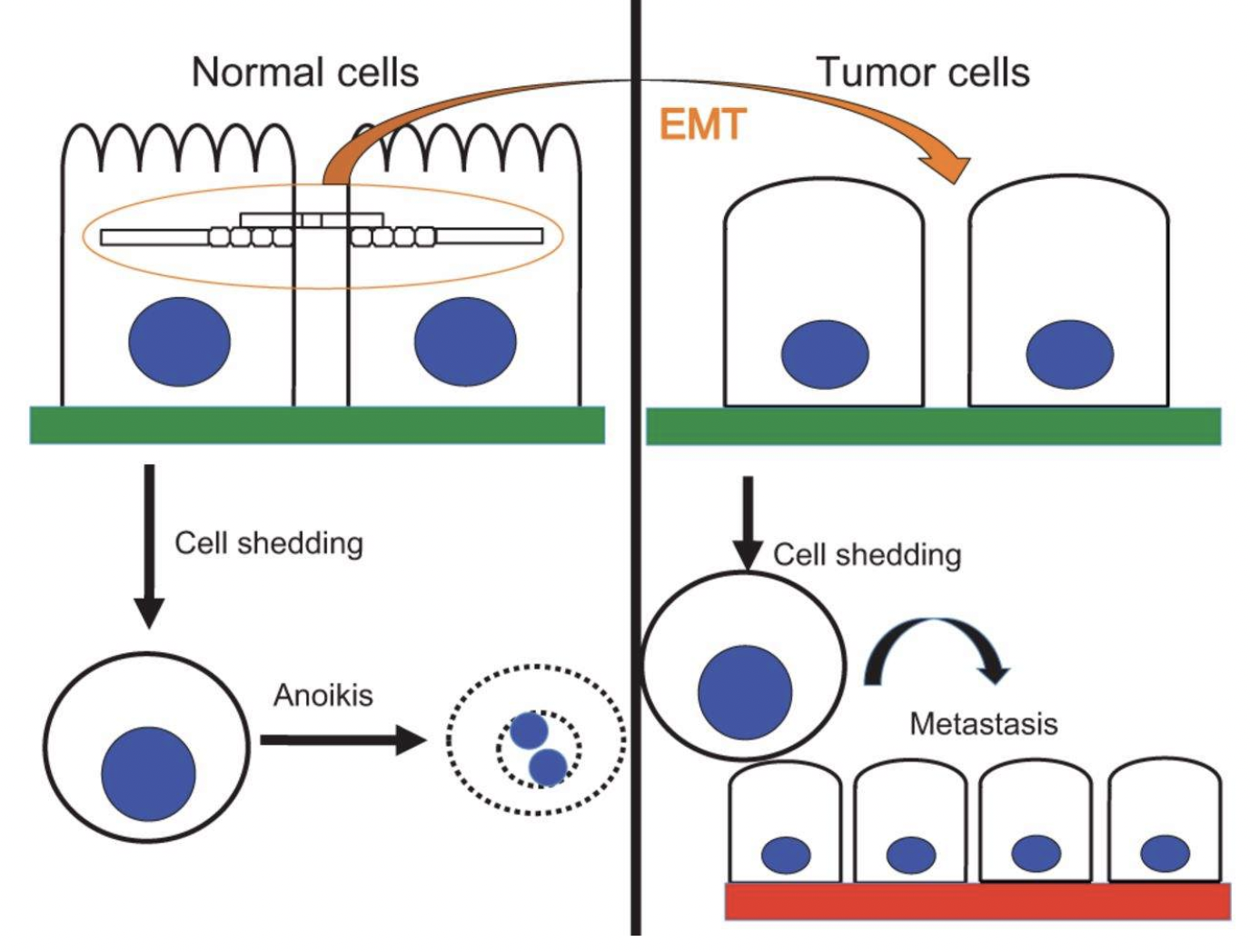
What mechanisms allow cancer cells to avoid anoikis?
Hyperactivation of RTKs
Modulation of apoptotic signalling
Cytoskeletal rearrangement sensing
Oxidative stress protection
Autophagy (lysosomal self-digestion)
Activation of EMT
How does RTK hyperactivation allow cancer cells to evade anoikis?
Cancer cells secrete growth factors which constitutively activate pro-survival signals including PI3K, Ras-Erk, or Rho-GTPase
Overexpression of the RTK ErbB2 (which inhibits pro-apoptotic protein Bim) while maintaining EGFR expression
How does apoptotic signalling modulation allow cancer cells to evade anoikis?
