PCL381 Final
1/157
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
158 Terms
Genotoxic vs non-genotoxic carcinogens
Genotoxic → carcinogens interact with DNA, resulting in mutations
No threshold to toxicity
Most require metabolism
Non-genotoxic → carcinogens modify gene expression, do not damage DNA
Have a threshold to toxicity
Usually function during tumour promotion stage
Species and tissue specificity
How does cancer arise?
Most cancers are monoclonal → arise from accumulation of mutations that provide proliferative / survival advantage → clonal expansion / tumour
Usually involves gain of function mutations in oncogenes or loss of function mutations in tumour suppressor genes
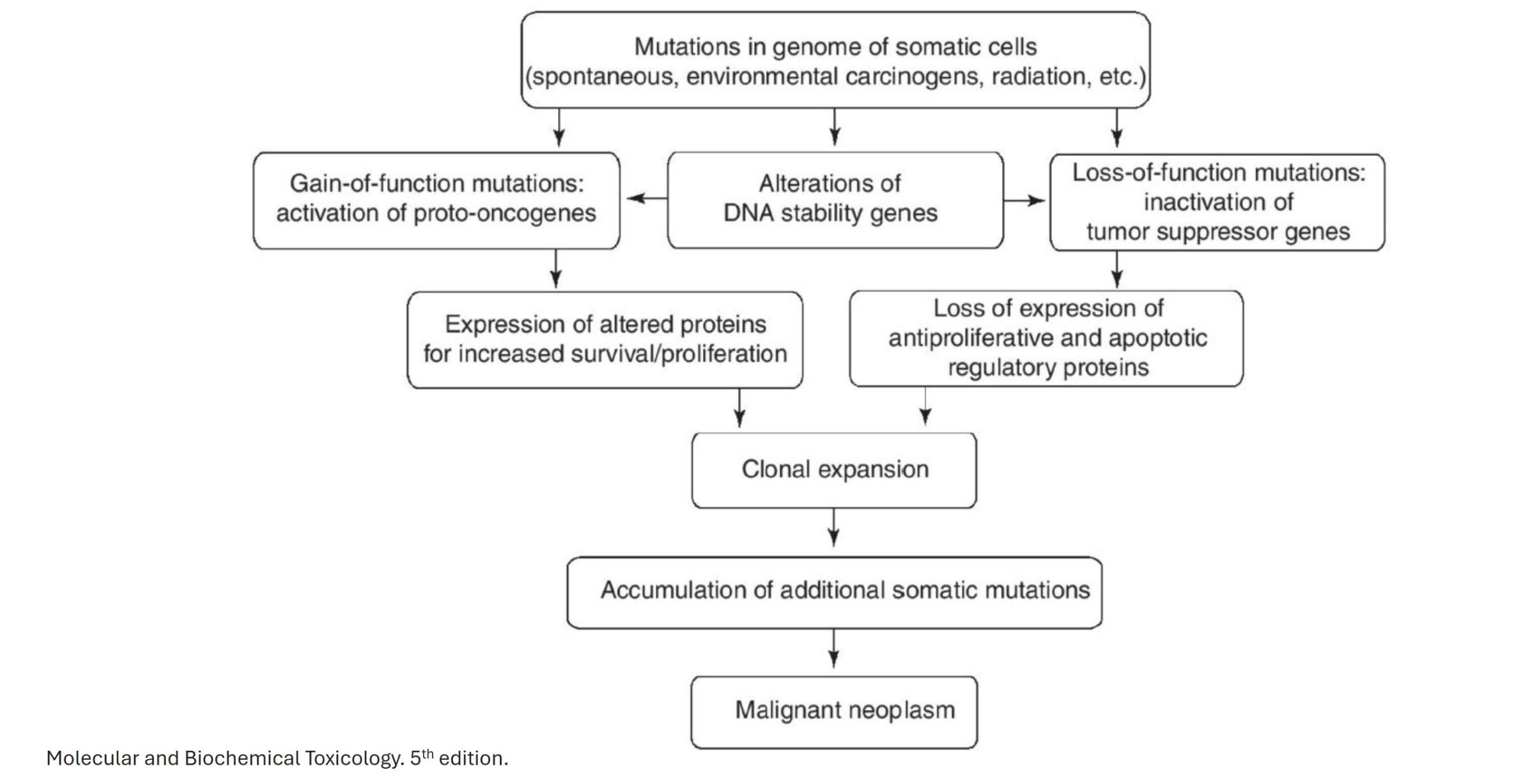
Describe the general mechanism of cancer arising from protooncogene mutation
Mutations in genome of somatic cells → alteration of DNA stability genes (DNA replication, cell cycle, DNA repair) → gain of function mutations: activation of protooncogenes → expression of altered proteins for increased survival/proliferation → clonal expansion → accumulation of additonal somatic mutations → malignant neoplasm
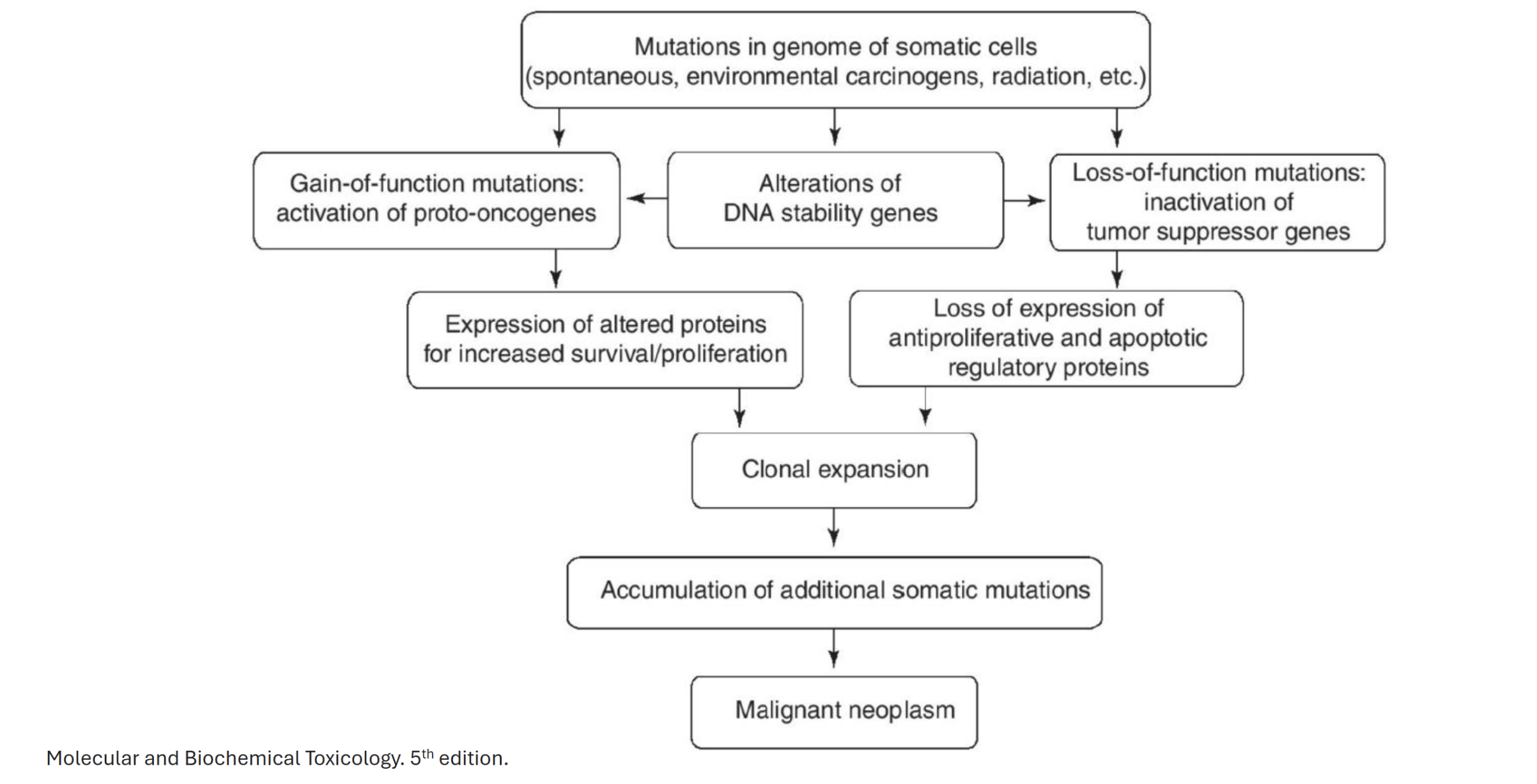
Describe the general mechanism of cancer arising from tumour suppressor gene mutation
Mutations in genome of somatic cells → alterations of DNA stablity genes (DNA replication, cell cycle, DNA repair) → loss of function mutations: inactivation of tumour suppressor genes → loss of expression of antiproliferative and apoptotic regulatory proteins → clonal expansion → accumulation of additional somatic mutations → malignant neoplasm
Clastogen
Mutagens that cause gross chromosomal rearrangements
Give an example of chromosomal rearrangement leading to cancer
Philadelphia chromosome → associated with chronic myeloid leukemia
Chromosome 9 and 22 rearrangment contains a BCR-ABL1 fusion gene → encodes constitutively active tyrosine kinase signaling protein
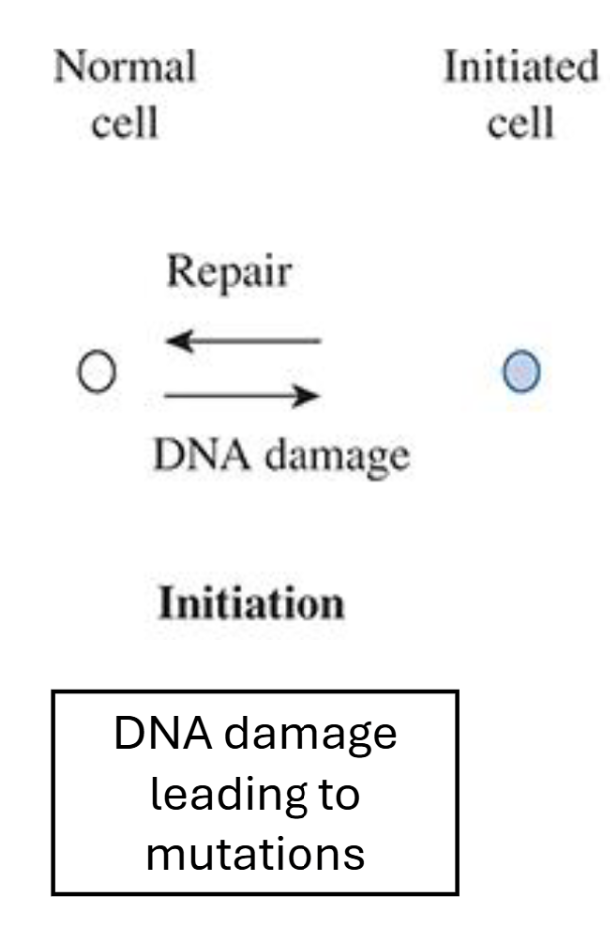
Describe the initiation stage of the Multistage Model of Carcinogenesis
Genotoxic agent modifies DNA → mutation
Irreversible process, however DNA repair mechanisms can repair
One cell division is necessary to “lock in” mutation
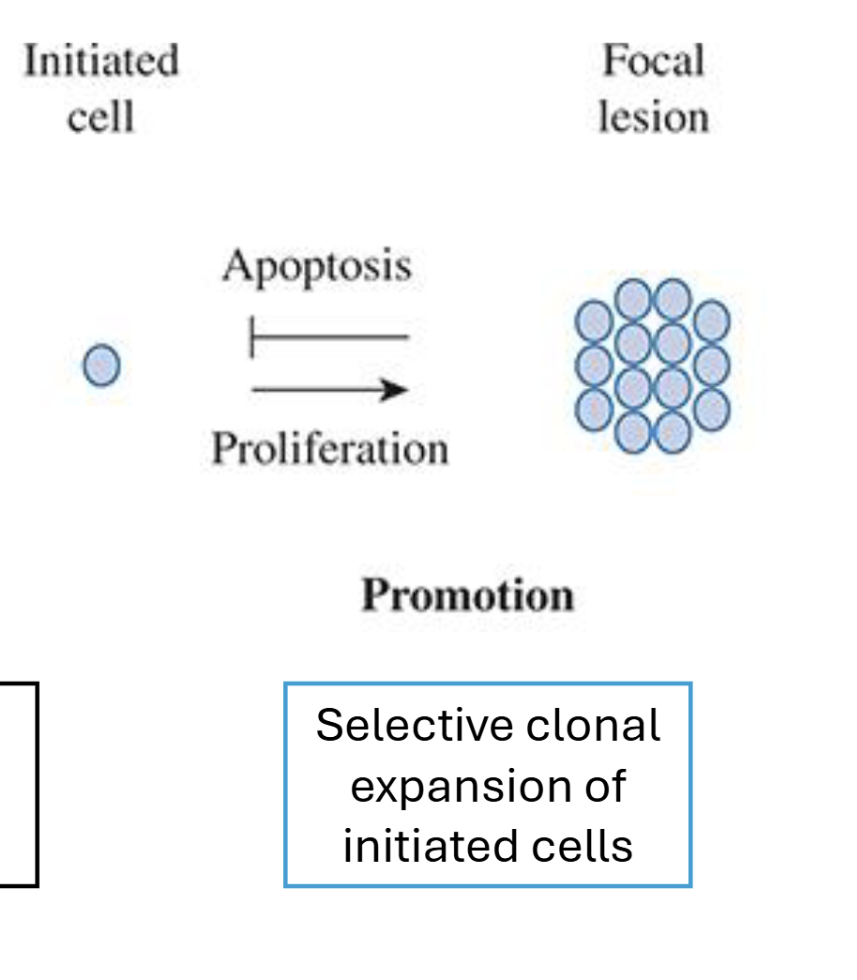
Describe the promotion stage of the Multistage Model of Carcinogenesis
Selective expansion of cells
No direct DNA modification or mutation involved
Multiple cell divisions necessary → clonal expansion of initiated cell population
Reversible via apoptosis of cells
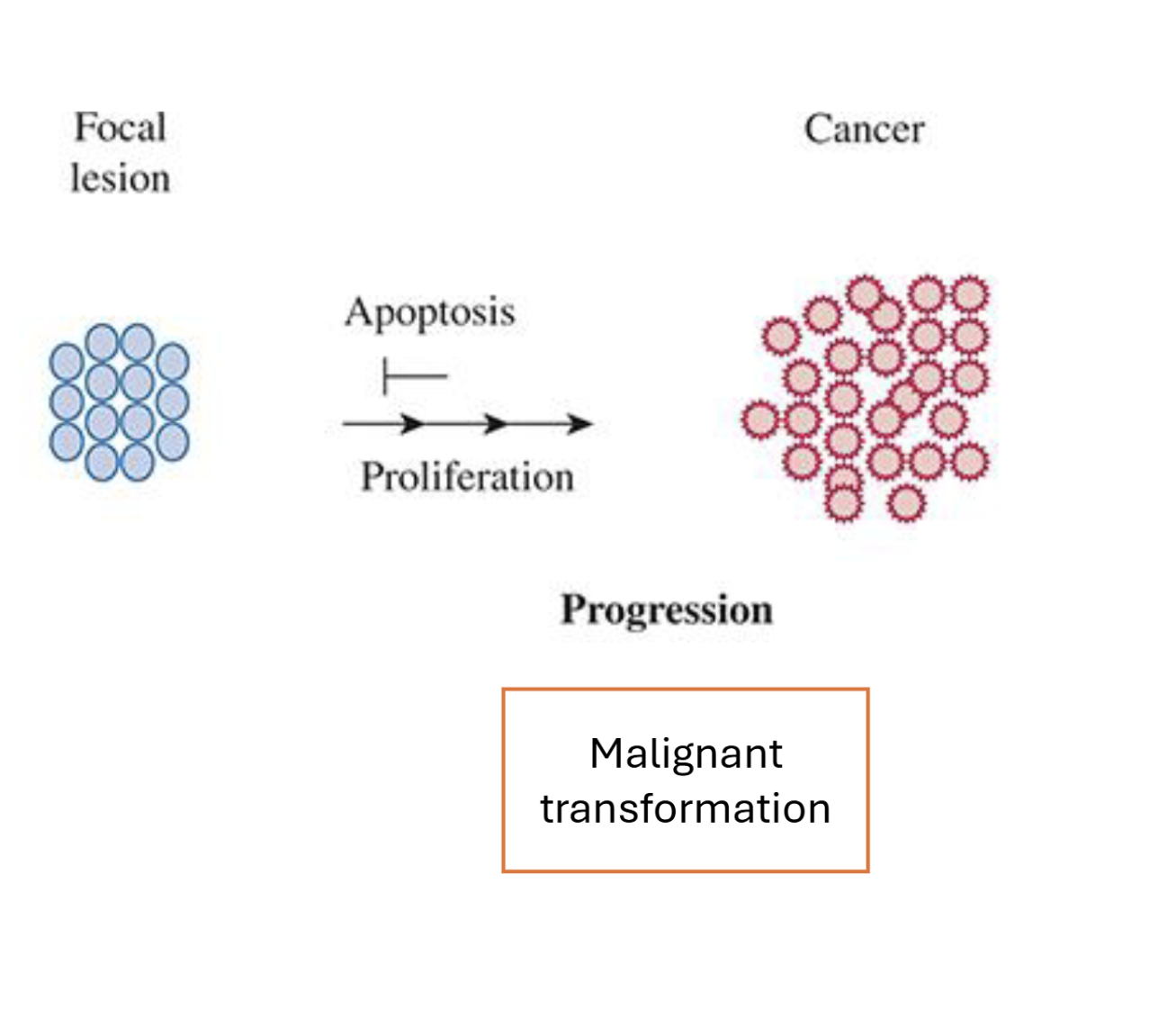
Describe the progression stage of the Multistage Model of Carcinogenesis
Malignant transformation
More mutations acquired (or chromosome disarrangement)→ no longer reversible
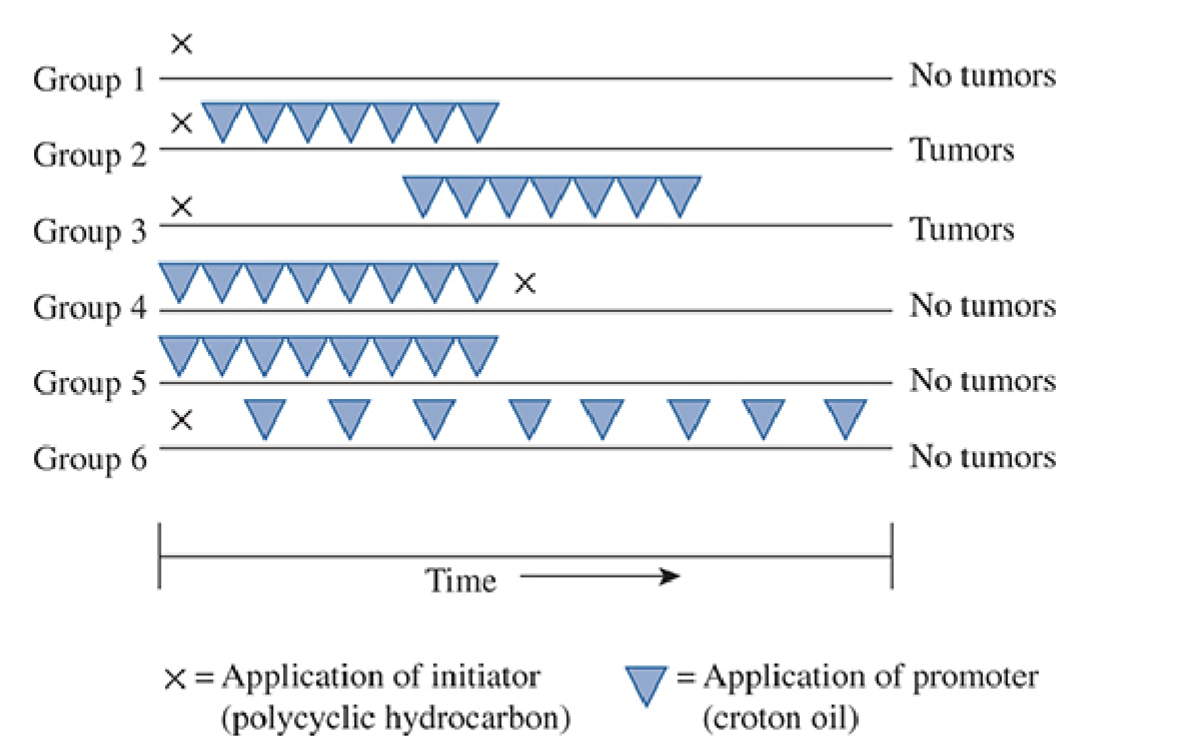
Describe what is being shown in the image
Application of an initiator causes DNA damage which is irreversible, but not on its own
Promotion (stimulation of cell proliferation) is non-genotoxic and reversible; required after initiation to form tumours
Promoter must be applied many times → single application not sufficient
Describe the procedure and mechanism for studying multistage carcinogenesis in vivo
Commonly use mouse skin model
Initiation via subcarcinogenic dose of carcinogen (eg. DMBA, targetting Hras1)
Repeated application of tumour promoting agent (eg. TPA)
Progression of skin cancers (papillomas) → squamous cell carcinoma
What may differentiate the significance of a pro-carcinogen that is transformed in the body into the ultimate carcinogen between a carcinogen not requiring transformation
Metabolic activation
Tissue specific toxicity
Distribution of metabolic enzymes (eg. CYP 1A1, 1A2, 2E1, and 3A4 are found predominantly in the liver)
Dose response effects (dose may vary in different tissues, even in same organism)
Differences in response
Eg. P450 polymorphisms
Describe the metabolism of Benzo[a]pyrene into its ultimate toxicant and possible detoxification mechanisms
