Patho Hematologic Disorders
1/62
Earn XP
Description and Tags
Patho exam 1- lecture 4
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
63 Terms
plasma
the aqueous component of blood&about 90% water, contains __ proteins: albumin, globulins, fibrinogen, clotting factors and complement proteins
albumin
most abundant plasma protein. carrier molecule and regulates movement of water and solute through capillaries—
– ↓ ___ results in ↓ ability to pull water into vessels
– range: 3.5-5
globulin
plasma protein responsible for immune response. contains antibodies-IgG, IgA, IgM, IgD, IgE
– gamma__ are the most abundant
order of antibodies=
most abundant to least= IgG,A,M,D,E
– (Germs And Microbes Dont Exist)
fibrinogen
plasma protein, most abundant clotting factor (hemostasis), precursor to fibrin clot
true
(T/F) clotting factors are involved in hemostasis and complement proteins are involved in immune response
lymphoid progenitors
produced in bone marrow from Pluripotent stem cell. it produces lymphocytes- a)T-cells, b) B-cells, c) Natural Killer cells
myeloid progenitors
originates from Pluripotent stem cell and produces all other cell types: Erythrocytes, Platelets, Granulocytes (Neutrophil, Eosinophil, Basophil), and Monocytes/Macrophage
erythrocytes
red blood cells, function is tissue oxygenation (due to hemoglobin carrying)
– normal range= 4.2-6.2 mil/mm3
erythropoietin
function is the development of RBCs in bone marrow
– peritubular cells in the kidney release __ after some hypoxia. the __ stimulates bone marrow to make RBCs
– requires B12 and folate btw
hemoglobin
function is to increase oxygen carrying capacity for blood. made up of 2 pairs of proteins (2 α globins and 2 β globins) and 4 hemes.
– each heme can carry 1 O2 molecule; thus a saturated __ has 4 O2 bound.
rbc destruction process
old rbcs are destroyed by enzymes
– heme and globin are separated
– globin is broken down into amino acids &recycled
– heme breaks down into iron and porphyrin. iron is recycled.
– porphyrin is reduced to bilirubin (which can be conjugated in liver [jaundice..] or excreted in urine)
jaundice how?
Conditions that ↑ destruction of RBCs (hemolysis) cause ↑ release of porphyrin and ↑ bilirubin – more than liver can manage → ___.
leukocytes
white blood cells! function: defend against infection, cancer, and remove debris from cellular damage.
– contains Granulocytes (neutrophils, eosinophils, basophils) and Agranulocytes (monocytes/macrophages, lymphocytes, B and Tcells)
neutrophils
most numerous of the WBCs-60%
function: first responders to an infection and chief phagocytic cells of early inflammation
eosinophil
granulocyte and only 3% of WBCs. Function: mildly phagocytic cells—they destroy parasites+worms and are prevalent in allergic reactions
basophil
granulocyte and <1% of WBCs. Function: induce inflammatory response and activate T lymphocyte
lymphocyte
about 30% of WBCs. Function:
1. CD4+ T lymphocytes mature into T-helper cells—
a) create memory B and T cells
b) activate B lymphocytes which mature into plasma cells and make antibodies
c) recruit CD8+ T lymphocytes
2. CD8+ T mature into cytotoxic T cells (which kill cancer and virus infected cells)
monocytes/macrophages
function: garbage disposal cells. they ingest unwanted materials and act as antigen presenting cells (-present antigens to CD4+ cells to initiate adaptive immunity)
– monocytes in the blood, macrophages in tissue
thrombocytes (platelets)
cytoplasmic fragments w no DNA. Function: hemostasis.