Cancer cells activate anti-apoptic Bcl-2 proteins
How does cytoskeletal rearrangement sensing allow cancer cells to evade anoikis?
Cytoskeletal rearrangement sensing allows cells to monitor their adhesion to the ECM
Cancer cells can undergo epigenetic silencing of adhesion related genes like the Src family member p66Shc, causing Ras hyperactivation and RhoA inactivation
How does oxidative stress protection allow cancer cells to evade anoikis?
Oncogenes like ErbB2 bypass metabolic stress-induced ROS production during cell detachment
Increased antioxidant responses are promoted by upregulation of heme oxygenase 1 (HMOX1)
How does autophagy allow cancer cells to evade anoikis?
Cancer cells initiate protective autophagy to allow themselves to become dormant during unfavourable conditions (eg lack of nutrients)
During detachment of cancer cells, activation of autophagy contributes to glycolysis and cell proliferation
How does EMT activation allow cancer cells to evade anoikis?
Upregulation of matrix metallopeptidases (MMPs)
What strategies could be used to promote intrinsic apoptosis in cancer cells?
Strategies to promote intrinsic apoptosis in cancer cells include interfering with autoprotective effects of anti-apoptotic members of the Bcl-2 family via:
Interference with mRNA function
Development of small molecule drugs to target specific proteins
Shut down gene transcription
How can interference with mRNA function promote intrinsic apoptosis in cancer cells?
Antisense oligonucleotides (ASOs) are used to interfere with the autoprotective effects of anti-apoptotic members of the Bcl-2 family
ASOs are short synthetic sequences of ssDNA which can bind to mRNA and enhance sensitivity to cytotoxic drugs
Oblimersen sodium (G3139, Genasense) is an ASO which is an anti-Bcl-2 mRNA agent used in advanced clinical settings
Currently being used in clinical trials for lymphoma
How can development of small molecule drugs to target specific proteins promote intrinsic apoptosis in cancer cells?
Small molecule inhibitors of the Bcl-2 family include those targeting histone deacetylase (HDACs) which regulate gene expression
Vorinostat and remidepsin are two currently approved HDAC inhibitors against refractory cutaneous T cell lymphoma (CTCL)
Other approaches include natural and synthetic BH3 mimetics to induce apoptosis by mimicking critical domains of pro-apoptotic members of the Bcl-2 family like Bax and PUMA
How can shutting down gene transcription promote intrinsic apoptosis in cancer cells?
Small molecule inhibitors can also target inhibitors of apoptosis (IAPs) by mimicking SMAC or via antisense-mediated interference of XIAP mRNA and protein expression
What are the early efforts in targeting extrinsic apoptosis and why have they had limited success?
Purified TNFα
To target one of the death receptors TNF
However this was too toxic for systemic therapy
Activating the second death receptor FAS
Also too toxic, causing apoptosis of hepatocytes
Targeting another death ligand TNFSF10 (aka Apo2L/TRAIL)
Had more success
TNFSF10 cognate proapoptotic death receptors DR4 and DR5 antibodies were created
These antibodies are better tolerated but resistance is common
Low efficacy in monotherapy or in combined therapy with conventional treatments
What are the disadvantages of targeting the PI3K pathway in cancer therapy?