Benzo[a]pyrene -(via CYP1A1)→ epoxide -(via epoxide hydrolase)→ diol -(via CYP1A1)→ BPDE (ultimate toxicant)
BPDE can be detoxified via GSH conjugation or epoxide hydrolase, however, bay region prevents epoxide hydrolase from binding and detoxifying
![<p>Benzo[a]pyrene -(via CYP1A1)→ epoxide -(via epoxide hydrolase)→ diol -(via CYP1A1)→ BPDE (ultimate toxicant)</p><p>BPDE can be detoxified via GSH conjugation or epoxide hydrolase, however, <strong>bay region</strong> prevents epoxide hydrolase from binding and detoxifying</p>](https://knowt-user-attachments.s3.amazonaws.com/8b2dc76e-d565-46f1-9fd5-63fbbbc69b1e.png)
Describe the mechanism of toxicity of Benzo[a]pyrene
Converted to ultimate carcinogen BPDE
Causes cellular damage via epoxide reactions with DNA and proteins
Molecular analysis of p53 mutations in lung tumours found the abundance of G:C to T:A transversions. What is the likely toxicant causing these mutations and how are humans exposed to it?
Occurence of G:C to T:A transversions increase with increasing exposure to cigarette smoke
Benzo[a]pyrene causes G:C to T:A transversions in DNA
Direct acting vs indirect acting alkylating agents
Direct acting → do not require metabolic activation (eg. alkylalkanesulfonates)
Indirect acting → require metabolic activation (eg. N-nitrosamines, DEN)
Common reaction sites of alkylating agents
Nucleophilic sites
Eg. N7 Guanine, N3 Adenine
What is the exposure route and mechanism of action of aromatic amines and amides
Exposed through cigarette smoke and other sources
Transformed via phase I metabolism to hydroxylated metabolites, which form adducts with proteins and DNA → liver and bladder carcinogenicity (eg. aniline, benzocaine)
Describe the general mechanism of chloroform or melamine induced carcinogenicity
Cytotoxicity induced by these chemicals leads to regenerative hyperplasia at high doses in vitro→ persistent regenerative growth, acquiring spontaneous DNA mutations → tumourigenesis
Role of PPAR in carcinogenesis
PPAR regulates cell growth and DNA synthesis → PPAR-alpha agonist exposure may promote tumours
Describe a mechanism of receptor-mediated carcinogenesis
PPAR binding → increased DNA synthesis / altered cell growth
ER binding → oral administration of 17-beta-estradiol in mice increases incidence of mammary tumours; DES administration increases risk of mammary and cervical cancers
Why is dietary intake of cholate/choline important in context of carcinogenesis?
Choline/cholate is a precursor for methyl groups in the body
Methylation affects expression of genes → focal methylation of tumour suppressor genes (silenced) or global hypomethylation resulting in demethylation of oncogenes (expression) may contribute to carcinogenesis
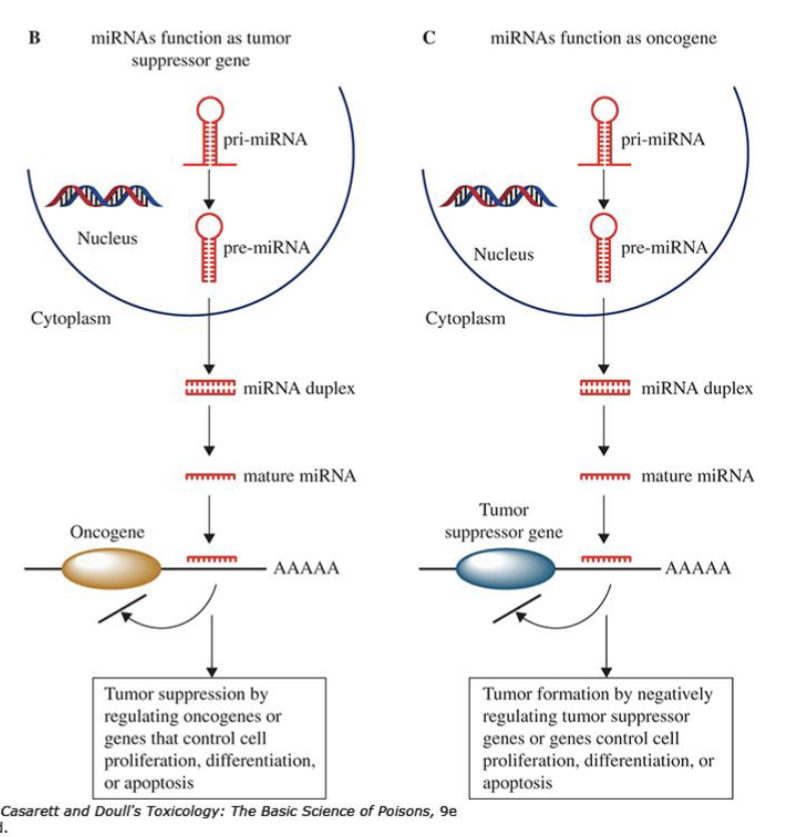
How can microRNAs have a dual function in gene expression regulating cell growth/replication
miRNAs can act as tumour suppressors (by degrading/repressing oncogenes) or as oncogenes (by degrading/repressing tumour supressors
How does the immune system regulate cancer?
Cold tumours - increase in immunosuppressive cells (Tregs), exclusion of CD8+ cells and NK cells
Hot tumours - increase in CD8+ and NK cells and suppression of Tregs
Immunotherapies usually target hot tumours because they express specific antigens
Why may oxidative stress be described as a non-genotoxic mode of action in carcinogenicity
Increases in ROS can lead to oxidative DNA damage
Oxidative stress can drive cell growth (via NFKB or MAPK pathway)
How does ionizing or UV radiation cause carcinogenesis
Damages DNA by ionizing atoms comprising DNA or generating ROS via interaction with water
Eg. May form pyrimidine dimer from UV radiation → misread by DNA polymerase
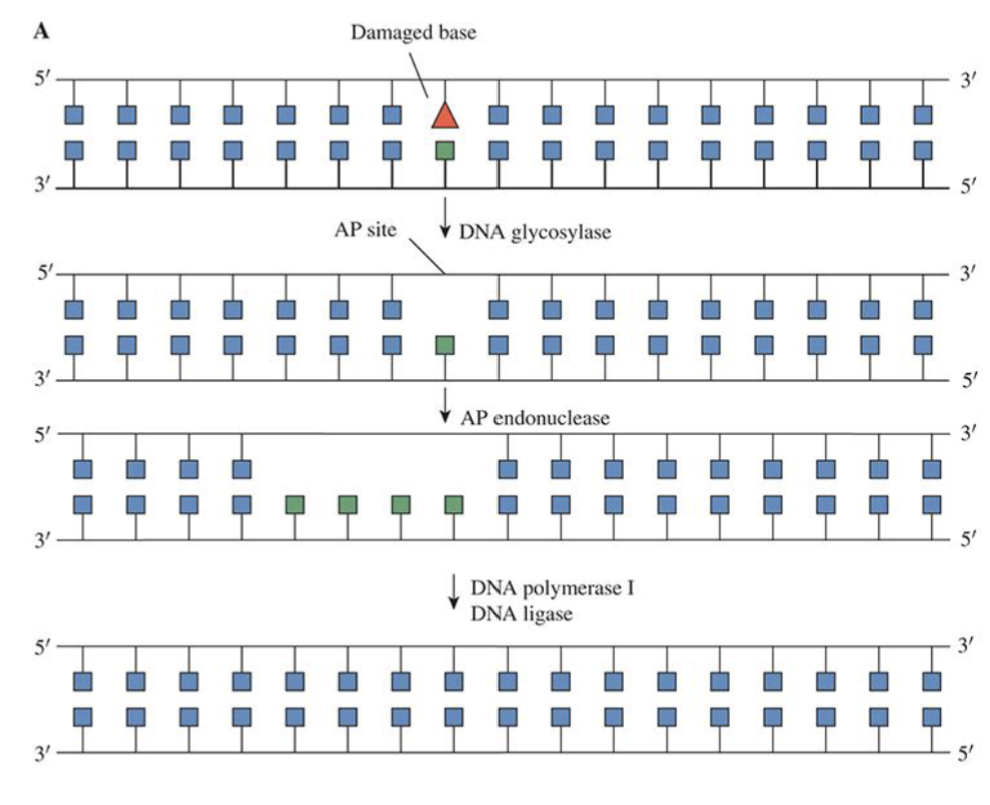
Base Excision Repair
Removal and replacing damaged bases
OGG1 recognizes and removes damaged bases → creates gap in DNA (AP site) → AP endonuclease cleaves gap → Gap filled by DNA polymerase and sealed by DNA ligase