– __ block an opening in a blood vessel. when activated, it releases chemical mediators to promote clotting
thrombopoietin (TPO)
produced by the liver and __ stimulates megakaryocytes in bone marrow to produce platelets
– regulation process: platelets contain TPO receptors and TPO binds to those receptors and then cant stimulate megakaryocytes. But when platelets ↓ , TPO has no place to bind and can then continue stimulating megakar
hematocrit
the measure of packed RBC cell volume when centrifuged
– Rule of 3: HCT = HGB x 3
labs for hematology

“Nobody Likes My Educational Degree”
White blood cell differential is the measure of the percent of each type of leukocyte=
≈ 60% Neutrophils
≈ 30% Lymphocytes
≈ 6% Monocytes/macrophages
≈ 3% Eosinophils
≈ 1% Basophils
Anemias
condition characterized by reduction in RBC volume or ↓ in quantity/quality of HGB
— often due to Blood Loss, Impaired RBC Production, ↑ RBC Destruction, or ↓ RBC production
acute blood loss
clinical manifestations of:
– Slow blood less: lost plasma replaced by the movement of water from tissue. few symptoms.
– Rapid blood less: ↓ BP, ↓CO (bc blood volume drops), ↑HR (reflex tachycardia)
iron deficiency anemia
anemia that occurs when Fe is depleted. Reduced Fe→ reduced HGB synthesis→ reduced erythropoiesis →small/pale cells
- causes: continuous low volume blood loss(periods) or pregnancy
chronic disease anemia
mild anemia in pts w/ __ __/inflammation.
- ↓RBC life span. Inflammation→ kidney injury→ reduced erythropoietin
B12 deficiency anemia
anemia due to inadequate/absence of B12 →abnormal synthesis of DNA, RNA, and Proteins. results are large weak RBCs that rupture
- causes: poor dietary intake, lack of intrinsic factor or transport protein for absorption, or excessive alcohol usage
true
(T/F) folic acid deficiency anemia has the same pathologic pathway as B12 anemia deficiency.
- folic acid is necessary for DNA and RNA synthesis
aplastic anemia
anemia characterized by hematopoietic failure (reduction in mature blood cells).
due to drug/chemical exposure which yields bone marrow suppression
sickle cell disease
recessive condition-point mutation in which glutamic acid is replaced by valine.
- result is atypical hemoglobin in RBCs which then precipitates out and stiffens cell.
- sickled cells now have shorter lives (hemolysis) and can obstruct blood vessels→ ischemia
sickle cell manifestations
Hemolytic anemia → hemoglobin release → jaundice
Vaso-occlusive crisis → pain from hypoxia/infarction (bones, lungs, brain, etc.)
Acute chest syndrome (pain, cough, dyspnea) = leading cause of death
Can cause stroke and ischemia of bone → bone necrosis
Sequestration crisis → sickle cells are removed and trapped in spleen → splenomegaly, shock
Glomerular disease → sickled cells damage glomerulus → renal failure
thalassemia
recessive autosomal disorder- impaired synthesis of one of the HGB chains. results in ↓oxygen carrying capacity
- hemolysis of immature fragile RBCs and splenomegaly
neutrophilia
Increase in the # of circulating neutrophils. Caused by inflammation, infection, or stress
- overload of neutrophils makes blood viscous→ thrombus can form or occlude a blood vessel
- when demand > supply, bands (immature neutrophils) are released
neutropenia
decrease in # if circulating neutrophils and caused by severe long infection, chemotherapy/rad therapy, or aplastic anemia.
- complications- recurrent, fatal infection
eosinophilia
increase in the # of circulating eosinophils and caused by worms, parasites, allergies/asthma, or collagen vascular diseases
- worms, wheezes, and weird disease^
- allergies/infections stimulate recruitment of __ and production from the bone marrow
infectious mononucleosis
benign self-limiting syndrome characterized by infection and death of B cells by Epstein-Barr virus.
- unaffected B cells then become plasma cells and produce antibodies against EBV
- fatigue, fever, pharyngitis, lymphadenopathy of cervical nodes
leukemia
malignant disorder of uncontrolled proliferation of leukocytes in bone marrow.
- overcrowded bone marrow→ reduced production/function of normal blood cells→ anemia
acute lymphoblastic leukemia
rapid proliferation of lymphocyte progenitor cells in blood and bone marrow.
- differentiation of cell stops. overcrowding of bone and reductes production/function of normal blood cells
- common in children. symptoms: fatigue,fever,anemia,infection,bone,pain
acute myeloblastic leukemia
rapid proliferation of neutrophil progenitor cells in blood and bone marrow.