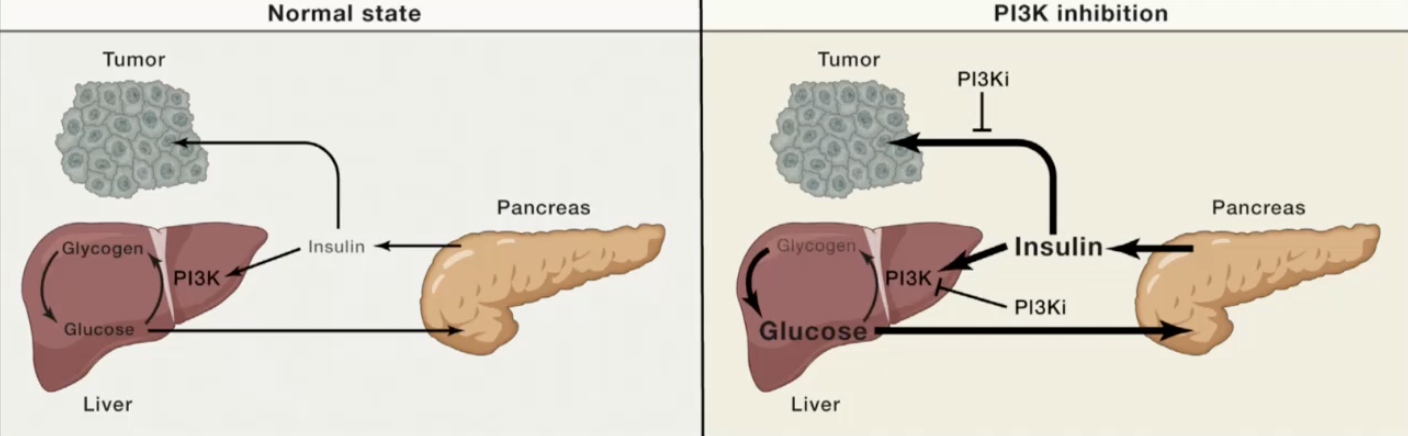
PI3K inhibitors perturb insulin signalling in the liver, causing it to release glucose which signals to the pancreas to release insulin that can reactivate insulin signalling in tumours, reactivating PI3K signalling and undercutting the efficacy of the PI3K inhibitors.
PI3K activity is important for insulin signalling and metabolism, so inhibiting PI3K could lead to decreased insulin sensitivity and diabetes.
PI3K also appears to be required for normal brain function, as decreased PI3K signalling has been associated with schizophrenia
How may cancer therapies bypass the disadvantages of targeting PI3K signalling in cancer therapy?
Drugs that inhibit PI3K signalling for a short period of time in normal tissues might have reversible or treatable side-effects
Tumour cells are often dependent on a hyperactive PI3K pathway, restoring normal levels of PI3K signalling to these cells could be sufficient to slow tumour progression
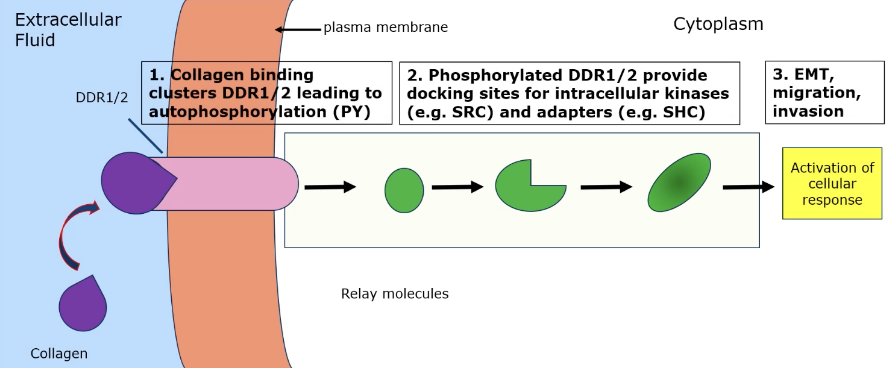
How can collagen binding lead to cancer properties?