Nucleotide excision repair
A fragment of nucleotides containing damaged lesion is removed and new DNA strand synthesized using undamaged strand as template
Applicable for bulky adducts and cross-linking lesions
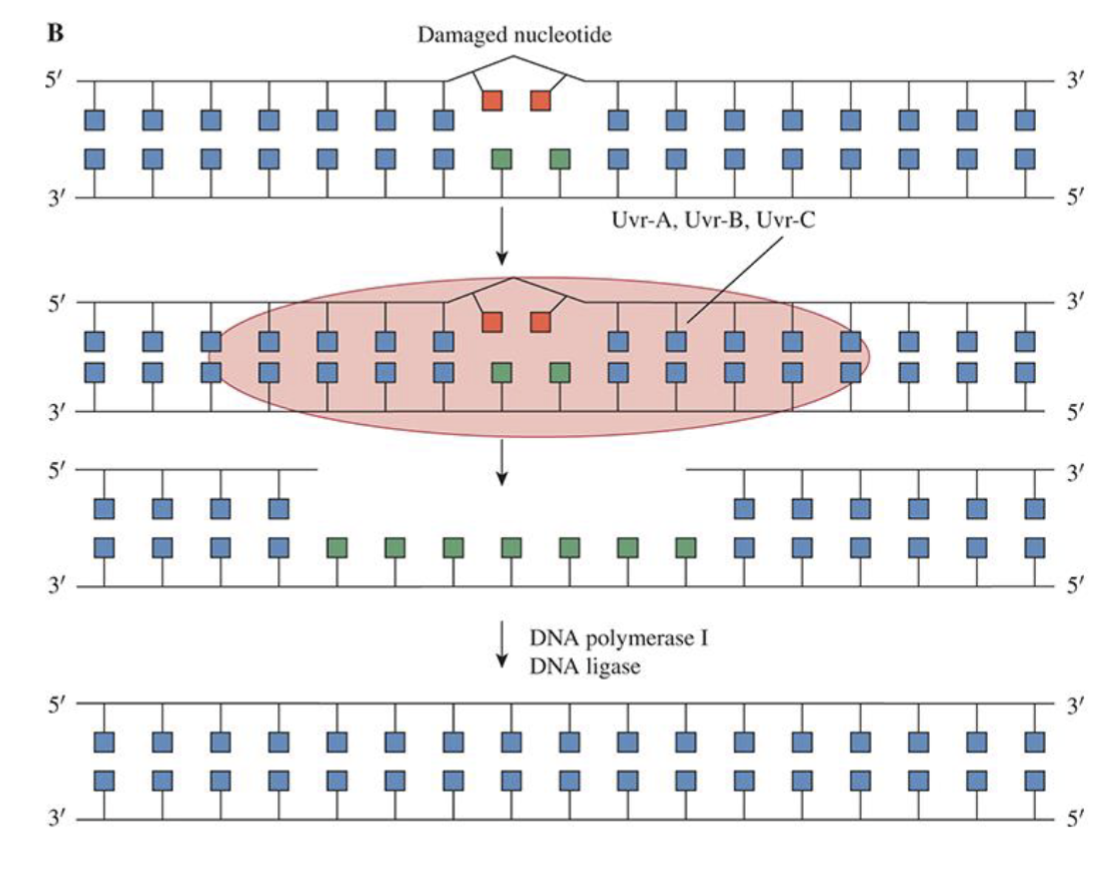
Mismatch Repair
Removal of mismatched bases and corrects insertion or deletion of short stretches of DNA
Mutation in MMR is a common cause of colorectal cancer
Double strand breaks
Caused by a variety of exogenous and endogenous sources
Need to be repaired quickly to avoid cell death, chromosomal abberations, mutations or cancer initiation
Assess the advantages and disadvantages of Ames Tests for detecting carcinogens
Ames test → plating bacteria unable to use histidine for energy and exposing to mutagen → test mutagenicity by seeing if they revert to use histidine for energy
Fast, cheap, sensitive (accurate), human/ethical
Not as relevant to human biology
Assess the advantages and disadvantages of Rodent Tests for detecting carcinogens
They can be relevant to human biology, but can be expensive, slow, not sensitive, and unethical
2 year rodent bioassay is gold standard → tests chronic administration of toxicant at max tolerable dose and at half that dose → assess tumour incidence in all tissues
Rodent carcinogenicity bioassays also respond to non-mutagenic carcinogens (promoters)
Functions of the liver
Detoxification
Metabolism (eg. T4 → T3, energy storage, bile, lipid turnover)
Storage (of minerals and fat soluble vitamins + vitamin B12
Immune function via sequestering of viruses and pathogen
Protein synthesis (eg. for blood clotting - prothrombin, or immune function - globulins)
Cholesterol production (precursor for sex hormones and vitamin D)
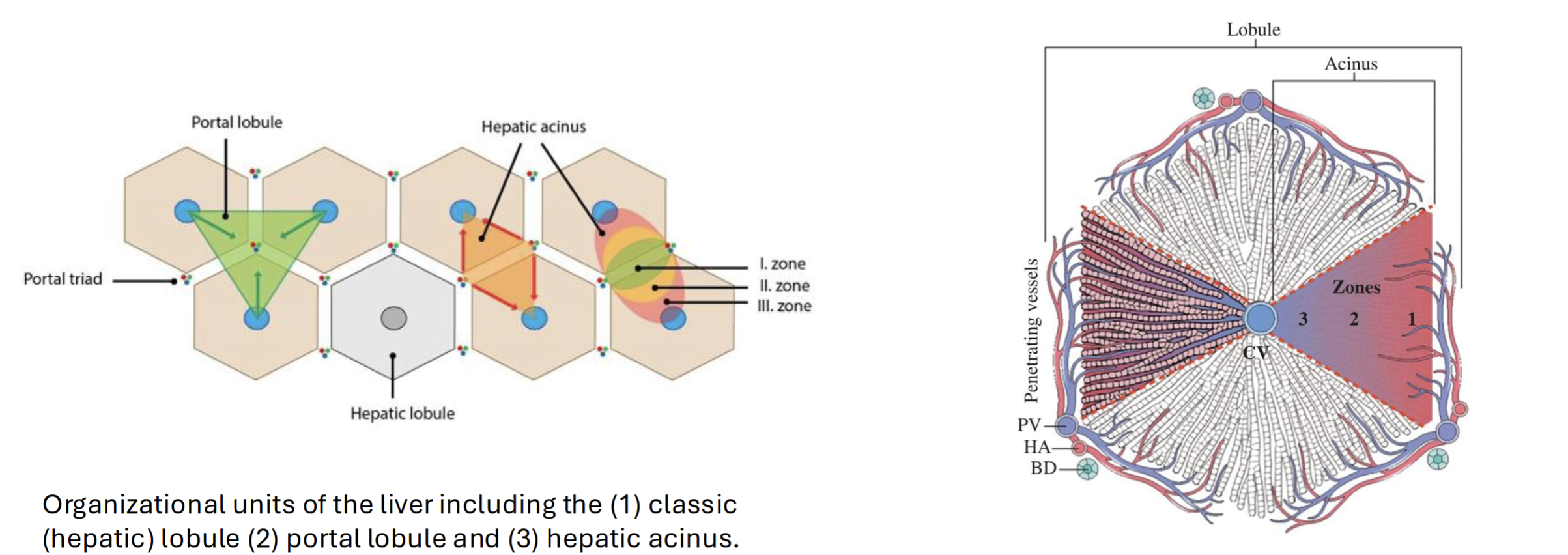
Anatomy of the liver
Divided into hexagons with central vein and portal tracts (consist of hepatic artery, portal vein, and bile duct)
Hepatic acinus (blood perfusion) moves from outside to inside in 3 zones, with the first being most oxygenated blood with most nutrients
Blood flows from portal tract to central vein, while bile flows from central vein to portal tract
Hepatocyte
Primary parenchymal cells in the liver
Responsible for variety of cell functions (metabolism, detoxification, immune cell activation)
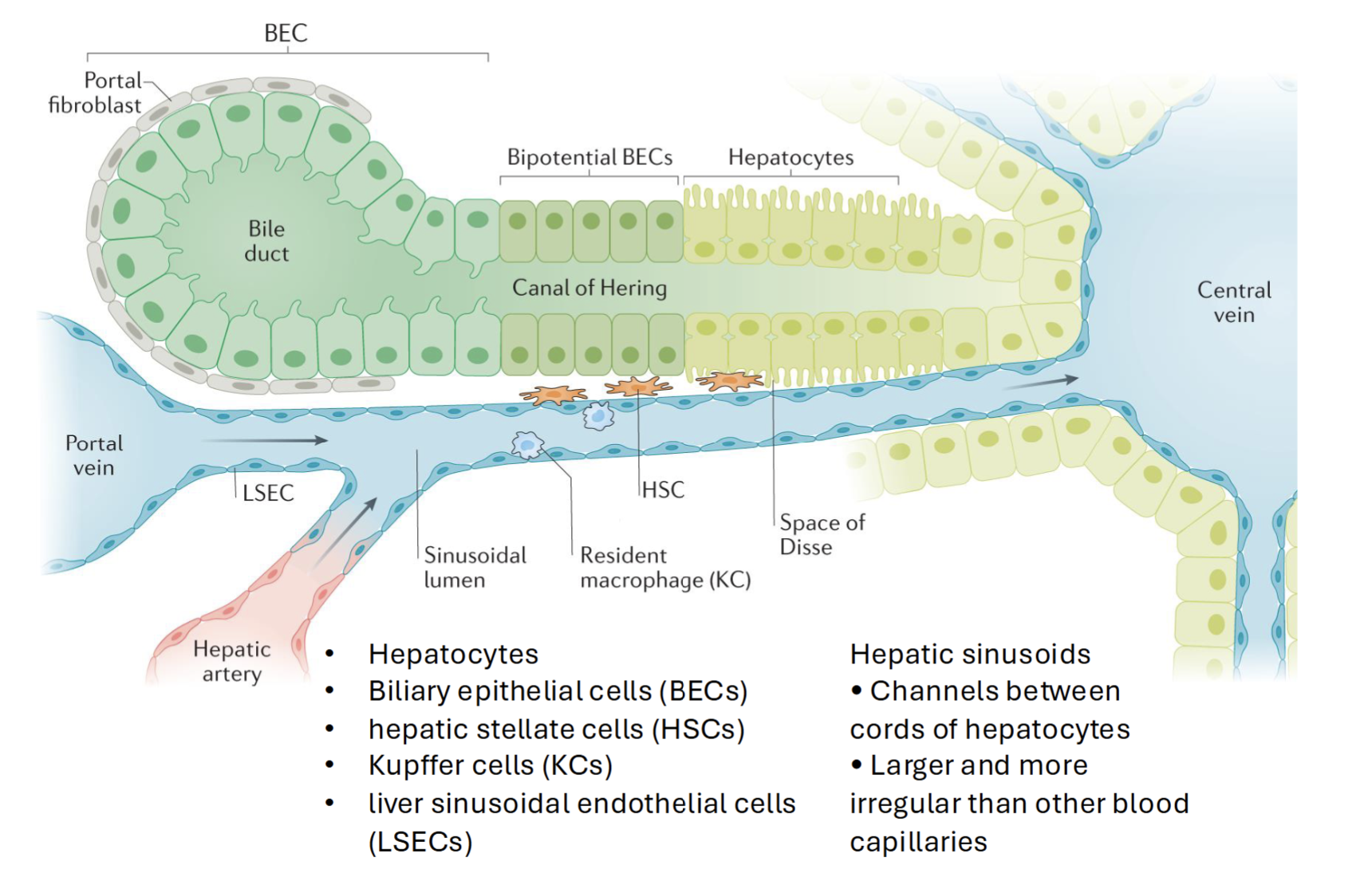
How do biliary epithelial cells and hepatocytes respond to toxic insult
BECs (parenchymal cells) differentiate to hepatocytes when hepatocyte proliferation is impaired and there is significant loss of liver mass (and vice versa)
Hepatocytes and BECs proliferate at high rates following acute liver injury → progressively lose regenerative capacity and become apoptotic/senescent in chronic liver disease
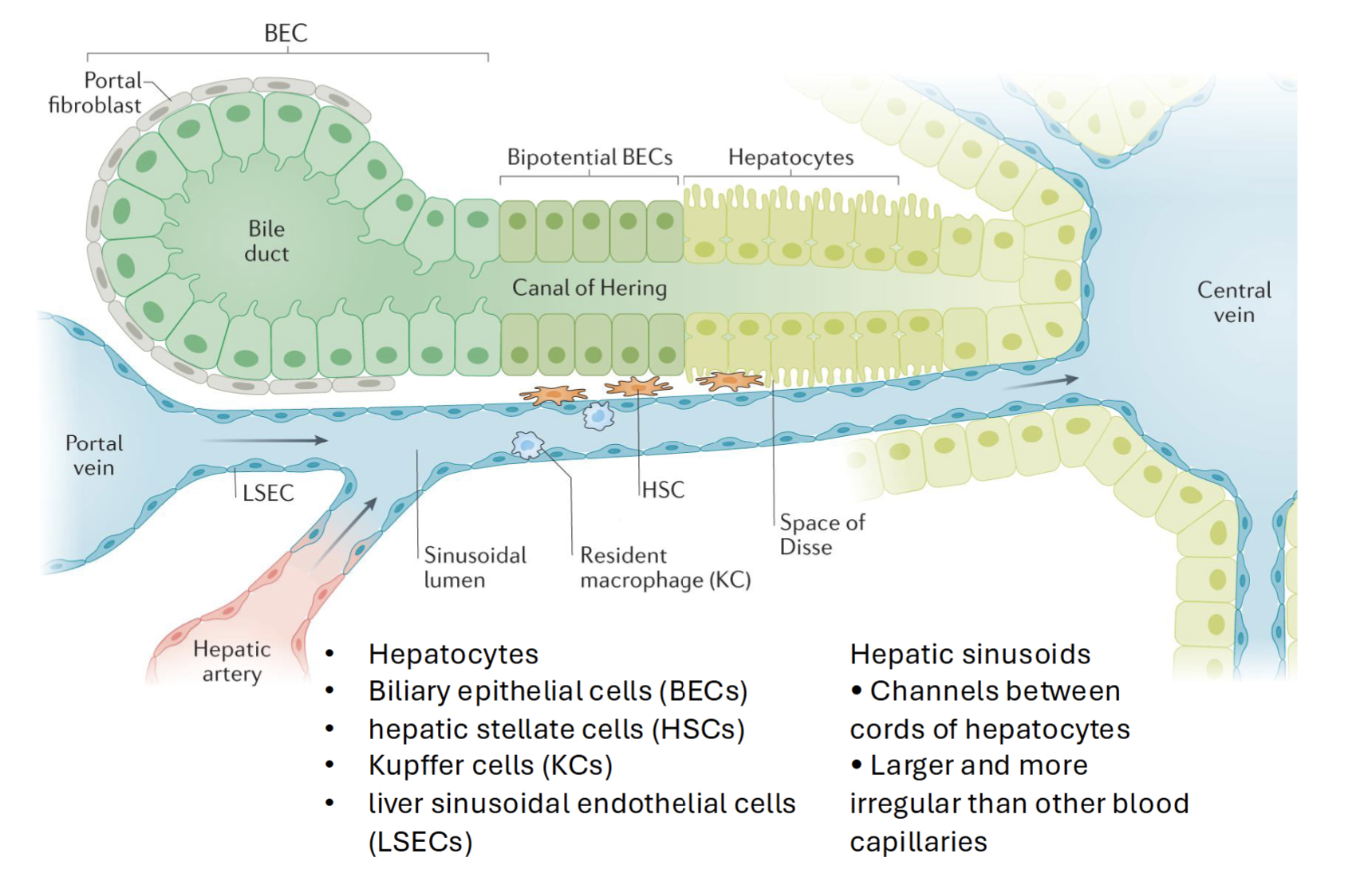
Kupffer cell
Resident macrophages, key players in acute and chronic liver disease
Eliminates damaged cells, prevents pathogen from spreading to circulation and clears metabolites and spent RBCs
Prevents movement of gut-derived pathogens
Zonated via cell surface markers
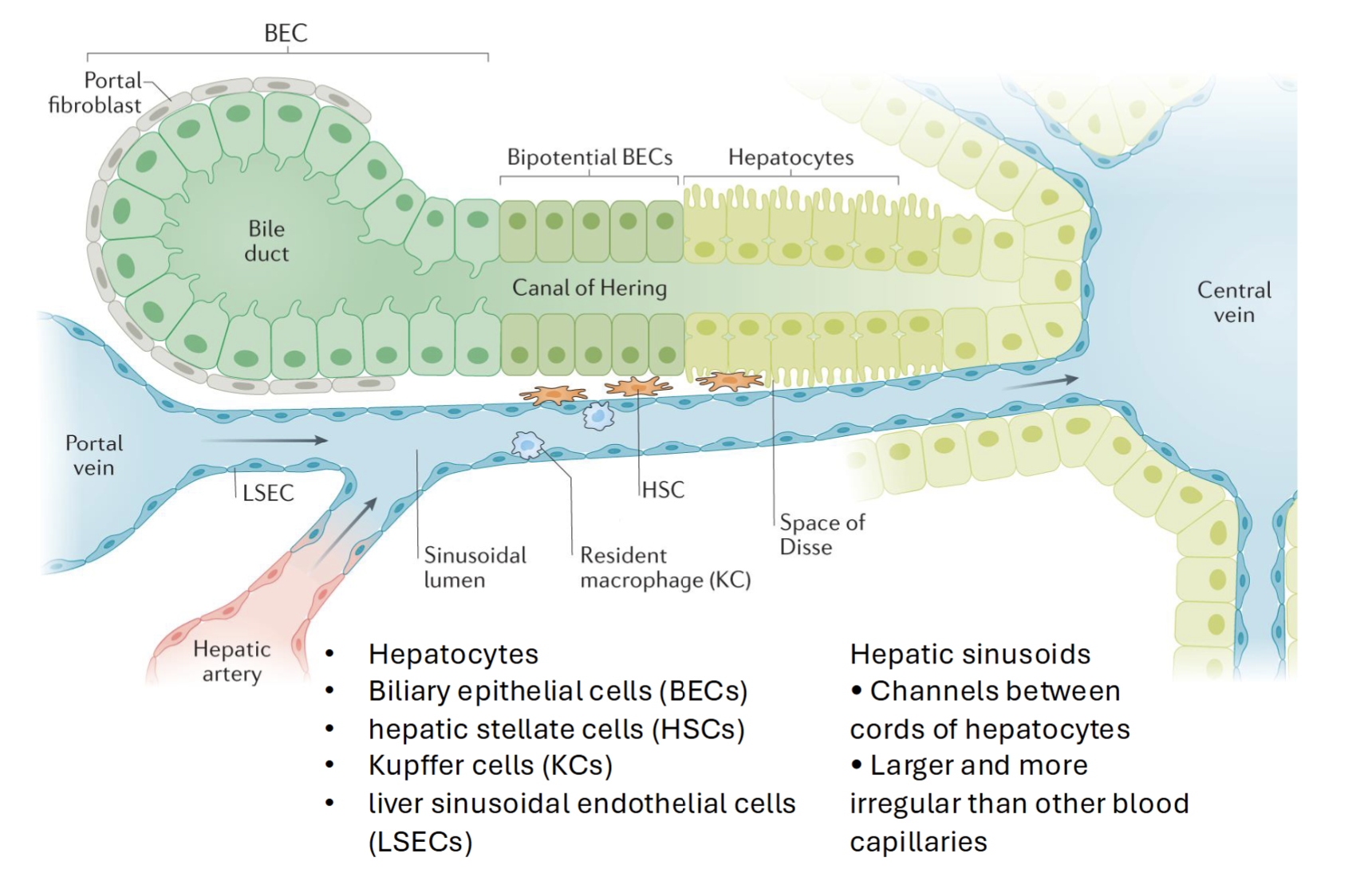
Hepatic Stellate Cells
Effectors in liver fibrosis
Dormant cells follow normal liver cell function
Activated by vitamin A into myofibroblast like cells (proliferate and migrate) → synthesize and excrete collegen, other matrix proteins, and smooth muscle actin
Liver sinusoidal endothelial cells
Lines smallest blood vessels in the liver (hepatic sinusoids)
Has pores - allows free diffusion of metabolites, proteins, drugs etc. into space of Disse → uptake by hepatocytes or HSCs
Scavenge lipoproteins, secrete cytokines and protanoids, NO, adhesion molecules
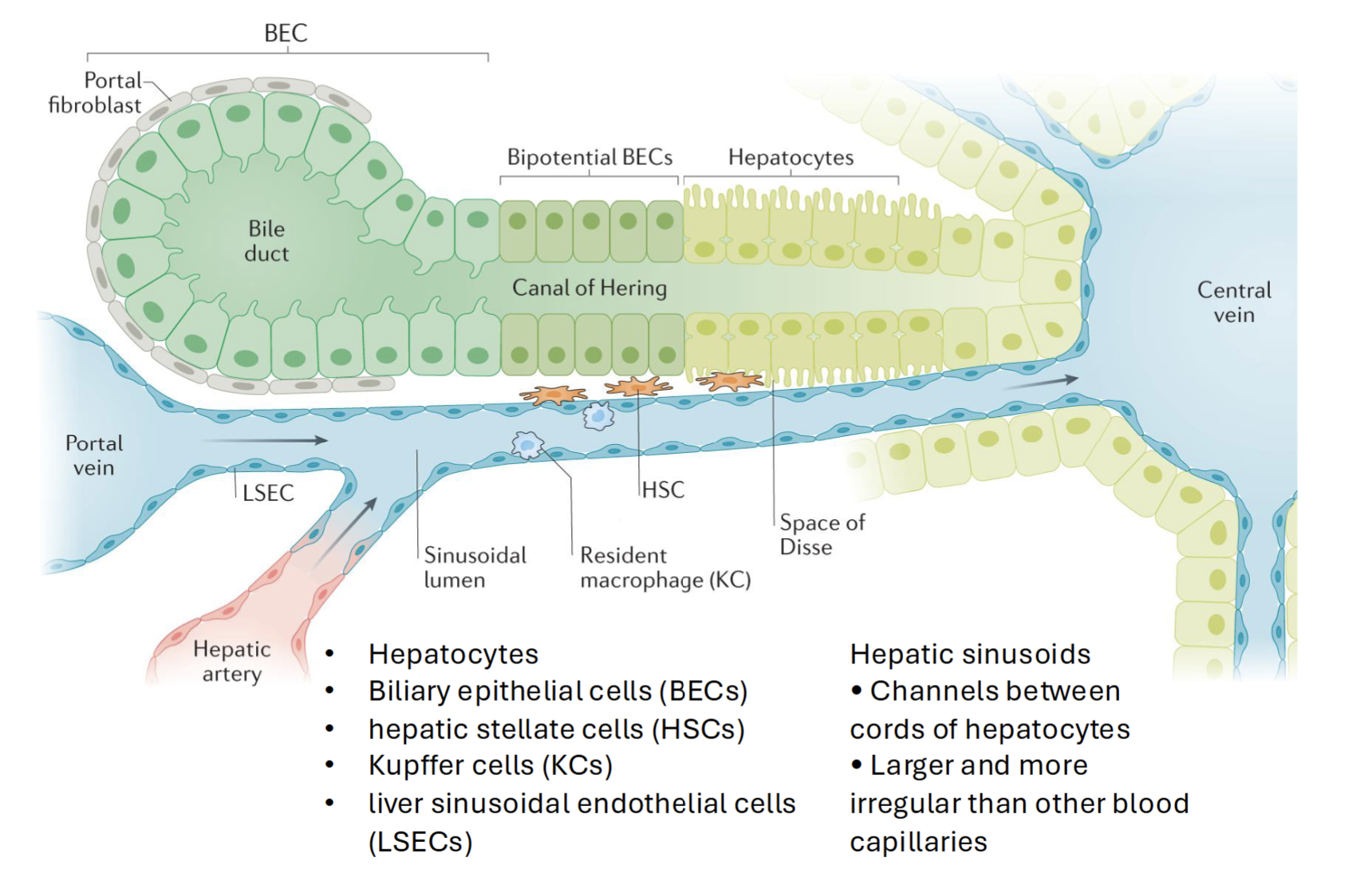
Acute vs Chronic Liver Injury
Acute liver injury (ALI) → severe acute hepatocyte necrosis without hepatic encephalopathy (loss in brain function in result of liver damage)
Chronic liver injury → Exhausted hepatocyte regenerative capacity → widespread hepatocyte senescence
