CHEM111 Module 6: Kinetics
1/51
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
52 Terms
how do thermodynamics and kinetics differ
thermodynamics looks at if reactions are favorable to occur (what happens)
kinetics looks at how fast reactions occur (does it actually happen)
for a reaction to be useful, it must take into consideration the thermodynamics AND the kinetics of the reaction
define reaction rate
the rate of change of concentration, with time
how do you calculate average reaction rate for a substance in a reaction?
what information must you use (equation included on formulae sheet)
what are the units of the answer
change in concentration of the substance (d[A])
stoichometry of the substance (a)
change in time (dt) - what area of the graph / part of the reaction do you want the rate for
units = mol L-1 s-1
how do rate laws differ from rates
rates tell you the change in concentration of a substance in a reaction over time, this is a quantity being measured
a rate law however is a law that defines a specific reaction, providing the parameters the reaction occurs at (an empirically found mathematical description of the reaction’s change in substance over time) allowing things to be calculated specifically for the reaction
this shows how reactant concentrations contribute to the overall rate of reaction (via partial orders), with consideration to T (reaction coefficient)
what information do you use to calculate rate laws
partial orders of the species in a reaction (how each contribute to make the overall order of the reaction - partial orders add to give the overall order of the reaction) - unitless, usually positive integers
k (reaction coefficient / constant) - is a proportionality constant, which varies with temperature
what are overall orders for reactions
what are the different options
these categorise the reaction into how their reactant concentrations are raised to the power of, allowing us to simplify the rate coefficient
zero-order reactions have the reactant^0
first order reactions have one reactant, with ^1
second order reactions have one reactant with ², or two reactants with ^1
how do different overall orders of reaction change the equation for rate laws (/ reaction coefficient)
0: rate = k (A is to the power of 0)
1: rate = k[A] (A is to the power of 1)
2: rate = k[A]² (for one reactant) = k[A][B] (for two reactants)
how do units of k change depending on the reaction’s overall order
why do these change
0: mol L-1 s-1
1: s-1
2: L mol-1 s-1
3: L² mol^-2 s-1
these change as different units cancel out, due to the different arrangement of equations
what experimental evidence suggests that reaction is…
0th order with respect to a reactant
1st order with respect to a reactant
2nd order with respect to a reactant
reaction rate is unchanged when intiial conc. of reactant is increases
reaction rate is doubled when initial conc. of reactant doubles
reaction rate is quadrupeled when initial conc. of reactant doubles
what is the initial rates method, why is this a good method
how is this helpful for finding the partial and overall orders for reactions
this method uses the concentrations and rate of the reactions initially
beneficial because rate is highest/largest so easiest to measure
no products present so no reverse reaction to complicate results
can alter initial concentrations to get more measurements (could provide different reaction rates)
this helps us find partial and overall orders, as we can look at…
if rate doubles when a reactant conc. doubles, it suggest its 1st order
if rate quadrupels when reactant conc. doubles, it suggests 2nd order (raised ²)
if unchanged, it suggests its 0 order for that reactant (wont contribute in the rate law)
what is the difference between differential and integrated rate laws?
differential rate laws are in differential form, looking at change in conc. with respect to change in time (a derivative)
this involved looking at rates of reaction, with the differential rate law
e.g. d[A] / dt = -k (0th order)
integrated rate laws are the integrated form of these differential rate laws
e.g. [A] = [A]0 -kt (0th order)
what happens if you plot experimental data of reactant conc. with time, for a 0th order reaction, and why?
it will have a linear fit, with slope = -k, and intercept = [A]0
this is because, the integrated rate law equation, is the equation of a straight line
therefore showing that in a 0th order reaction, there will be a constant decrease in reactant conc. with increasing time
what happens if you plot experimental data for change in reactant concentration over time for first order reactions, and why?
what is something you can do to change the appearance of this graphed data, to determine if the reaction is truly first order?