Collagen binding clusters DDR1/2 leads to autophosphorylation → phosphorylated DDR1/2 provides docking sites for intracellular kinases (eg SRC) and adaptors (eg SHC) → EMT, migration, invasion
Describe the Notch signalling pathway and its role in cancer
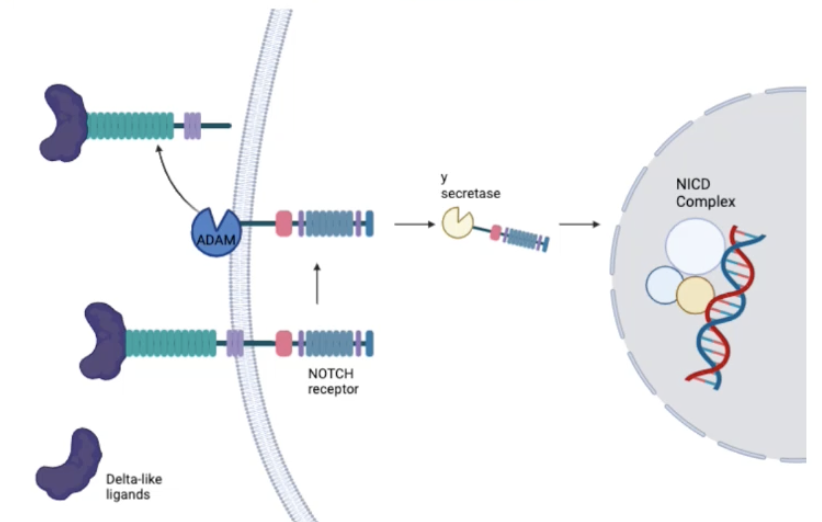
Notch signalling can be involved in cell survival/death, proliferation or apoptosis
Notch can act as an oncogene or tumour suppressor, playing an important role in cancers of the liver, pancreas, endometrium, uterus, ovary, prostate, bladder and colon
Delta-like ligands (eg. DLL1) bind to Notch receptors
There are four Notch receptors in mammals - Notch 1, Notch 2, Notch 3, Notch 4
Notch receptors bind to ligands on neighbouring cells which triggers a conformational change in the receptor
Ligand binding triggers a series of proteolytic cleavages in the transduction step
The Adam family of proteases catalyses the cleavage of the extracellular component of the receptor
The y-secretase enzyme catalyses the cleavage of an intracellular component of the receptor, releasing the Notch intracellular domain (NICD) into the cytoplasm
NICD complex translocates into in the nucleus where is associates with a transcriptional regulator complex
NICD complex activates the transcription of genes involved in cell fate determination and differentiation