Hepatocellular vs cholestatic liver injury
Hepatocellular → main injury at hepatocyte level
Liver cell necrosis
Cholestatic → main injury at biliary excretory system
Compare and contrast liver cell regeneration in response to acute vs chronic injury
Acute injury → Some hepatocytes are damaged, triggers inflammation from some macrophages and prorestorative function from others
Chronic injury → BEC expansion, loss of hepatocyte capacity to regenerate (senescence)
Activated HSCs increase TIMP expression (binds fibrillary collagen; unavailable for MMP activation)
MMP activation usually leads to decreased fibrosis and collagen
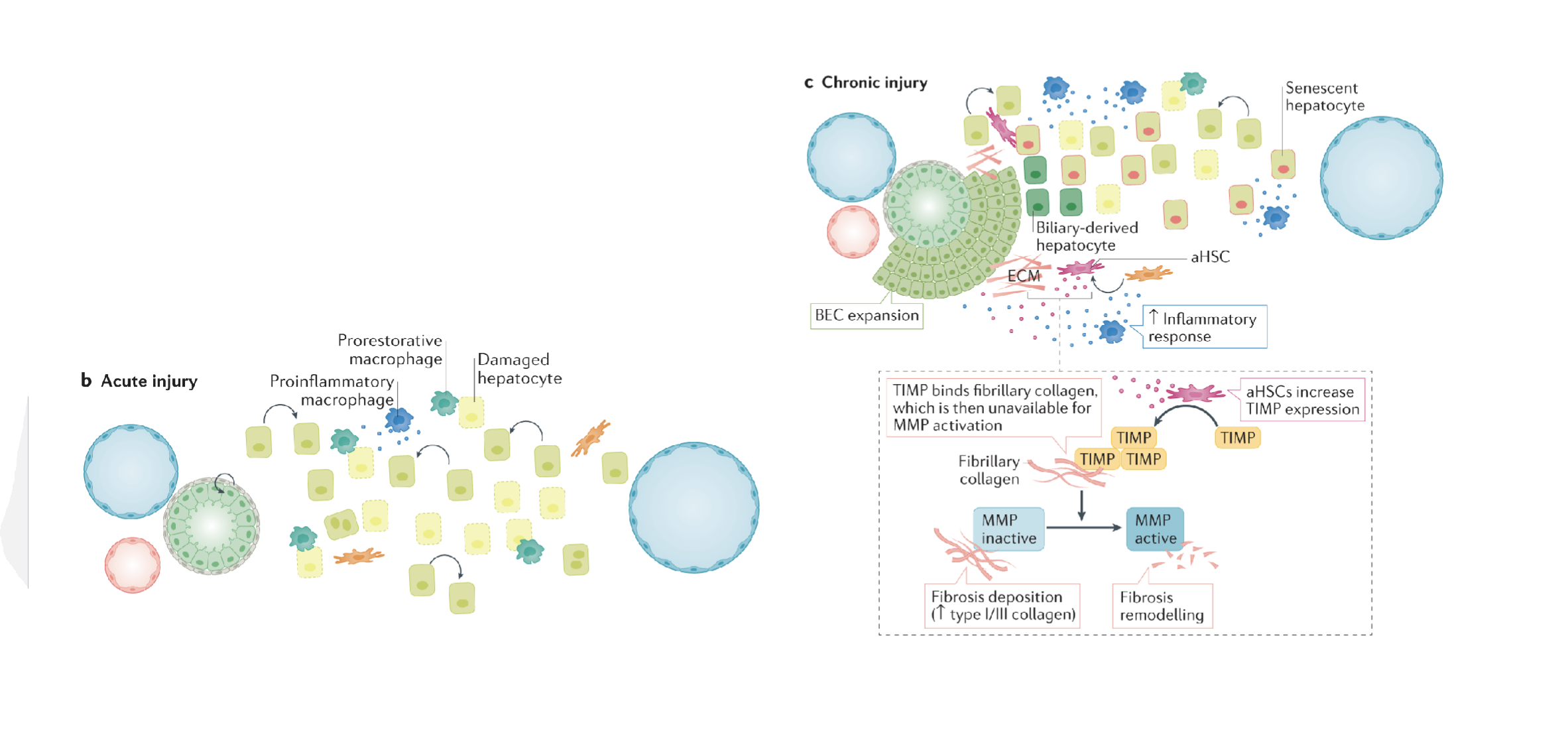
Describe the general pathological mechanism of liver fibrosis for acute liver injury
Toxic insult (eg. CCl4, acetaminophen, ethanol) → oxidative stress leads to injured hepatocytes (acute hepatic necrosis) → HSCs activated ,causing fibrosis
Describe the mechanism of toxicity in acetaminophen overdose
Converted into NAPQI by CYP enzymes → can conjugate with GSH or form APAP-protein adducts (initiates toxicity) →GSH depletion leads to inefficient detoxication → mitochondrial dysfunction and oxiditative stress → ALI
Describe the general pathological mechanism of liver fibrosis for chronic liver injury
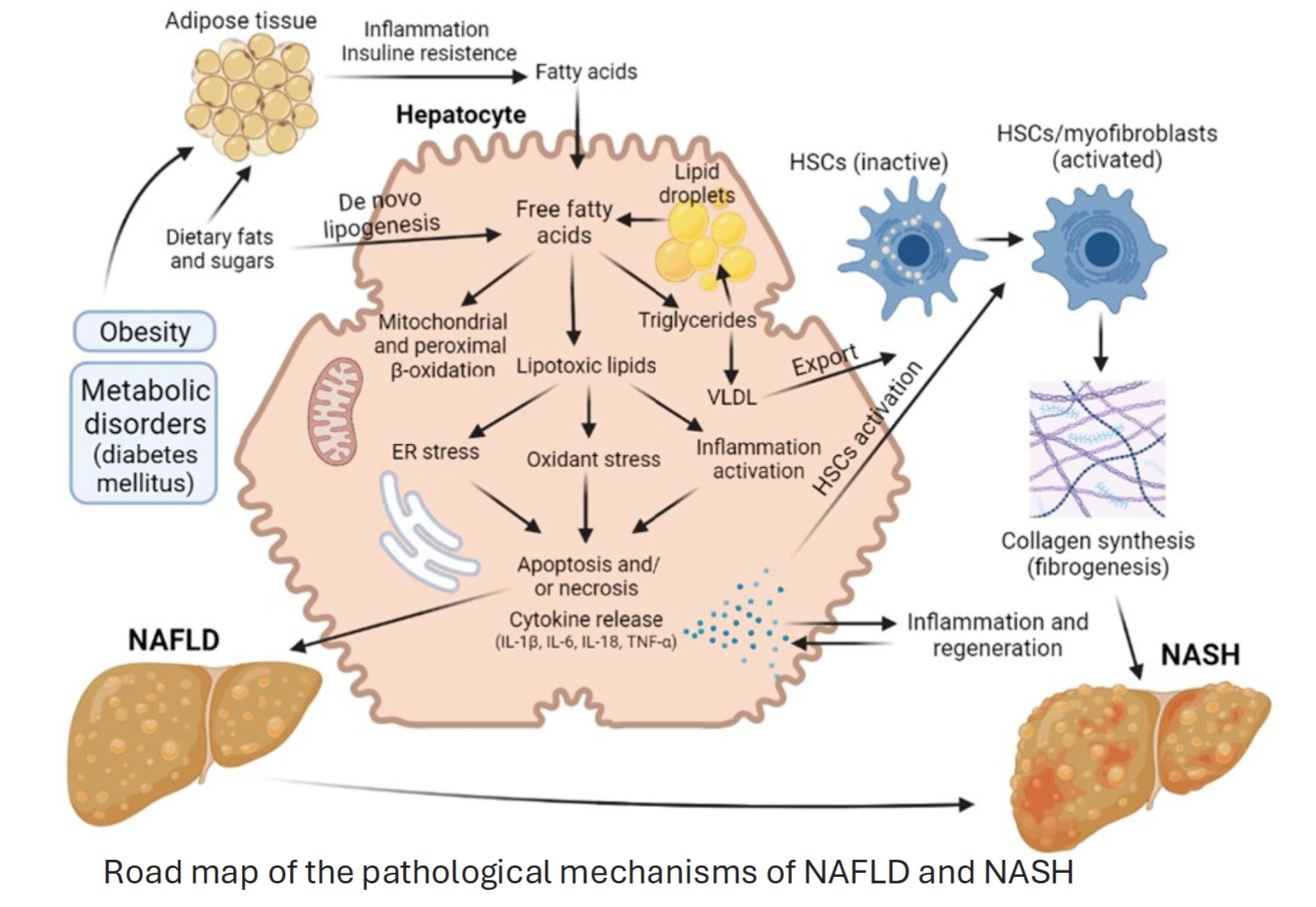
Conditions eg. obesity or metabolic disorders leads to deposition of fats into adipose tissue
Free fatty acids normally used for lipid storage, but too much results in synthesis of lipotoxic lipids→ oxidative stress and inflammation, resulting in cell death and cytokine release → NAFLD → may progress to NASH (more severe form)
Inflammation can also activate HSCs, causing fibrosis → NASH
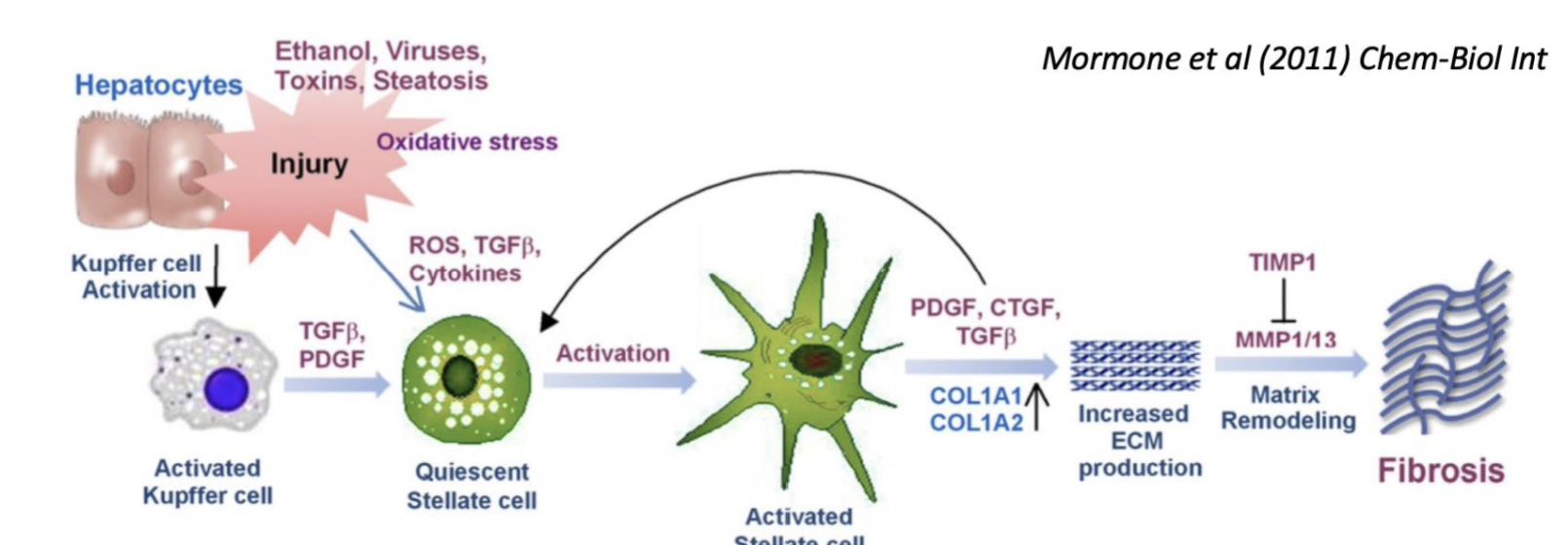
Describe the mechanism of toxicity of ethanol
Ethanol hepatocyte injury activates Kupffer cells → release cytokines to recruit inflammatory cells
Activation of HSCs → excess production and deposition of ECM proteins
Shift of HSCs towards fibril-forming collagens due to altered MMP expression → inhibition of normal degradation to these collagen types
Describe the molecular mechanisms of cholestasis
Mostly related to altered expression/function of transporters
Cell injury → accumulation of cholestasis causing chemical, bile acids, bilirubin, other bile components
Bile salts → increase proinflammatory gene expression in hepatocytes
Describe the mechanism of toxicity of sinusoidal endothelilal cells induced by Galactosamine
Loss of small proes in SECs and appearance of large gaps → leakage of blood cells into space of Disse → accumulate blood in liver → blood neutrophils damage hepatocytes, hypovolemic shock
What is a therapy that can combat sinusoidal epithelial cell damage induced by Galactosamine
MMP inhibitors
MMP usually activates TNF-alpha → recruitment of neutrophils to site of damage
Describe the mechanism of toxicity of Gadolinium chloride
Selective toxicity to Kupffer cells (due to phagocytic ability and degree of exposure of macrophages to circulating GdCl3)
Phagocytosis of circulating crystallized GdCl3 by Kupffer cells → lysosomal redissolution of GdCl3 → disrupts Ca2+ dependent processes → Kupffer cell depletion → reduced Kupffer cell derived mediators (eg. eicosanoids, cytokines, neutrophil chemoattractants, NO, ROS)
General functions of the kidney
Regulate bone mineral metabolism (via Vitamin D)
Regulate red blood cell production
Excretion of metabolic waste
pH regulation
Regulate blood pressure
Why is the kidney particularily susceptible to toxicity
High blood flow to kidney → relatively high amounts of toxicant in systemic circulation delivered to kidney
Process involving concentrating urine may lead to potential toxicants being concentrated in tubular fluid
Non-toxic concentration of chemical in plasma may reach toxic concentrations in kidney
Distribution of drug metabolising enzymes may result in bioactivation
What are causes for Acute Kidney Injury and what is used to detect it?