- differentiation of cell stops
- overcrowding of bone and ↑ in number of immature neutrophils makes blood more viscous (thrombosis/occlusion may occur)
chronic lymphocytic leukemia
slow development of fully differentiated lymphocytes (b cells) in blood & bone marrow
- transformation of fully differentiated but immature B cell, cells cannot mature into plasma cells or make antibodies. no bone marrow crowding
- common in adults. symptoms: usually asymptomatic, lymphadenopathy, high risk of infection
chronic myelocytic leukemia
slow proliferation of fully differentiated granulocytes and other myeloid cells
- Philadelphia chromosome is present in most cases and is thought to initiate CML
- myeloid cells are not mature and dont function right—overproduction of immature blast cells of several myeloid types (no bone marrow crowding)
multiple myeloma
plasma cell cancer → masses in bone marrow and lytic bone lesions
- excessive proliferation of malignant plasma cells which produce M protein—this makes blood viscous and ↑ infections
- also produces Bence-Jones protein (nonfunctional and toxic to renal tissue)
- myeloma cells promote activation of osteoclasts→ bone reabsorption and absorbs calcium from it (↑Ca2+ in blood)
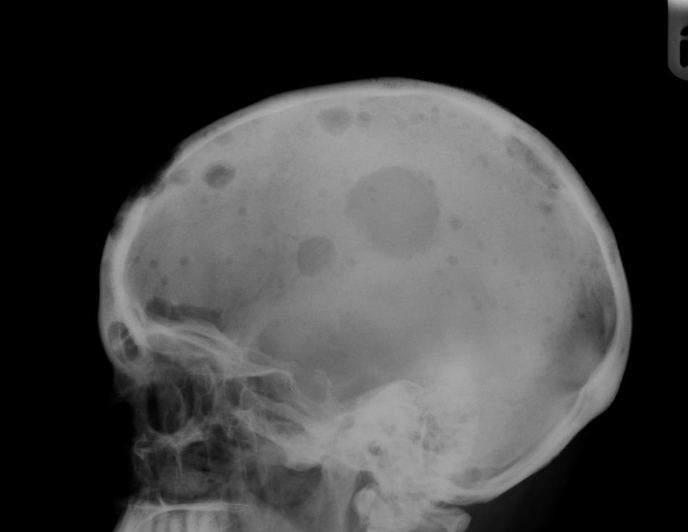
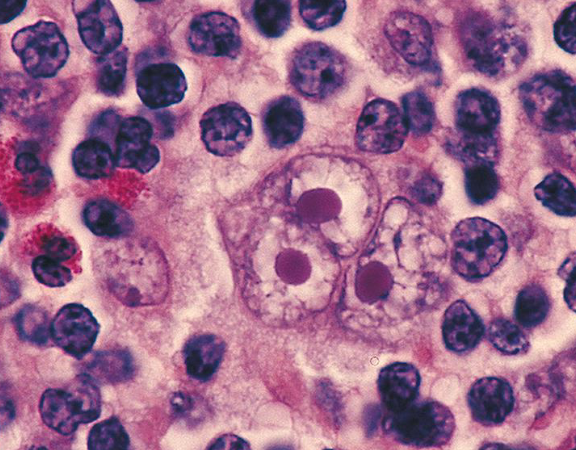
hodgkin lymphoma
neoplasm that develops from malignant lymphocytes