Notch signalling is often associated with suppression of differentiation and maintaining cells in the undifferentiated state
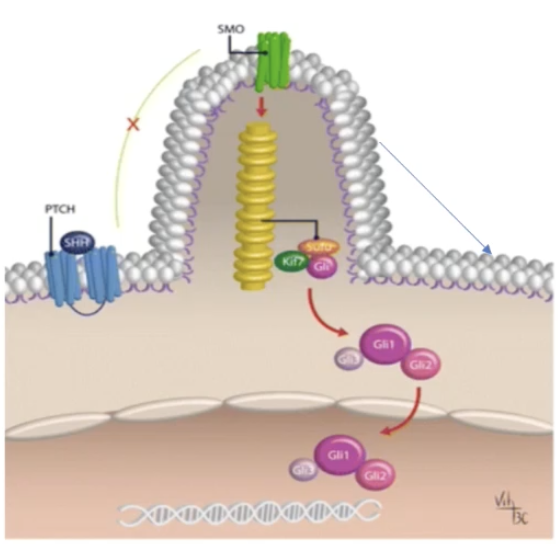
Describe the Hedgehog signalling pathway and its role in cancer
Hedgehog ligand (Hh) binds to the Ptch1 receptor and the Hh-Ptch1 complex is subsequently internalised and degraded in lysosomes
Ptch1 inhibits Smo, which gets disinhibited by Hh binding to Ptch1
Smo travels to the tip of the primary cilium and signals to Sufu to release Gli activator (GliA)
This causes the release and nuclear translocation of Gli1/2 proteins which increase transcription of target genes
This pathway mediated cell proliferation, differentiation and tissue patterning
Dysregulation of Hh signalling can cause cancer
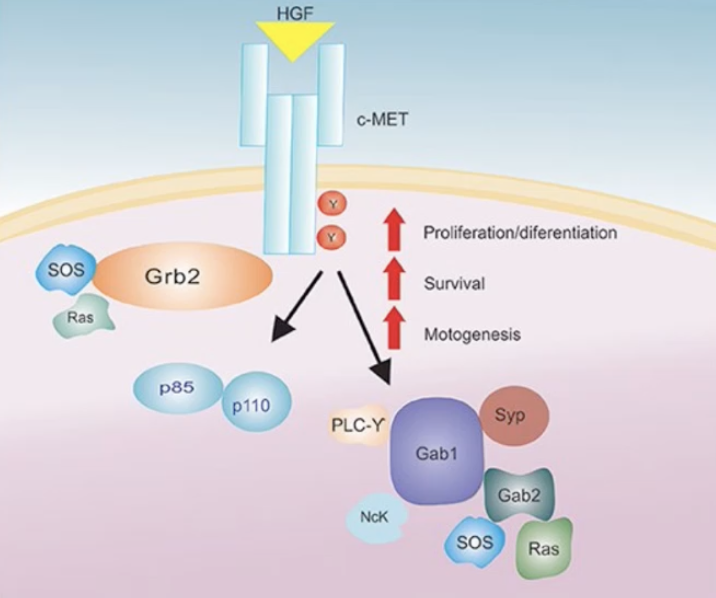
Describe the hepatocyte growth factor (HGF) signalling pathway and its role in cancer
HGF finds to the c-MET receptor which causes the homodimerisation of two tyrosine residues in the catalytic loop of the tyrosine kinase domain of the receptor
This leads to the phosphorylation of intracellular tranducers eg. PI3K, Grb2, Sos and Ras
The HGF/c-MET axis activation is associated with various biological responses implicated in tumorigenesis (eg. cell proliferation, angiogenesis, metastasis and cell survival) and it can cause resistance to some cancer therapies
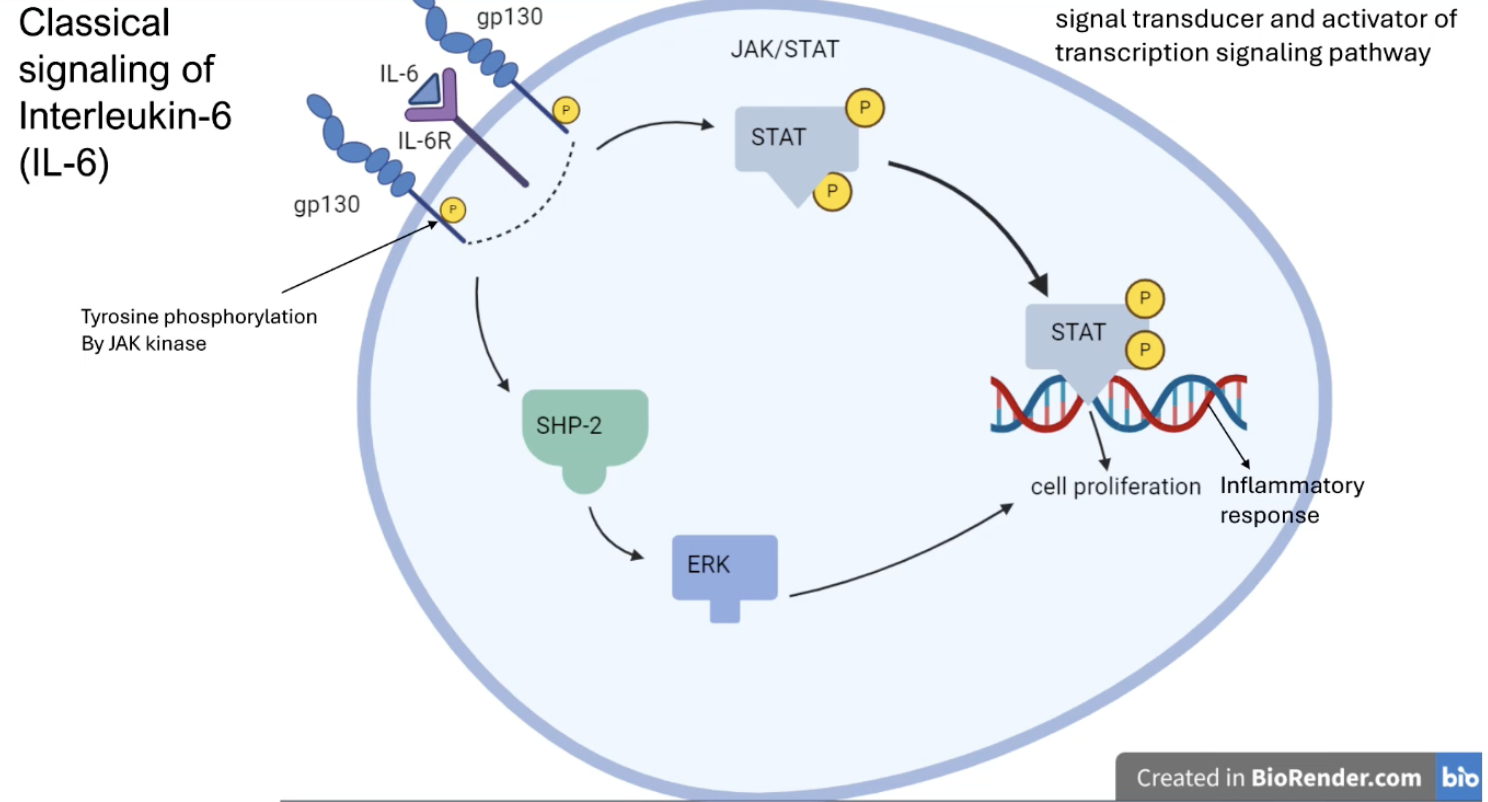