AKI → sudden loss of kidney function as a result in extent of serum creatinine increases or changes in urine output
Decline from GFR →
Prerenal factors (eg. intravascular volume depletion)
Postrenal factors (eg. bladder obstruction)
Intrarenal factors (eg. glomerulonephritis - glomerular inflammation)
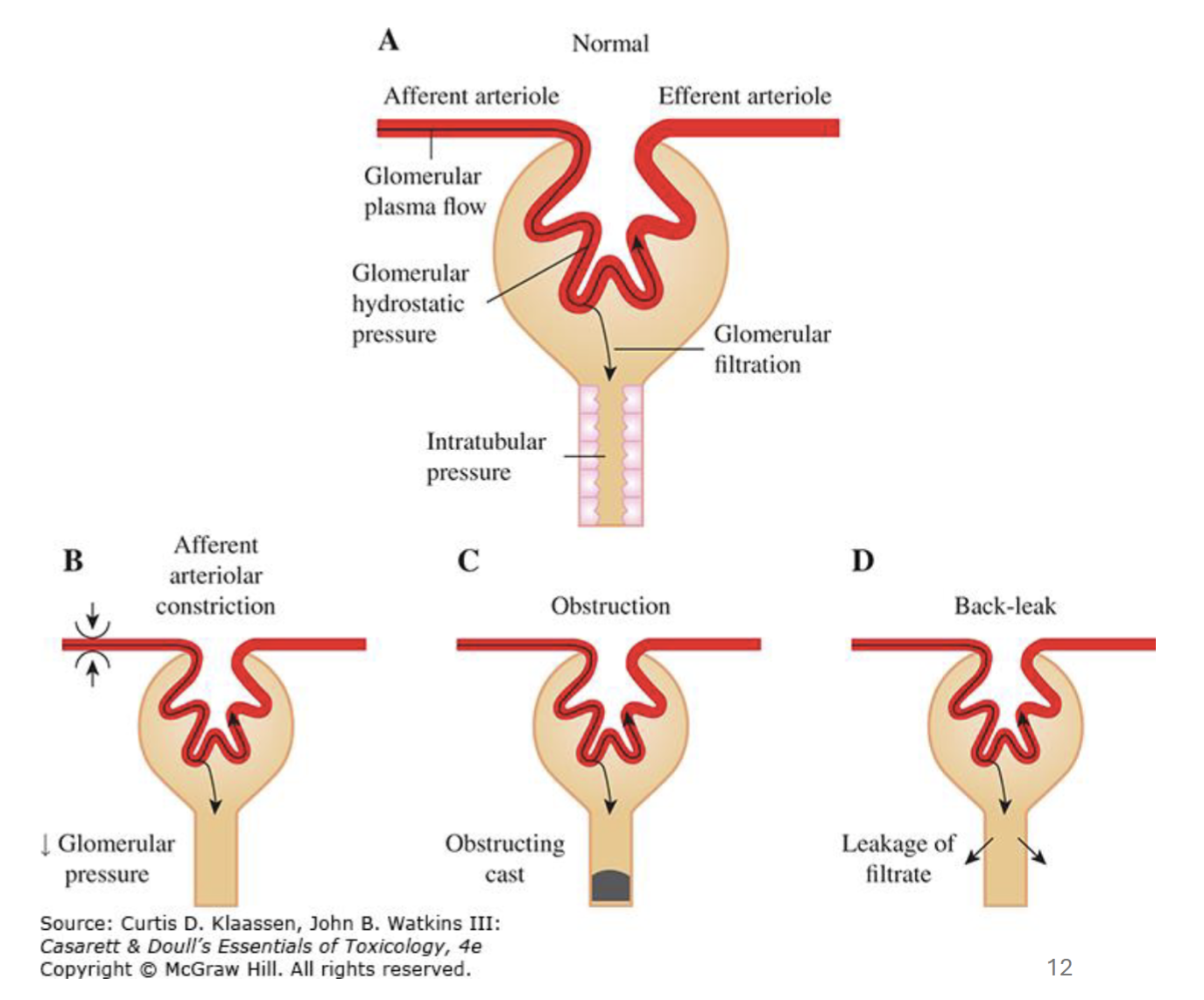
Glomerular filtration rate depends on…
Adequate blood flow to glomerulus
Adequate glomerular capillary pressure
Glomerular permeability
Lower intratubular pressure
Describe what causes chronic kidney disease
Prolonged exposure of toxicants
Previous AKI → persistent low-level injury or inflammation after AKI can lead to CKD or make kidney susceptible to more damage
“Initial kidney injury triggers secondary processes that contribute to CKD development.”
Explain what is meant by this phrase and describe an example
Kidney first may undergo AKI, which makes the person more susceptible to CKD
Eg. In nephron loss, other nephrons try to increase filtration in response to loss of nephrons to maintain homeostasis, but this may be maladaptive
List 5 markers of kidney damage and their significance
GFR → low GFR indicates CKD or AKI
Serum creatinine → high levels indicate impaired kidney function
Proteinuria → indicates kidney damage
Blood urea nitrogen → elevated levels may suggest kidney dysfunction
Cystatin C → elevated levels can indicate kidney damage
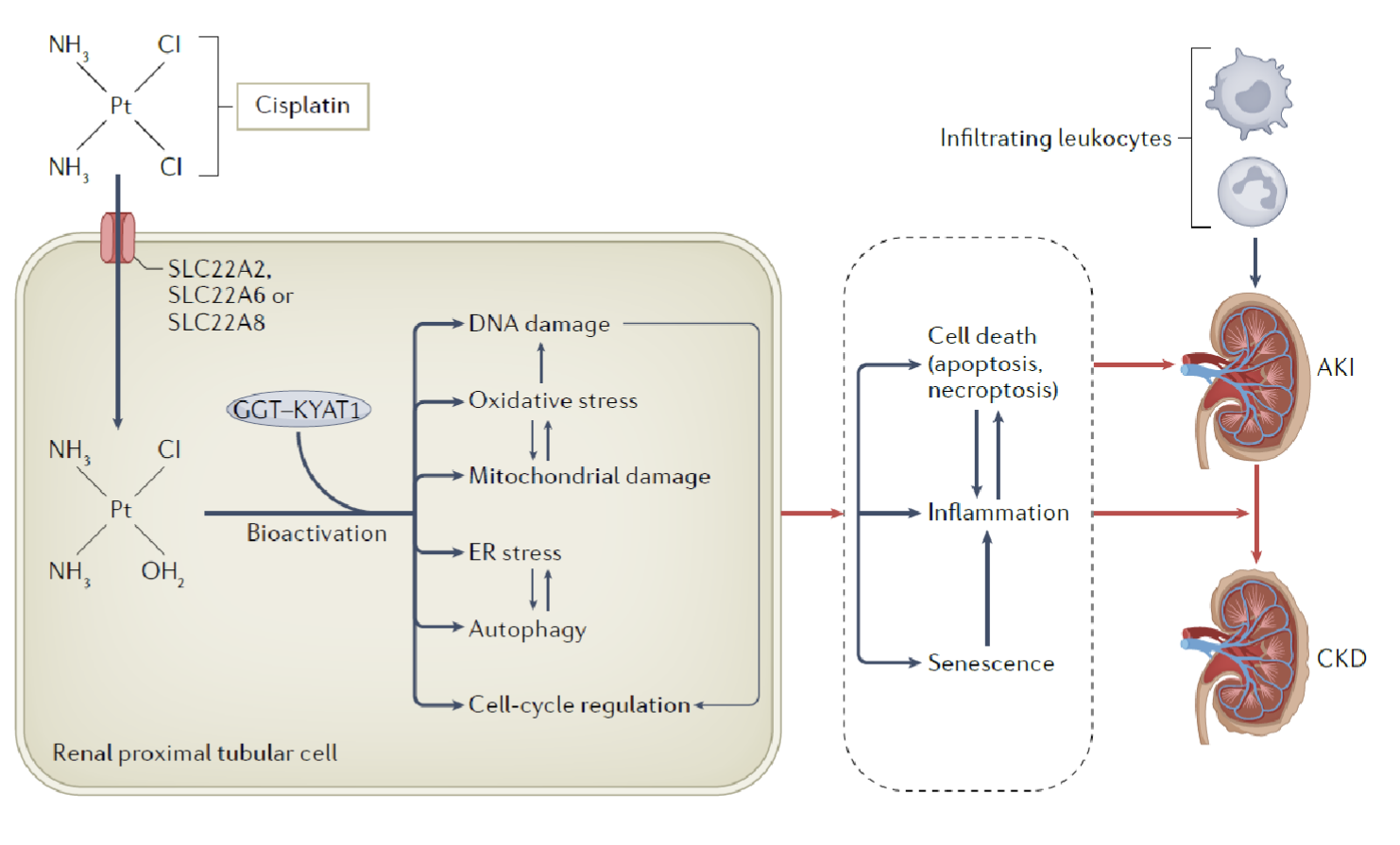
Cisplatin pathophysiology and mechanism of toxicity
Reaches 5-flold concnetration in renal tubular cells compared to plasma
Uptaken by SLC transporters into the cell, where it is bioactivated
Bioactivated cisplatin is an alkylating agent → ROS, ER stress, activation of inflammatory and apoptotic pathways → AKI/CKD
What actions are taken to decrease nephrotoxicity from cisplatin?
Cisplatin nephrotoxicity is dose-limiting adverse effect → need to reduce dose or stop treatment
Typically chemotherapeutic → switch to less nephrotoxic agent, but may compromise efficacy of drug
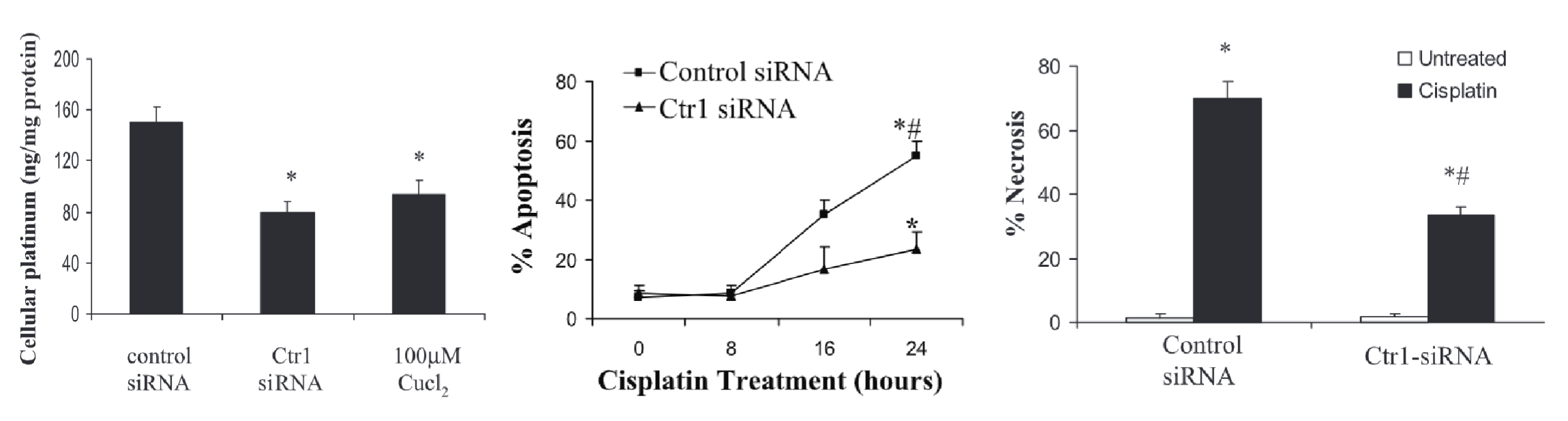
Describe the role of Ctr1 in cisplatin toxicity. What treatments are available to combat mechanism of toxicity?