the graph will not be linear, it will show a curve with exponential decay
this is because the integrated rate law for first order reactions is not the equation for a straight line
since integrated first order rate law involves e^-kt, you can take the natural log (ln) of both sides of the equation, includng reactant concentration with time, which when plotted will provide a linear relationship
the slope of this will = -k, and the intercept = loge[A]0
how do you calculate a slope from linear data on a graph
what might this help you find
calculate rise (change in reactant concentration between the two points) divided by run (the time in seconds this time frame goes for)
if a 0th order reaction, -slope = k
if a 1st order reaction, loge of concentrations plotted will have -slope = k
how does a first-order differential and integrated rate law, change for gas-phase reactions
they simply replace concentrations of reactants (e.g. [A]) with partial pressures (e.g. pa)
exponential decay when plotting pa with time, and linear when loge of pa plotted - same as usual first-order reactions
do the units and magnitude of k change for gas-phase reactions
no! is the same
does time units change the magnitude of k
yes! make sure to do this accurately
(e.g. 60 seconds vs 60 minutes (3600 seconds)
what is the half-life of a reaction
what equation represents this
the time required for a reactant to reach half of its initial concentration
(for first-order reactions) use: t1/2 = (1/k) ln(2)
so k = ln2 / t1/2
so t1/2 = ln2 / k
in these equations, k and t1/2 are interchangeable, (for first order reactions only), so k can be reported as t1/2 instead
this provides more of an intuitive view, as t1/2 is more visualisable, it must take 2 half lifes to get ¼ of intial conc, 3 half lives to get 1/8 of initial conc, and so on
how many half lifes technically gets you to completion
how many half lives gets you to 1/8 initial conc.
10 to completion
3 to 1/8 initial conc, 2 to 1/4
what is a lifetime of a reaction (first order)
what is its equation
what is its symbol
how does it relate to k
a lifetime is the time taken for a reactant to reach 1/e of its initial value - which is the average time it takes for a reactant molecule to react
denoted τ
at t = τ, [A] = [A] / e
so k = 1 / τ (gained from rearranging the first-order rate law)
compare half-life and lifetimes of a reaction
half lives tell you the time taken for a reactant to reach ½ its initial conc.
lifetimes tell you the time taken for a reactant to reach 1/e its initial conc., which is equal to the average time taken for a reactant molecule to react
therefore, lifetimes tell you more useful information, as they provide a point of average reaction time, wheras half lives just go to infinite saying how long for half of initial reactant to be used, then half of that to be used, and so on
what does [A]0 mean
concentration at t=0, so initial conc, of a reactant
how would you determine concentrations of reactants, after some elapsed time, given the initial concentrations (for a first order reaction)?
first consider the change in mols for the conc (denoted x), based on reaction stoichometry of the reaction
so reactants final conc is -x multiplied by stoichometry (e.g. -2x) while for products it is x (e.g. 2x), as reactants decrease while products are formed
then use the integrated rate law, which becomes
[A]t = [A]0 e-kt (for a first order reaction at time t)
then subsitute in known information (k is often provided) to find the unknowns
what is an elementary reaction
how do these combine to form complex reactions
an elementary reaction is a reaction that occurs as written, reactants → products directly
complex reactions (most chemical reactions) instead involve a number of elementary steps (the reaction mechanism) that add to give the overall reaction (the reaction equation)
in complex chemical reactions, what are intermediates
these are chemicals that appear in the reaction mechanism (in an elementary step), but not in the overall reaction
this is due to them playing a role in the reaction, but being created and used up so are not written in the equation
what are the different molecularities of elementary steps
what are the rate laws for each
unimolecular = 1 reactant molecule
rate = k[A]
bimolecular = 2 reactant molecules (2 options)
rate = k[A]²
rate = k[A][B]
termolecular = 3 reactant molecules
rate = k[A][B]²
what are rate laws for elementary reactions based on
why does this only apply to elementary steps, and not complex reactions
just based on conc. of reactants (rate = k[A][B]², etc), as their partial orders = stoichometric coefficients
however for complex reactions, multiple elementary steps must be considered, so their rate laws are not as simple (e.g. rate determining step)
note - be careful assigning rate laws without knowing if a reaction is elementary / complex
what is a rate-determining step in a reaction
how may this affect a reaction’s rate law
this is an elementary step in a complex reaction, that is much slower than the other steps, therefore it determines the overall rate of the reaction
this causes the overall rate law to be simply determined by this rate-determining step, it becomes the rate law for this individual elementary step
how would we know a reaction is complex, based on its rate law
if the rate law, and partial orders of the reactants, don’t correlate to the overall reaction stoichometry
e.g. doesnt contain all reactants / partial orders
or if there is an intermediate chemical detected while the reaction is occurring, which isn’t present in the overall reaction equation (or in its reactants / products formed)
how does collision theory explain…
how elementary reactions occur
how rate laws of bimolecular reactions are dependant on molecule concentrations
how reaction rates are often greater at higher temperatures
how termolecular reactions are rare
by collision of particles, resulting in reaction
because collision frequency, and number of collisions (contributing to reaction rate) - is influenced by concentrations of molecules (double the conc. will double the rate, etc)
collision frequency increases with higher temp, as molecules move faster, thus more reactions occur
3-body collisions are unlikely
what relationship does the Arrhenius equation describe
what theory aids in understanding this?