Describe the classical IL-6 signalling pathway and its role in cancer
IL-6 binds to IL-6R receptors and gp130 on the cell membrane which causes them to get phosphorylated
Phosphorylation of the receptors activates the JAK/STAT and the SHP-2 pathway
Phosphorylated STAT and Erk (activated by SHP-2) translocates to the nucleus and urpegulates genes that mediate cell proliferation and inflammation
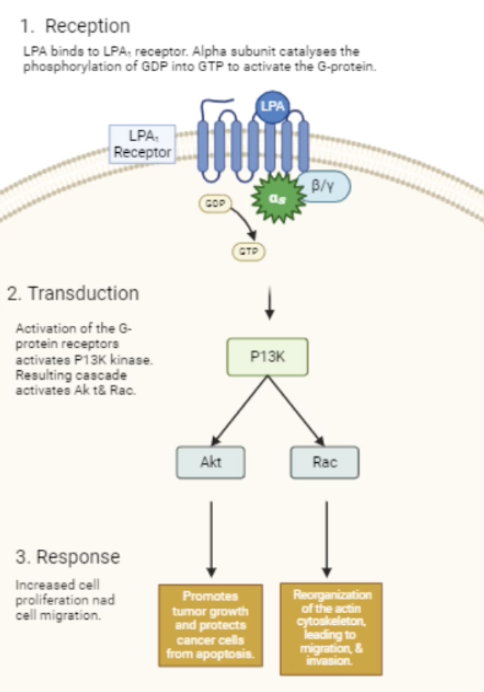
Describe the LPA signalling pathway and its role in cancer
LPA binds to the LPA receptor which is a GPCR, allowing it to activate a G protein by converting GDP to GTP
Resulting inhibition of adenylate cyclase initiates the PI3K pathway which then activates the Akt and Rac pathways
Akt pathway promotes tumour growth and protects cancer cells from apoptosis
Rac pathway reorganises the actin cytoskeleton and promotes cell migration and invasion
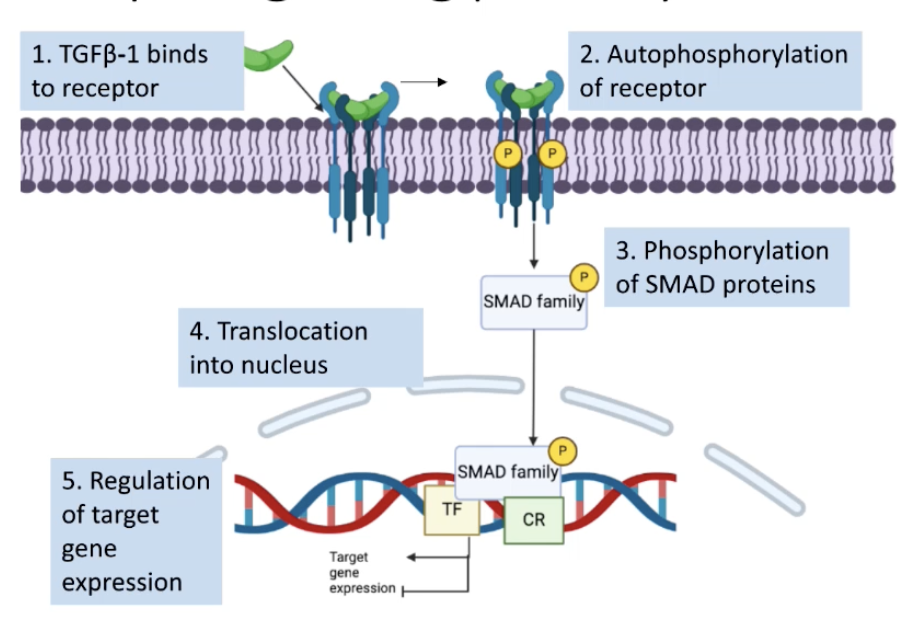
Describe the TGFβ-1 signalling pathway and its role in cancer
TGFβ-1 binds to Type I and Type II TGFβ-1 receptors
The type II receptor gets activated which phosphorylates the type I receptor
Type I receptor activation leads to phosphorylation of SMAD family of proteins which gets translocated into nucleus
Phosphorylated SMAD proteins bind to DNA and regulate target genes involved in cell proliferation and apoptosis evasion by recruiting transcription factors and chromatin remodelling proteins