Ctr1 (copper transporter) → can uptake cisplatin into cells
Addition of Ctr1 siRNA (KO) or Cu2+ (competitive inhibition) decreases uptake of cisplatin into renal tubular cells and decreases % cell death after cisplatin exposure
What is the role of megalin and cubulin in gentamicin toxicity
Gentamicin is endocytosed via megalin and cubulin complex, forming endocytic receptor (multifunctional; including transport of aminoglycoside antibiotics)
Describe the mechanism of toxicity of aminoglycosides eg. gentamicin
Endocytosis via endocytic receptor (megalin and cubulin)
Endosomes traffic gentamicin to mainly lysosomes, but also Golgi and ER
Inhibition of protein synthesis and folding → ER stress and unfolded protein response
Leads to calcium release → activation of calpain → activates caspases → apoptosis
May also act on mitochondria if released into cytosol → Cytochrome C release → apoptosis
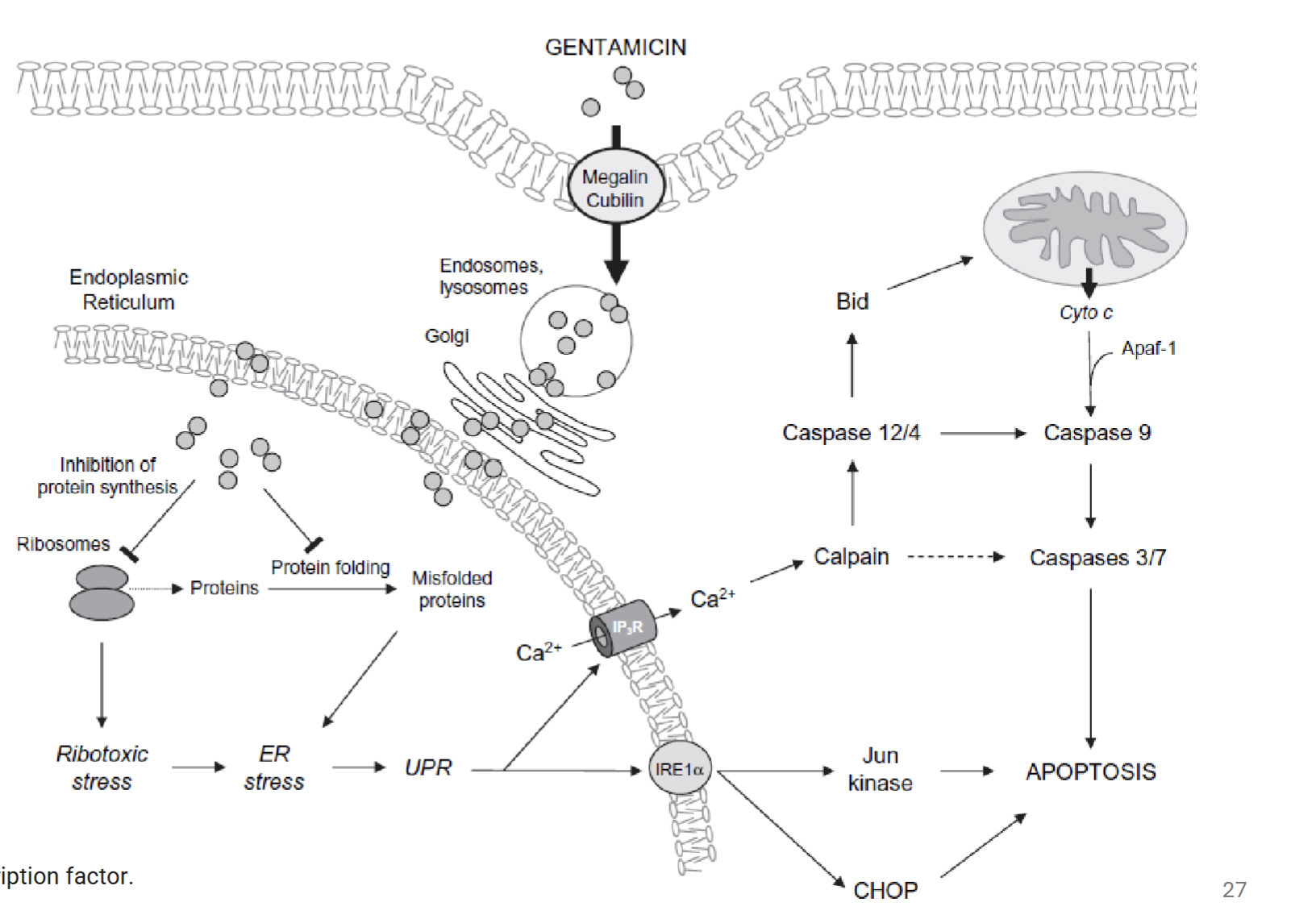
“Cytosolic concentration of gentamicin is a critical factor in its tubular cytotoxicity.” Explain what is meant by this statement
When gentamicin concentration exceeds threshold in lysosomes, Golgi, and ER, their membranes are destabilised (phospholipidosis), causing release into cytosol
Gentamicin acts on mitochondria, triggering release of Cytochrome C and other apopototic proteins; cell energy status impaired
What are physiological consequences of tubular epithelial damage
Toxic injury leads to cell death → loss of concentration gradient, leading to backflow in tubules
Death of cells detaches them from basal layer → macrophages try to clear debris → tubular obstruction → increased luminal pressure → backflow?
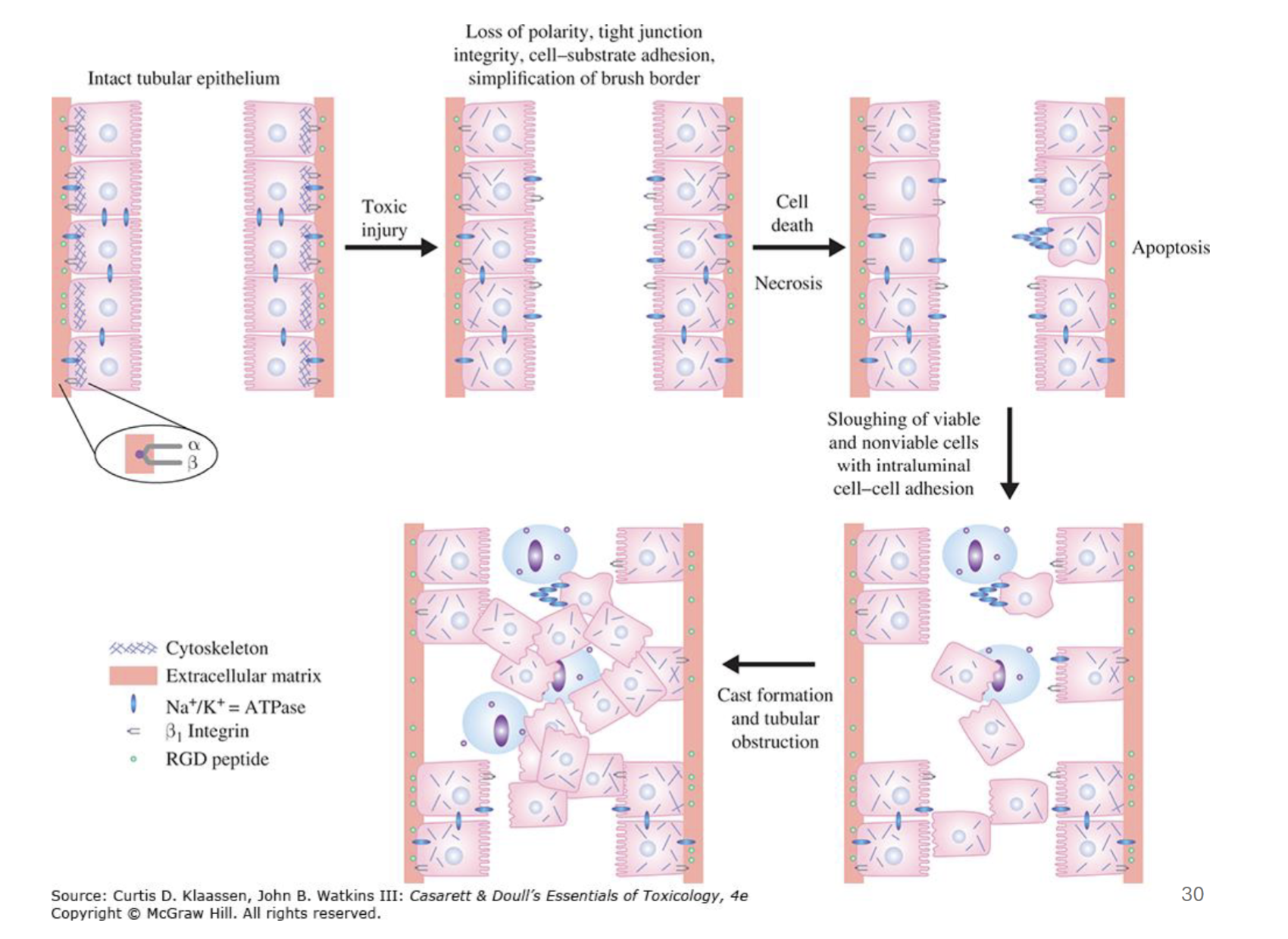
Why is gentamicin described to have multiple mechanisms of toxicity?
Can cause cell death via ER stress and/or mitochondrial toxicity
May also inhibit cell membrane transporters
Cell death also associated with activation of CaSR → bone regulation, other physiological processes, etc.
Inflammation and ischemia (decreased blood flow) may amplify cytotoxic effects of gentamicin
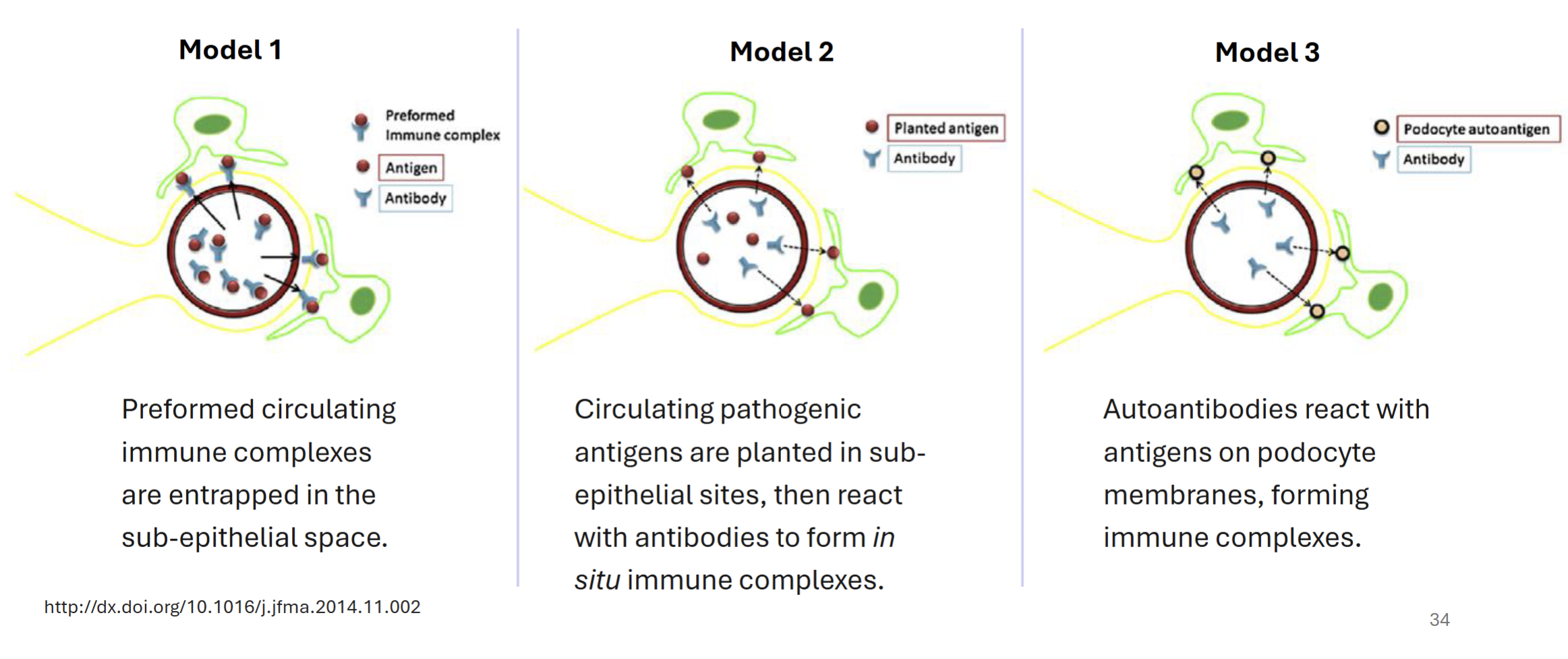
Compare and contrast the different models for D-penacillamine induced membranous nephropathy
All models involve subepithelial deposition of immune complexes (immune mediated) → abnormal pordocyte membranes → thickened glomerular membrane, leading to difficult filtration
Model 1 - preformed antigen-antibody complexes deposit into sub-epithelial space
Model 2 - circulating pathogenic antigens planted in subepithelial sites, react with antibodies to form immune complexes
Model 3 - autoantibodies react with antigens on podocyte membranes, which forms immune complexes
Autoantibodies against phospholipase A2 receptor and myeloperoxidase are common triggers
Aristolochic acid (AA) mechanism of toxicity
Renal uptake via OAT transporters (AA-I) → bioactivation to form nitrenium ion → DNA/protein adducts
Renal dysfunction, characterized by tubular dysfunction, proteinuria, and interstitial fibrosis
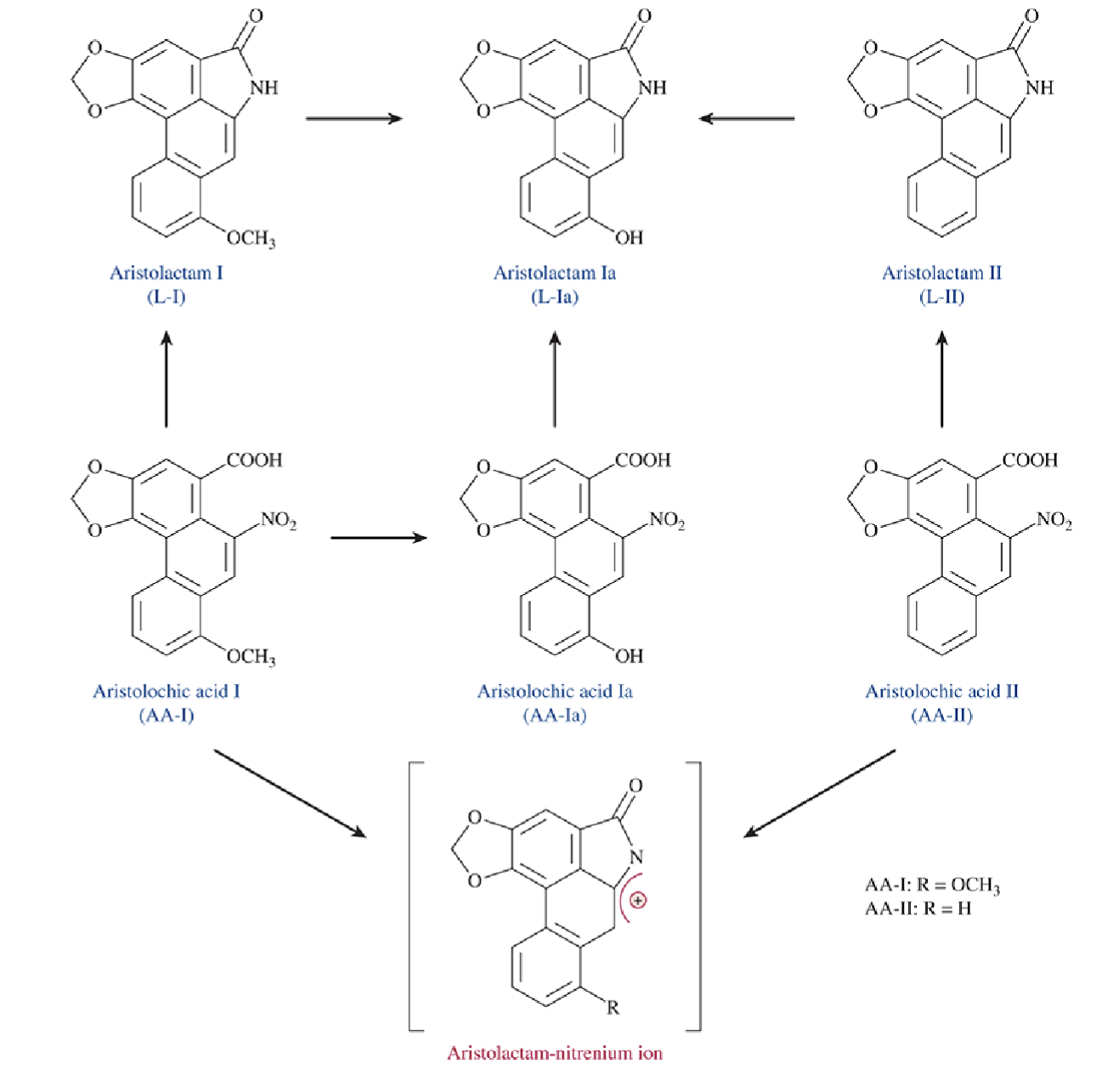
Describe the differences between AA-I vs AA-II induced nephrotoxicity
AA-I produced tubule necrosis and interstitial fibrosis while AA-II did not
Both produced similar levels of DNA adducts
Nephrotoxicity of AA-I may not be due to DNA adducts and damage
Describe the mechanism of toxicity of Sulfamethoxazole (SMX)
SMX metabolized to N-acetyl-sulfamethoxazole (NASM)
Crystalization of sulfonamide crystals leads to tubular lesions → occurs rapidly after drug introduction and improved after discontinuation of drug
Functions of Type I vs Type II alveolar cells
Type I → gas exchange
Type II → produces surfactant for alveoli
Based on the positioning of the lung and circulation surrounding it, what implications can be drawn in a toxicological standpoint?