- reed-sternberg cell is the malignant lymphocyte, spreads to adjacent nodes, inflammatory cytokines release symptoms: enlarged, painless lymph nodes
non-hodgkin lymphoma
group of neoplasms arising from lymphoid tissue (usually B-cell neoplasms)
- chromosomal abnormalities→ activation of oncogenes→ inactivation of tumor suppressor genes→ ↑cellular proliferation
- symptoms: swollen, painless peripheral nodes and extranodal involvement
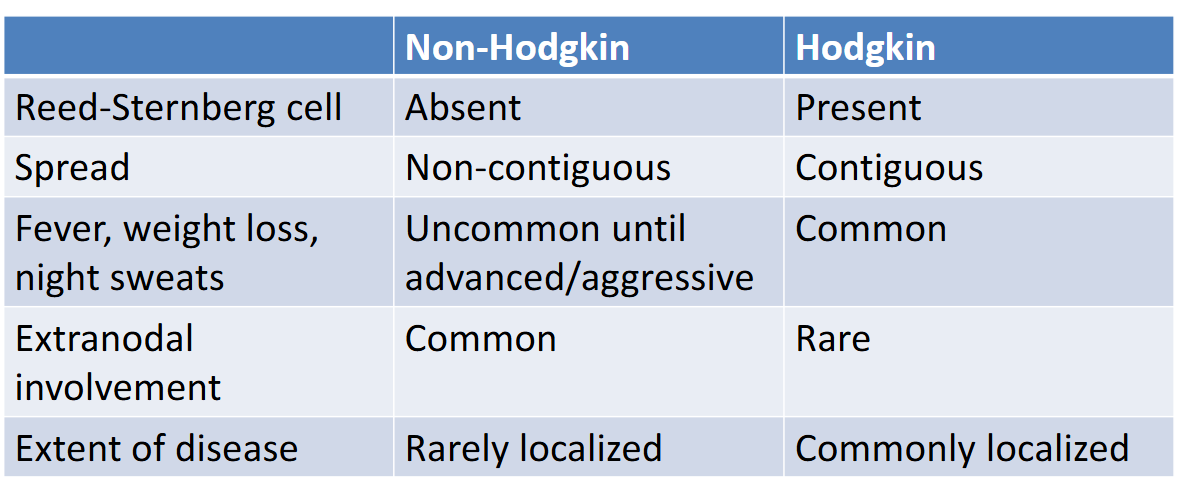
hodgkin vs non-hodgkin
hemostasis platelet response: Activation
step 1- platelets are recruited to damaged endothelium by thromboxane
- platelets are activated by inflammatory mediators and change shape and express receptors on their surface
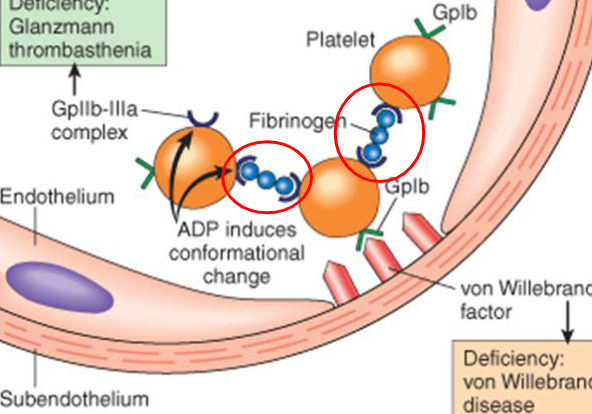
hemostasis platelet response: Adhesion
step 2- von Willebrand factor is produced by endothelial cells and binds to collagen in subendothelium. vWF also binds to receptor on the activated platelets—which is now attached to subendothelial
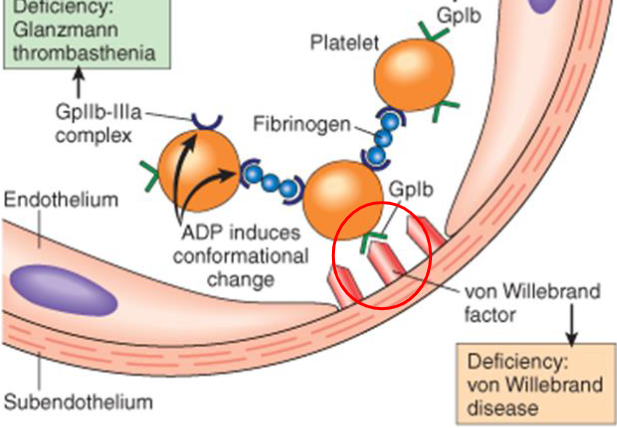
hemostasis platelet response: Aggregation
platelets bound to subendothelial collagen send chemicals to recruit more
- fibrinogen binds to the Gp IIb/IIIa receptors of adjacent platelets and theyre bound together forming a hemostatic plug
hemostasis- clotting factor response
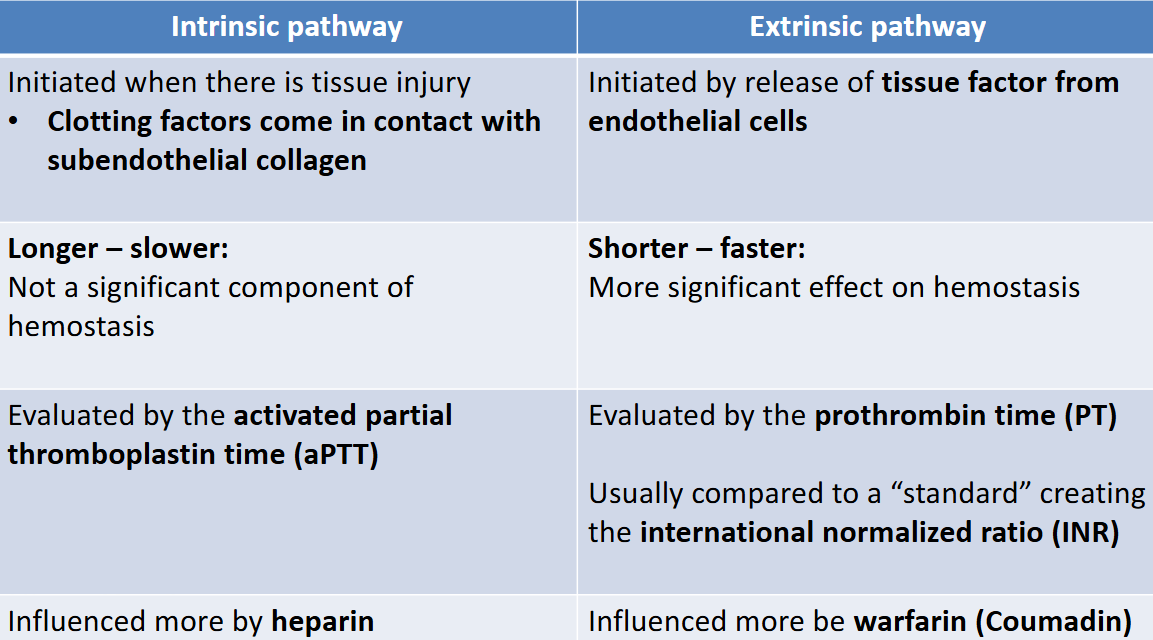