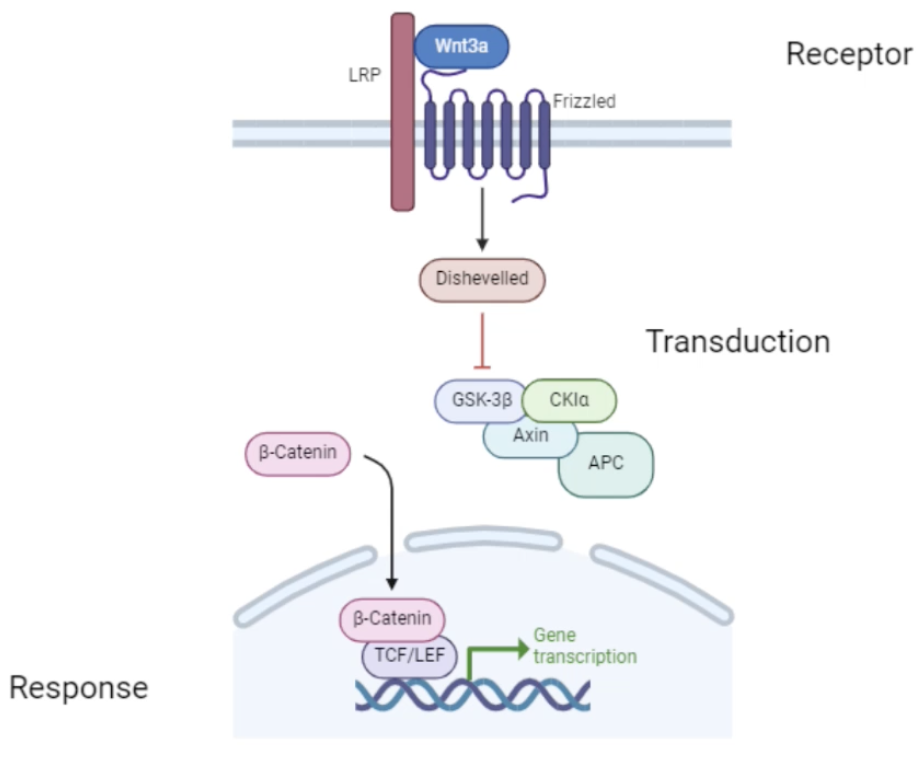
Describe the Wnt3a signalling pathway and its role in cancer
Wnt3a binds to the Fz receptor and the LRP co-receptor
Activates Dsh protein which degrades a destruction complex which normally phosphorylates β-catenin and marks it for degradation
In the presence of Wnt3a, β-catenin can accumulate in the cytoplasm
β-catenin translocates into the nucleus where it interacts with Tcf/Lef transcription factors which promote the expression of genes involved in cell proliferation, differentiation, embryonic development and tumorigenesis
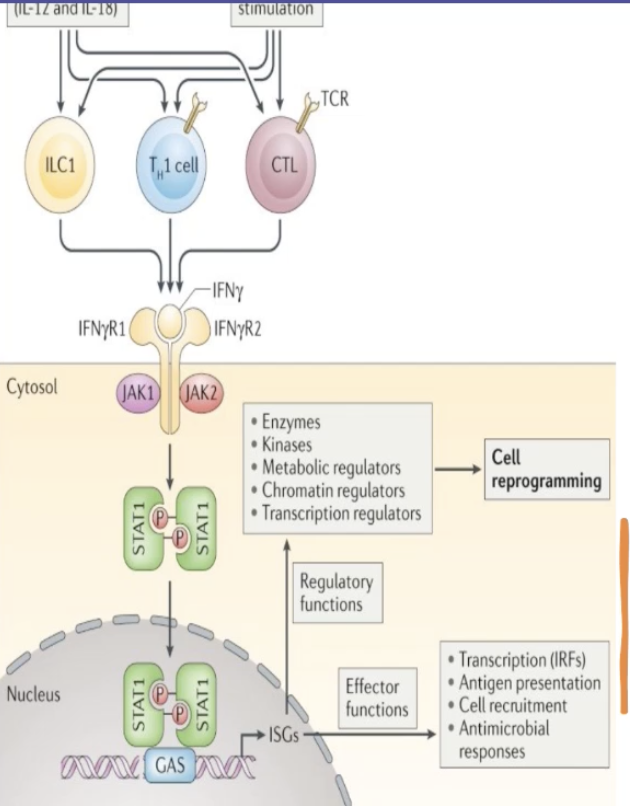
Describe the IFNy signalling pathway and its role in cancer
Innate-like lymphocytes and adaptive lymphocytes produce IFNy in response to cytokine and antigen stimulation
IFNy acts on its receptor to induce rapid and transient Janus kinase (JAK)-signal transducer, activator of transcription (STAT) signalling and interferon-stimulated gene (ISG) induction
INFy receptor is a tyrosine kinase-associated receptor which recruits JAK
STAT1 transcription factor is phosphorylated and dimerised and can bind to the gamma activated site (GAS) in the promoter region of target genes
Induces expression of IFNy regulating genes
What therapeutic approaches could be considered to target EGFR signalling pathways and why might they have limited success?