Due to the circulatory system, the lung is one of the first contacts of IV drugs
How does solubility of a gas affect its ability to deposit into the respiratory tract?
Highly soluble gases (eg. SO2 or formaldehyde) do not enter beyond nose in nasal breathing
Relatively insoluble gases (eg. O3, NO2, reach smallest airways and alveoli
Very insoluble gases (eg. CO, H2S) do not dissolve in mucus, pass through respiratory tract and are taken up by pulmonary blood supply
What are 6 facctors taht affect toxicity of inhaled materials
Solubility
Size (particle size)
Absorption
Site of deposition
Bioactivation
High blood flow
What are key pulmonary defenses of the airways and alveoli against toxicants?
Airways → mucus, cillia (mucociliary escalator)
Alveoli → macrophages (scavenge particles and toxicants)
Responses of the respiratory system to injury
Airway reflexes (triggering of airway irritant receptors)
Bronchoconstriction and airway hyperactivity
Triggered by irritant gases with moderate solubility
Can prime autonomic responses → threshold for ACh-mediated bronchoconstriction is lowered
Acute lung injury and pulmonary edema
Alveolar cell damage
Inflammatory cell influx, activation of NF-KB
Surfactant disruption → edema
Tickening of capillary barrier → impairs gas exchange
How does nasal irritation to toxic compounds occur?
Via irritant receptors (eg. TRPA1) → binding triggers nerve firing → tickling, itching, pain
Many volatile irritants need to be activated by CYP enzymes → bioactivation to electrophiles, which can be found in nasal tissues
Naphthalene-induced irritation mechanism
What can inhibit the irritation mechanism?
Naphthalene is bioactivated by CYP enzyme → binds to TRPA-1 → causes “braking response” (brief pause before taking next breath due to irritation in mice)
Addition of Metyrapone can inhibit the braking response to naphthalene (a CYP inhibitor)
Chronic Obstructive Pulmonary Disease
COPD → characterized by progressive airflow obstruction
Includes bronchitis (airway) component and alveolar (emphysema) component
What is a risk factor for COPD
Poverty → COPD mortality decreases with higher income → differences in exposure and treatment
Describe characteristics of the emphysema component of COPD
Airflow obstruction, leading to surtness of breath, decreased forced exhalation volume in 1 second
Characterized by abnormal enlargement of airspaces distal to terminal bronchiole with no fibrosis
Swollen and hyperinflated lung no longer effectively exhchanges O2 and CO2
Pathogenesis of emphysema
Stress induces pyroptosis/necroptosis, leading to inflammation, continuous activation of macrophages
Dysregulation of normal macrophage-mediated repair following repeated chemical insult → pathological cycle of progressive emphysema
Macrophage-mediated destruction of ECM in emphysema
Cigarette smoke → activates T cells → induction of macrophage production of MMPs via CD40 stimulation
Cigarette smoke also inactivates HDAC2 → NF-kB mediated transcription of inflammatory cytokines (TNF-alpha, IL-8) and MMP (protease)
MMPs and neutral elastase degrade eachother’s inhibitors (TMP and A1AT) → altered ECM degrading capacity (more ECM destruction) → emphysema
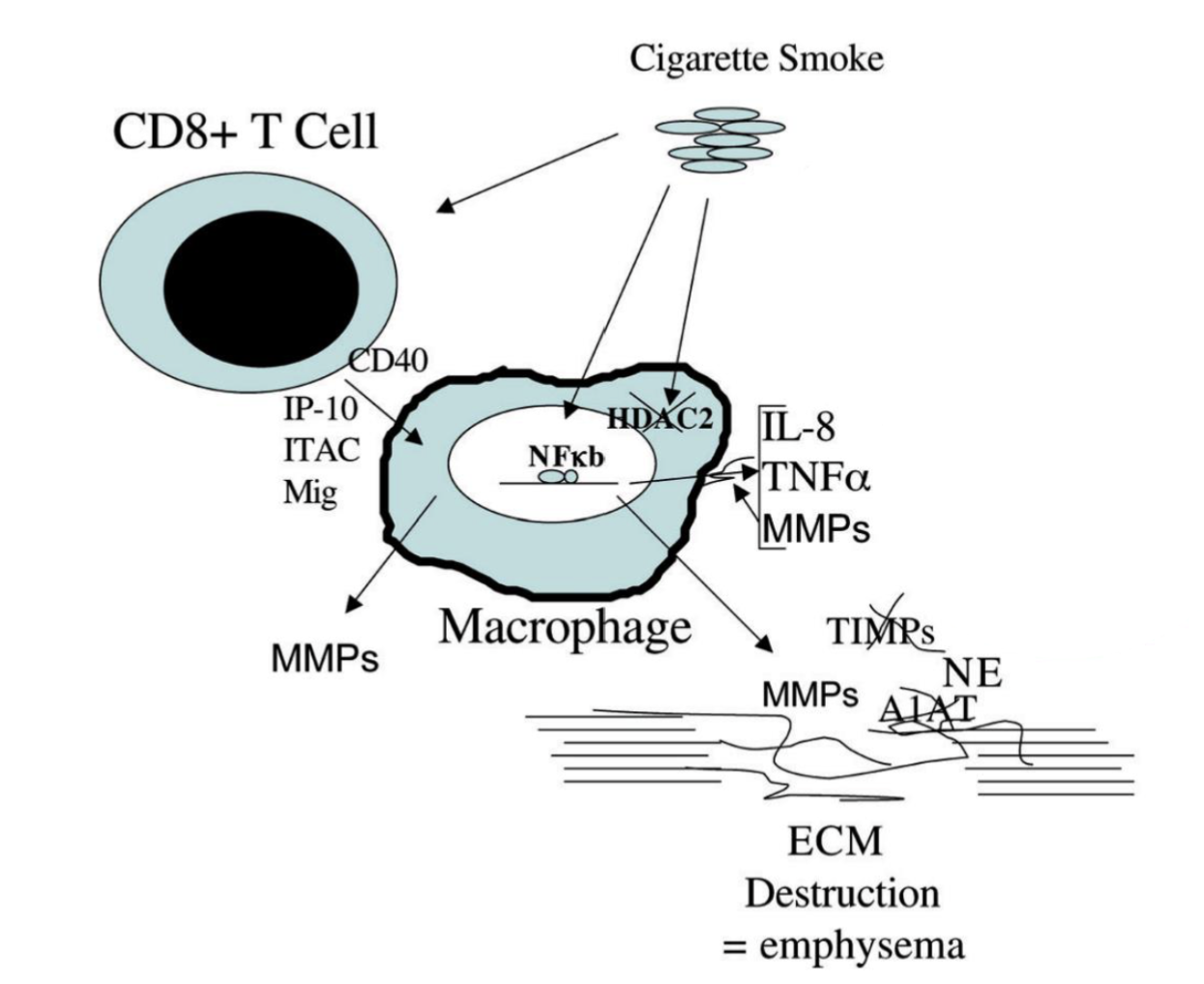
Describe characteristics of pulmonary fibrosis component in COPD
Progressive/irreversible destruction of lung archetecture associated with fibrotic scar tissue deposition (thickening of alveolar walls)
Leads to disruption of gas exhcange
Refractory to treatment
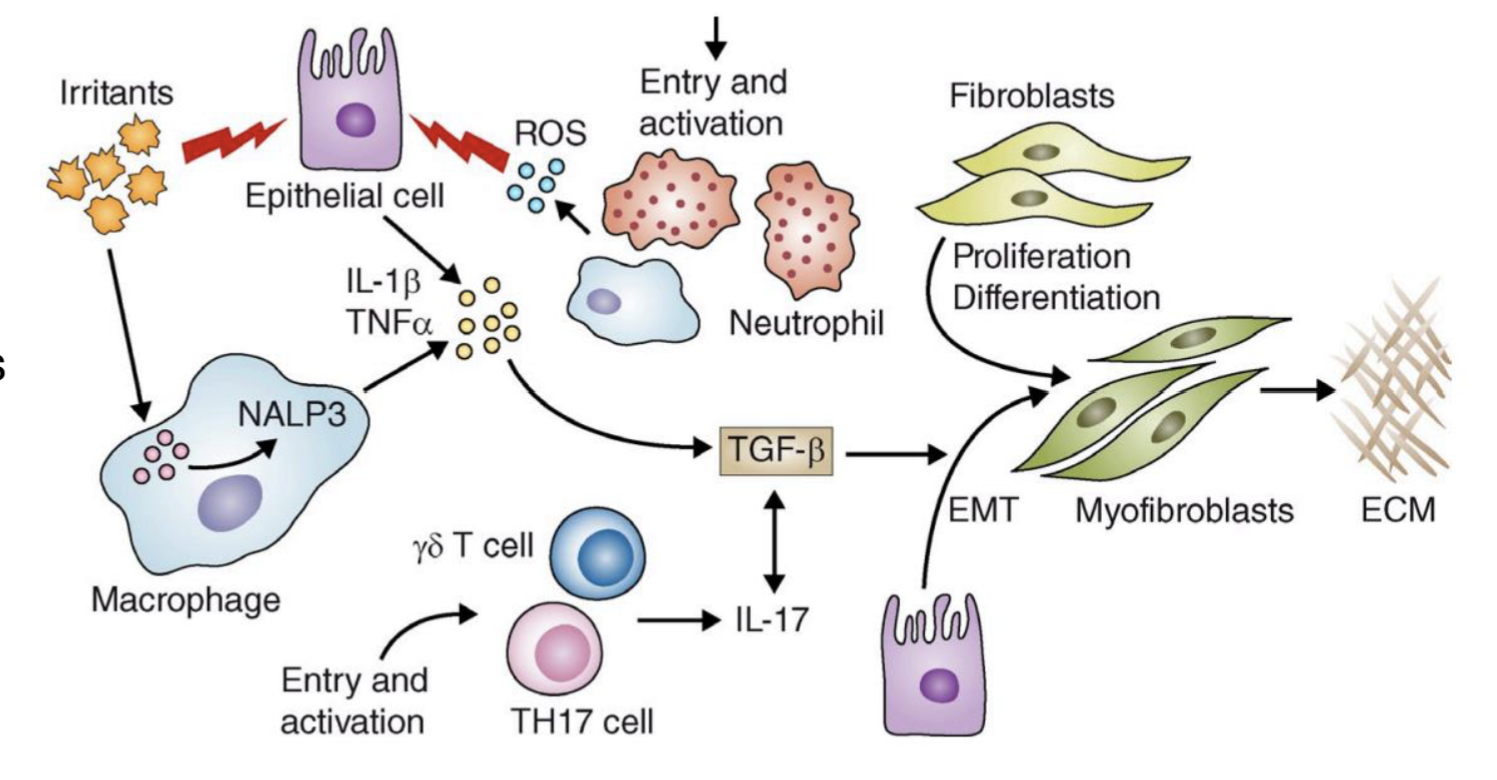
Describe the pathogenesis of pulmonary fibrosis
Irritants cause epithelial cell injury → detected by NALP3 inflammasome in macrophages → production of ROS, chemokines, and cytokines (eg. TNF alpha, TGF-beta?) → recruits leukocytes
TGF beta → induces epithelial-mesenchymal transition and formation of myofibroblasts → produce ECM
TGF-beta → differentiation of TH17 cells, exacerbates inflammatory response
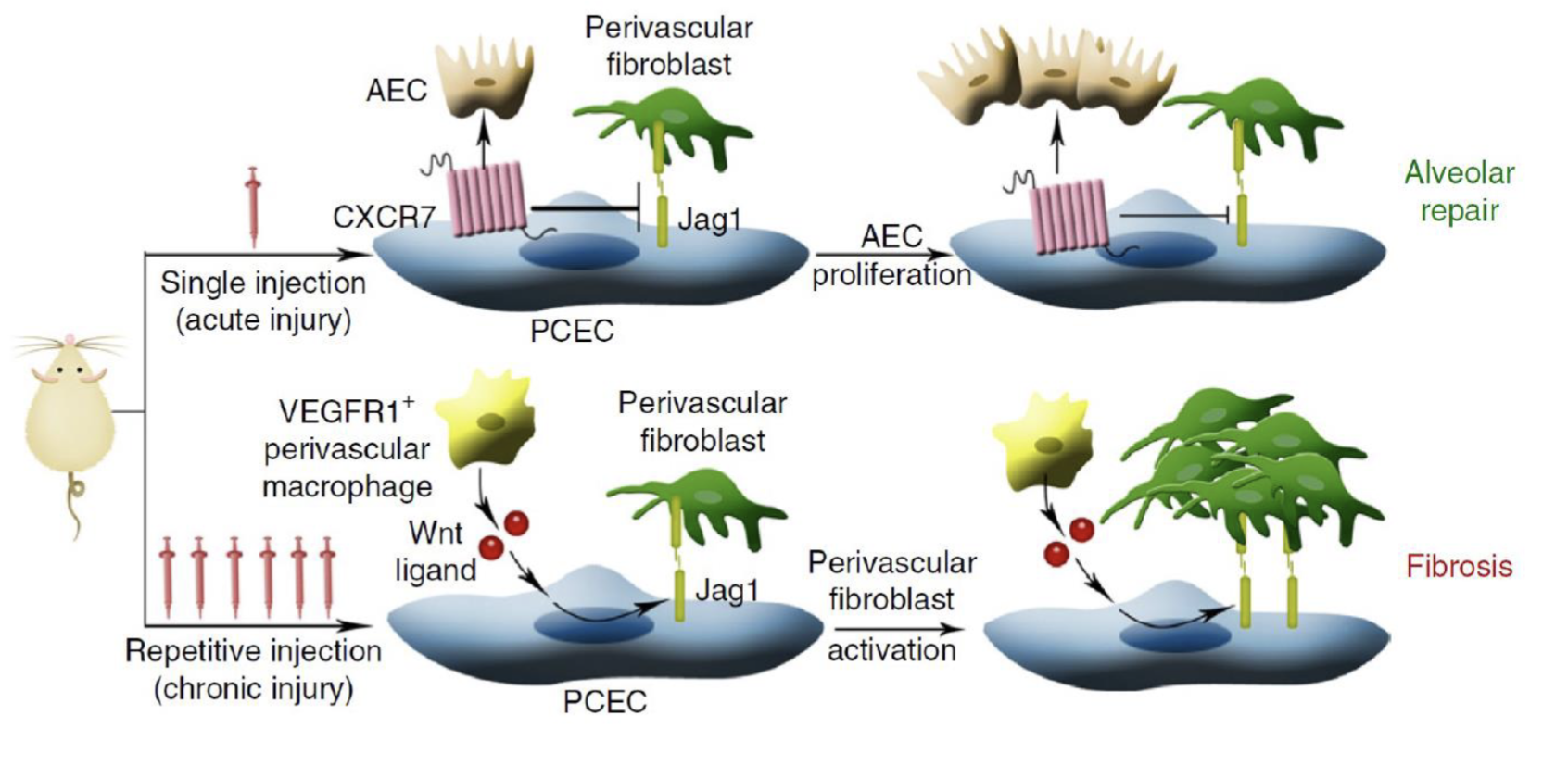
How do the molecular mechanisms of acute injury repair and chronic injury fibrosis differ?