the relationship of k (Rate coefficient) with temperature, as with most chemical reactions, k increases rapidly with increasing temperature, in a non-linear fashion (a curve)
Arrhenius equation allows this relationship to be made linear, taking the log of each side
this can be understood using the Transition State Theory
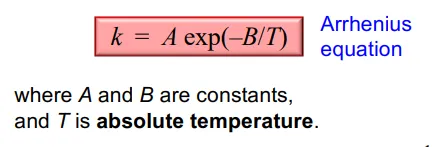
what kinetic theory do Reaction Coordinate / Energy diagrams display?
what kinds of graphs are these
for a collision between particles to result in a successful reaction, their kinetic energy must be sufficient to overcome the activation barrier (input energy required for a reaction occur)
this is shown using energy as a function of reaction progress
it shows the parameter Ea (activation energy)
if the kinetic energy of collision (Ek) is greater than the activation barrier, it is successful and the reaction occurs (And the opposite)
similarities and differences between endothermic and exothermic (elementary) reactions in terms of Ea
similar: they both require collisions with a certain amount of energy, to overcome the activation barrier for a reaction to occur
different: the activation barrier is typically smaller for endothermic reactions
different: the products have lower energy than reactants in exothermic, and higher enery in endothermic
describe the Transition State Theory
the tip of the activation barrier (the middle between products/reactants), is transition state (between products and reactants)
Ea is the energy required to break the bonds in the reactant, in order for bonds to be formed between reactants to create the products
we must get through the transition state (pass Ea barrier) to get to the end
all reactions have these, often more complex with more bonds breaking / forming (but always requires energy)
how does k correlate to P (Ek>Ea), for a collision
how does this link to temperature
k and rate of reaction, depends on the probability that a collision will be successful (have sufficient energy Ek to overcome Ea barrier)
as temperature increases, the distribution (Maxwell Boltzmann dist.) of Ek of molecules in a reaction, flattens out (so a higher area of the distribution, has Ek > Ea)
along with Ea of collisions, what else must be considered to use the Arrhenius equation (and relate k to collision theory)
orientation effects and the steric factor
successful collisions also require reactants to have the correct orientations relative to each other, particles must hit the right particle they specifically react with (in a certain cone of area)
steric factor refers to the probability of correct orientation
how do kinetic theories transform the Arrhenius equation, for usability (form a theoretical relationship form this empirical one)
what functions make this up
these reveal the Arrhenius constants (A&B) to be
collision frequency x steric factor = zp = A (frequency factor)
p(Ek>Ea) = exp(-Ea / RT) = B (activation energy)
using collision theory, Ea and orientation of particles, for reactions to occur
how do the collision theory and Arrhenius equation apply for complex reactions
use the z/p/Ea for the rate-determining step (an elementary reaction), to find k
after putting theoretical ideas into the Arrhenius equation, how does it reveal how k is temperature dependent
what final equation does it provide
the things that make up the Arrhenius equation (-Ea/RT & A) are temperature dependent, therefore the k they equal, also must be temperature dependent
this creates the curved relationship we see of rate coefficient (k) with increasing temperature
this also makes sense considering collision theory
treating A as a constant provides us with the final Arrhenius equation in the formulae sheet
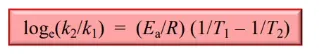
what can you calculate using the Arrhenius equation
what approach should you use
what units should be used
Ea (provided k1, k2, T1, T2)
k(x) at T(x) (provided Ea, and other k & T)
corresponding T for given k (also provided Ea and other k & T)
to calculate, input the known data, and rearrange the equation
units combine for J mol-1, temperature in K, time in s
describe how a reaction coordinate diagram changes for complex reactions
transition states
intermediate troughs
rate determining step
these just become a combination of multiple diagrams for elementary steps, for the ones that make up the reaction mechanism
the peaks of the graphs are each transition state, and the Ea required to overcome the specific bond forming / breaking to transition from reactants → products
the troughs after these peaks, are the intermediates formed (products of the previous step, reactants for the next, used up so not in reaction mechanism)
the step with the higher Ea, is the rate determining step (Elementary reaction)
what is a catalyst
how do these differ from intermediates
species that increase the rate of reaction, by providing an alternate complex mechanism reaction pathway, with lower Ea
but aren’t used up (must partake, so are consumed and regenerated), therefore distinct from intermediates (formed then consumed)
how does the presence of a catalyst change the reaction coordinate diagram
what functions does it change, what functions remain the same
it creates more steps on the coordinate diagram (together with less Ea in total), in order to consume and regenerate the catalyst (AND do the original reaction) - therefore reaction is definitely complex
reactants / products keep the same deltaH (change in reaction energy) as before, the catalyst only reduces the Ea part of the coordinate diagram (lowers this energy)
therefore only kinetics change (rate coefficients) NOT thermodynamics (state functions / heat / deltaH, etc)
how does a catalyst affect equilibrium
a catalyst does not affect equilibrium!