Anti-EGFR antibodies - to prevent receptor binding (not specific to tumour cells)
EGFR kinase inhibitor - to prevent kinase activation, targets variants with constitutively active kinase domains
Peptide vaccines - targets the variant sequence that causes constitutive kinase activation
However there are downstream components which may be amplified or mutated which negates the effects of the therapy as there can be a wide range of mutations throughout the signalling cascade
The further upstream you target, the more likely that there will be other factors downstream that lead to redundancy, causing resistance and lack of efficacy
Combination therapies may be more effective by targeting more than one component of the signalling pathway
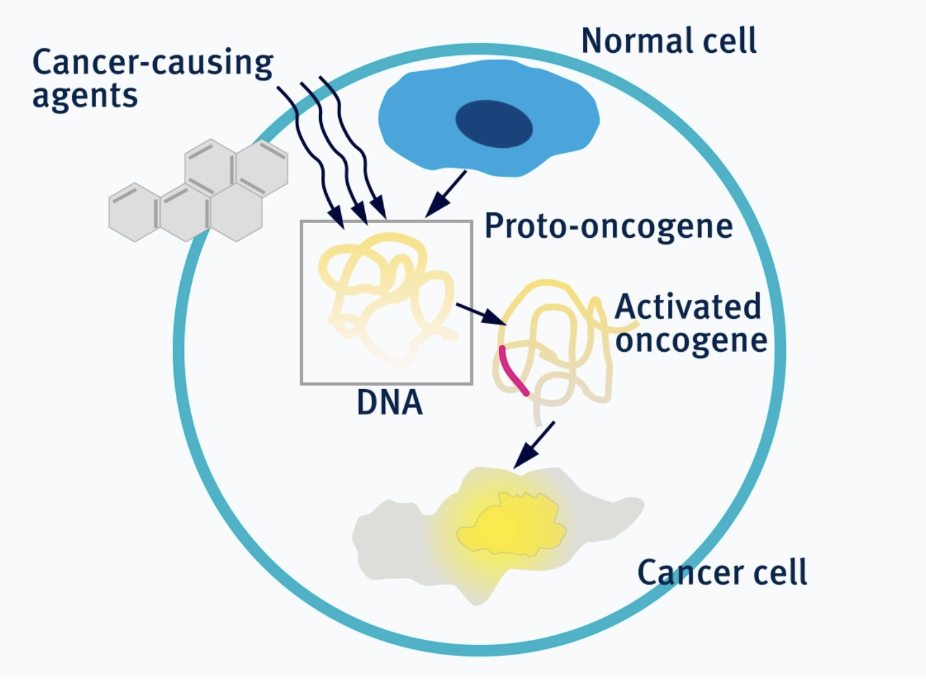
proto-oncogene
Proto-oncogenes are genes that typically promote cell growth and when mutated or when copy number is increased, it can be constitutively activated which causes uncontrolled cell growth
oncogene
a gene encoding for a protein that can promote oncogenesis when activated or overexpressed
oncogenesis/carcinogenesis
a process by which healthy cells transform into cancer cells
oncovirus
cancer-causing virus
retrovirus
a class of RNA viruses that produce dsDNA copies of their genome and integrate them into the chromosome of the host cell