Acute injury:
CXCR7 promotes alveolar epithelial cell proliferation → repairs alveolae
Chronic injury (repeat injury):
Recruits VEGFR1+ macrophages
Wnt/Beta-catenin → of Jag1
Jag1 → Notch on fibroblasts →vpromote fibrosis
Describe the pathogenesis of Immune Hemolytic Anemia (IHA) and its physiological consequences
Destruction of red blood cells mediated by IgG or IgM antibodies that react with the RBC surface antigens → complement activation and acute intravascular RBC lysis
Results in symptoms associated with anemia, jaundice (hemoglobin broken down into bilirubin), and hemoglobiuria
Can be initiated by drug dependent or drug independent antibodies (eg. antibody to drug, antibody to mainly membrane, and antibody to drug and membrane)
Eg. Cephalosporin antibiotics are responsible for many IHA cases
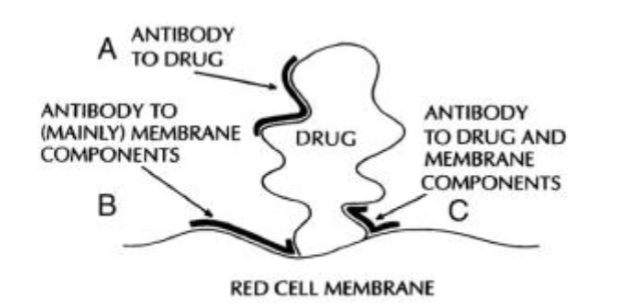
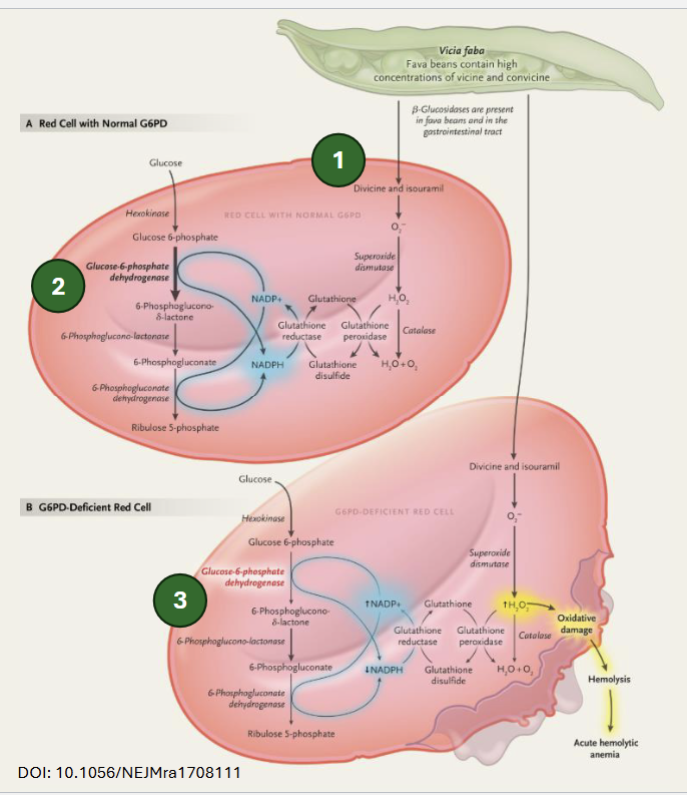
Describe the mechanism of toxicity of fava beans
Non-immune hemolytic anemia
Fava bean glycosides are hydrolyzed into divicine and isouramil → can generate ROS (ie. H2O2), which is sequestered by GSH in cells with normal G6PD activity (NADPH regenerates GSH)
In cells with reduced G6PD activity, NADPH production is limited → insuffiient regeneration of GSH → oxidative damage → hemolysis
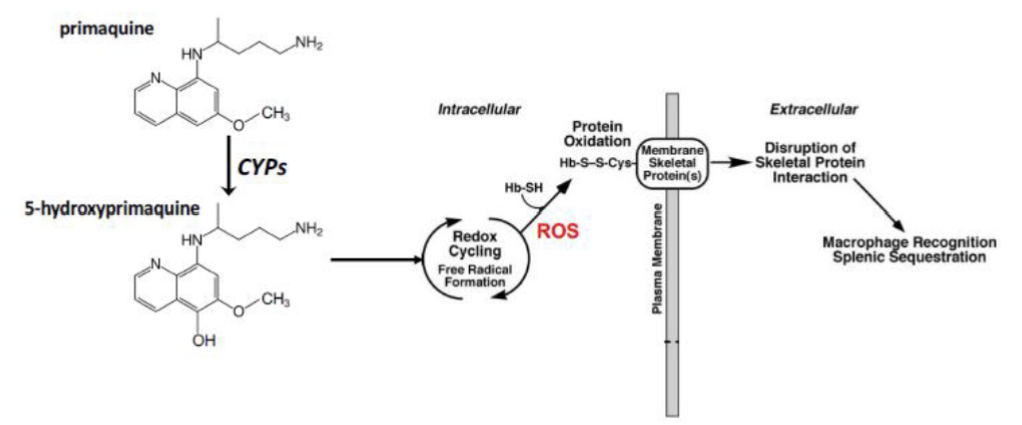
Describe the mechanism of toxicity of Primaquine
Oxidized by CYP enzymes to 5-hydroxyprimaquine → redox cycling to produce ROS → attack hemoglobin-sulfhydryl groups → Hb-thiyl radicals → binding of radicals to Cys in membrane skeletal proteins → disruption of skeletal protein interaction is recognized by macrophage → destruction of cells in the spleen
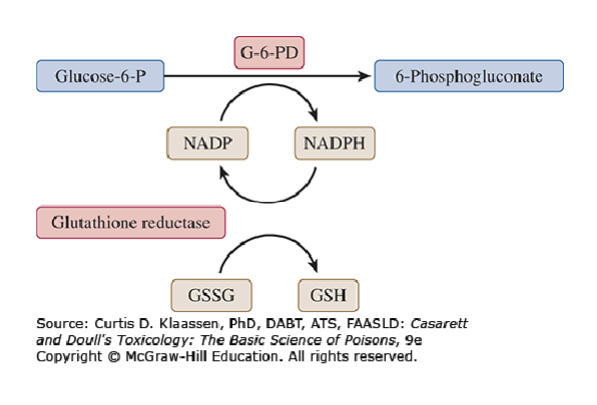
Role of G6PD in erythrocyte protection
G6PD produces NADPH from NADP (rate limiting enzyme)
NADPH is used by glutathione reductase to maintain intracellular levels of GSH
What demographics affect primaquine-induced hemolytic anemia
Demographics surrounding G6PD polymorphisms
G6PD is highly polymorphic
G6PD deficiency is more prevalent in malaria endemic regions, where it has an evolutionary advantage
G6PD deficiency more common in males since it is inherited on X chromosome → femalse can be heterozygous and have intermediate deficiency
Physiological characteristics of Megaloblastic Anemia
Related to deficiencies in cobalamin (vitamin B12) or folate (vitamin B9) → can be drug induced
Also drugs that interfere with DNA synthesis → cells cannot progress to M stage of cell cycle → cekks grow but do not divide → megaloblasts
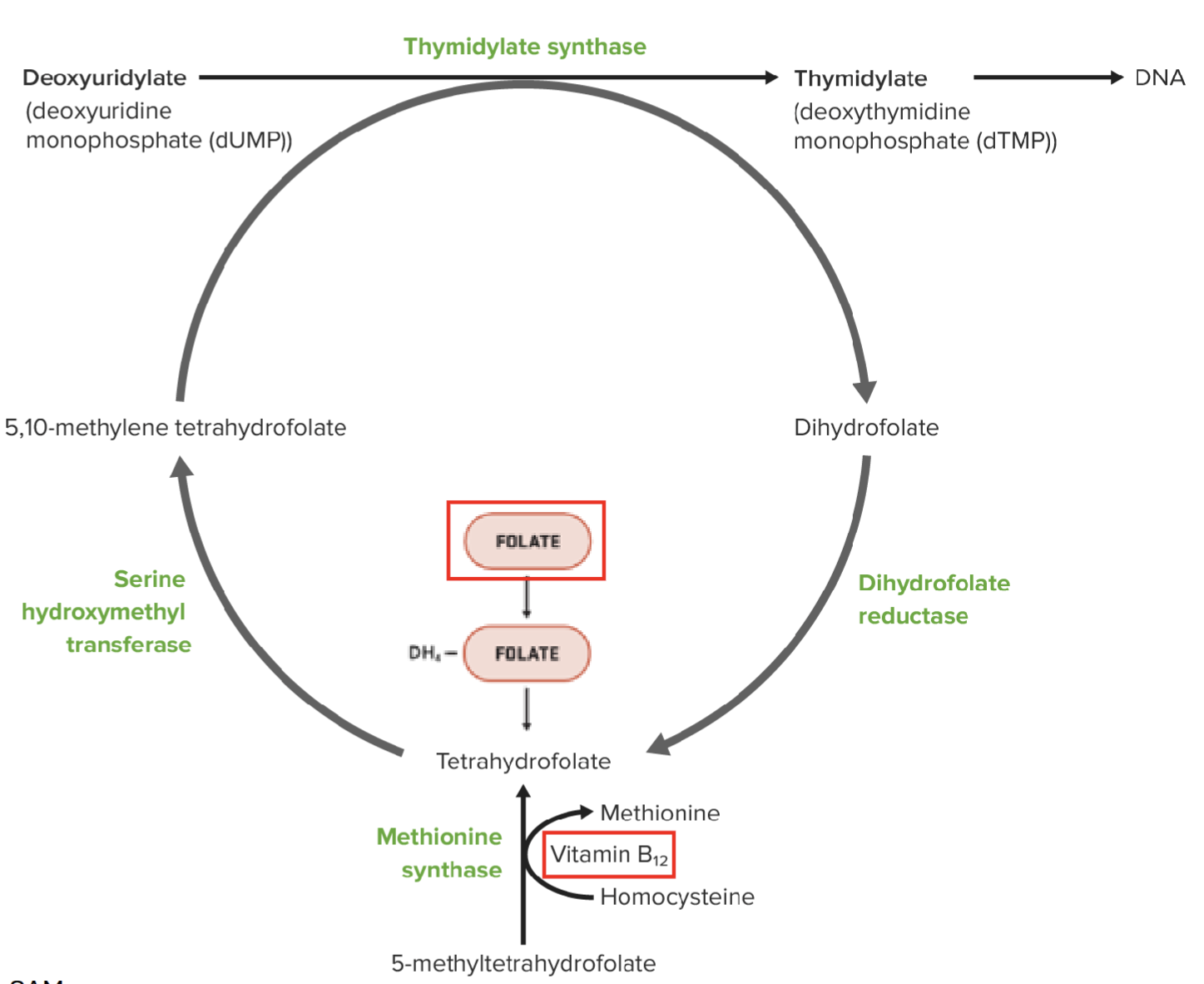
“Megaloblastic anemia can be associated with pyrimidine synthesis”
Describe what is meant by this statement and the mechanism
Vitamin B12 plays a key role in synthesis of TH4-folate, which is required for the synthesis of dTMP by thymidylate synthase
Vitamin B12 is needed to convert 5-methyltetrahydrofolate to TH4-folate → required in pyrimidine synthesis → DNA synthesis
Deficiency in thymidylate synthase results in buildup of dUMP → uracil is wrongly incorporated into DNA
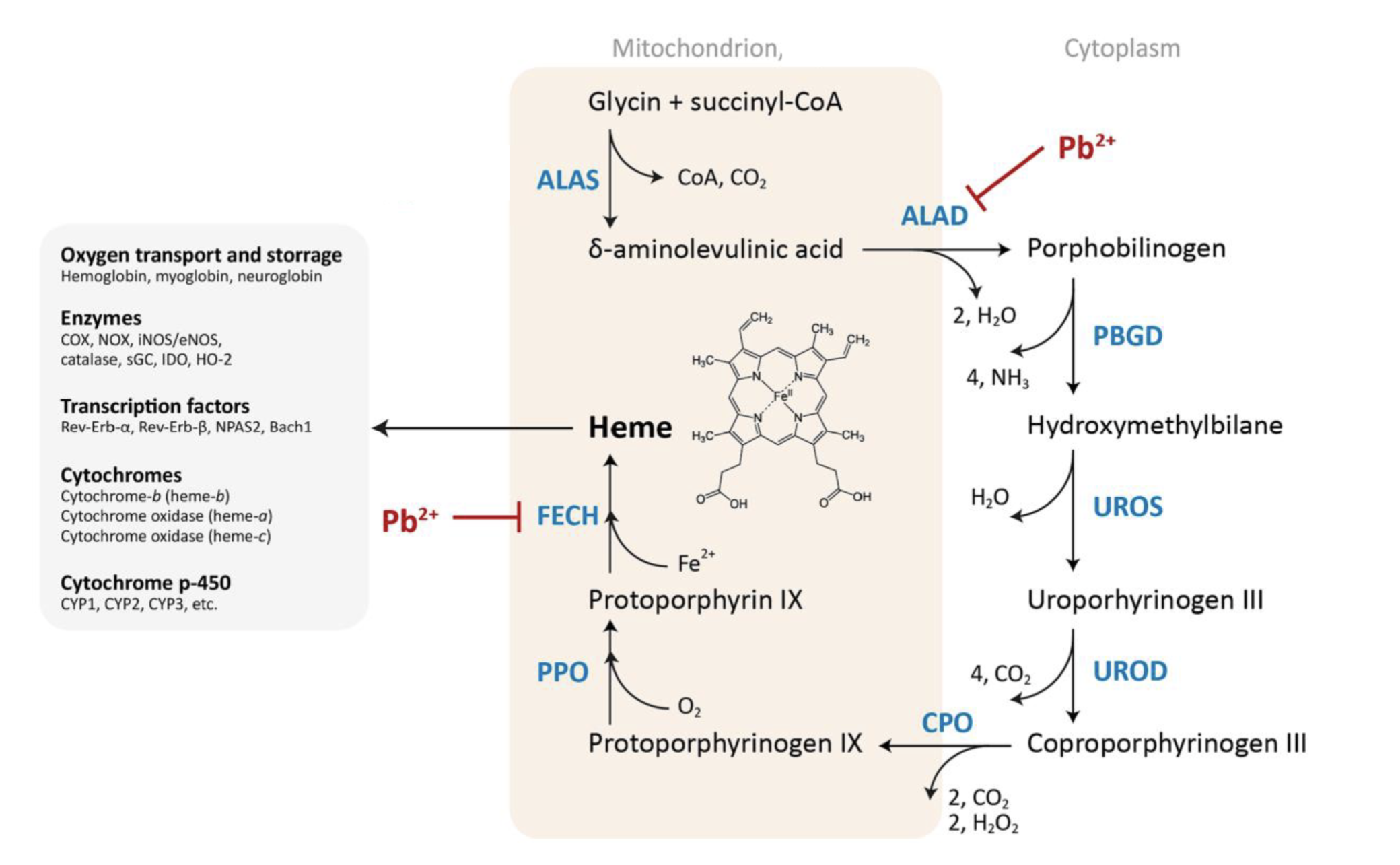
Describe the pathophysiology of sideroblastic anemia
Impaired synthesis of heme, leading to accumulation of non-heme iron around mitochondria in a ring in erythroblasts (RBC progenitor) → ring sideroblasts
Describe the mechanism of toxicity of lead causing sideroblastic anemia
Pb2+ inhibits ferrochelatase (FECH) → involved in last step of heme synthesis, inserting iron into porphyrin ring → inhibition results in buildup of iron in mitochondria
Also inhibits aminolevulinic acid dehydratase (ALAD) → results in build up of delta-aminolevulinic acid in mitochondria, which is toxic
Describe the pathophysiology of aplastic anemia
Complete failure of hematopoiesis
Blood-forming stem cells replaced by fat cells
Pancytopenia → reduction in circulating RBCs, WBCs, and platelets
Reticulocytopenia → reduced RBC-forming reticulocytes
Can be caused by xenobiotics or immune causes
Describe the pathophysiology of immune thrombocytopenia
Reduced thrombocytes due to antibody-triggered destruction by phagocytosis → impairment in blood clotting
Caused by drugs that bind covalently and act as haptens on cell surface proteins of platelets or by exposing novel epitopes of platelet surface proteins, or by increasing protein binding affinity of circulating naturally occuring antibodies
Drug dependent antibody binding
Describe drug dependent antibody binding and provide an example of a drug
Drug binding to antibody alters the conformation of the antibody, allowing it to bind to an antigen with higher affinity (eg. in immune thrombocytopenia, higher affinity to platelet cell surface antigen)
Eg. Quinine → binds and reconfigures CDRs of DDAbs, allow them to bind with high affinity to platelet surface proteins
Describe the pathophysiology of neutropenia and how it can be treated
Reduction of circulating granulocytes, most easily observed by counting neutrophils
Can be immune mediated or caused by xenobiotics (eg. Clozapine, clopidogrel)
Can be treated by G-CSF
Describe the mechanism of toxicity of Clozapine
Induces neutrophil apopotosis via reactive metabolites produced by peroxidative metabolism → can cause neutropenia
In presence of activating system (peroxidase + H2O2), induces apoptosis at lower dose
Describe the mechanism of toxicity of Clopidogrel
Reduces colony formation from mmyeloid precursors, enhanced by metabolism by CYP3A4
Concentration dependent toxicity
Can be reversed by addition of CYP3A4 inhibitor (eg. ketoconazole)