this is because thermodynamic state functions aren’t changed, only kinetics are
therefore rate increases and equilibrium is reached faster, but equilibrium’s position (favoring forwards/reverse) is unchanged, as this rate increase will equally increase the reverse reaction
name the 3 types of catalysis
homogenous catalysis
heterogenous catalysis
enzyme catalysis
distinguish between homogenous and heterogenous catalysis
homogenous
reactants and catalysts are in the same phase (either everything is gas, or everything in solution)
heterogenous
reactants and catalysts in the same phase (reactants usually gas / solution, catalysts usually solids)
this provides a solid surface to catalyse the reaction (catalyic surface)
adsorbates are the intermediates, which are the molecules + the solid surface, at each step
can break bonds =
the reactant molecule weakly binds to solid (absorption)
the bonds between reactant are broken, while still bound to the solid (requiring less energy)
it then can easily seperats from the surface
can form bonds =
reactant weakly binds to solid
they dissociate from their original form and become atoms
these atoms then rearrange to form new bonds, and unbind as the product
what is the Haber-Bosch process
how is it done efficiently
the industrial process for ammonia synthesis
is thermodynamically favored (spontaneous) but kinetically slow
therefore a catalyst is used (heterogenous), originally Os, now Fe, along with high T & P
describe enzyme catalysis
occurs in biological systems
the catalysts are enzymes (large protein molecules), with one or more active sites, which catalyse the reaction (reactants = substrates)
this process is somewhat homologous to heterogenous catalysis, the active site providing this solid surface (specifically designed for the molecule to favor reaction)
E + S → ES → EP → E + P (adds elementary steps)
the intermediates are the enzyme-substrate & enzyme-product complexes
e.g. biological N fixation, using nitrogenous enzyme, with N2(g) + H2(g) (and ATP), to form NH2
name a common example of catalysis…
in nature
in modern day life
ozone hole formation at the poles is driven by catalytic reaction cycles of stratospheric Cl (resulting in O3 depletion)
car exhaust pipes contain catalytic converts, catalysing the conversion of undesirable gases (CO, NOx, unburnt hydrocarbons) → more environmentally friendly ones (which are pumped out
for taking measurements on kinetics of a reaction…
what do you measure
what do you change
what do you keep the same
concentration of reactants (or products?) as a function of time
ensure fixed T between trials (to not complicate measurement - influences k)
only change concentration between trials (Gather a variety of data)
this provides a graph of reaction rate
what kinetics measurement method would you use for slow reactions (lower reaction rate)
measurements of timescales of minutes (or longer) apart
solution-phase = may involve titrations (take out titration → work out conc. → plot point on graph → determine rate)
gas-phase = may involve mass spectrometer (open the gas to spectrometer → provides gas concentrations → plot point on graph → determine rate)
what kinetics measurement method would you use for fast reactions
make measurements on a timescale of seconds (or less) apart
solutions =
electromagnetic radiation (e.g. light) passed through reaction mixture → absorbtion detected and recorded (often by a computer, done quick) → Beer-Lambert law done to determine conc. → plotted on graph
electrochemical reactions (redox solutions & battery) =
electrodes in solution attached to voltmeter → voltage measured at intervals to computer → computer finds conc. → data plotted
gases =
reaction mixture open to pressure transducer → constantly measures pressure to computer → computer finds conc. from pressure (ideal gas law) → data plotted