a fibrin mesh will form to stabilize the platelet plug and the coagulation/clotting cascade occurs in 2 pathways
- extrinsic pathway(faster): initiated by release of tissue factor from endothelial cells
- intrinsic pathway: initiated when clotting factors come in contact w subendothelial collagen
These two converge in the activation of factor X which converts prothrombin into thrombin. Thrombin converts fibrinogen into fibrin
true
(T/F) during the clotting factor response, activated Xa converts prothrombin into thrombin. and thrombin is responsible for converting fibrinogen into fibrin
intrinsic vs extrinsic
antithrombin and thrombomodulin
regulates coagulation cascade- reduces thrombin function→ fibrinogen cannot convert into fibrin
thrombocytopenia
not enough platelets. results in ↓platelet production, ↑platelet consumption, and congenital conditions
- ↑platelet consumption can be caused by ITP, HIT, or TTP and ↓platelet production by viral infection/ chemotherapy/ nutritional deficiencies
immune thrombocytopenic purpura (ITP)
most common cause of thrombocytopenia (immune mediated platelet destruction)
- autoantibodies (IgG) produced against antigens on the surface of the platelet. macrophages bind to autoantibody and engulf the platelet
- purpura- red/purple discoloration. also bleeding can occur
thrombotic thrombocytopenic purpura (TTP)
disorder characterized by thrombocytopenia and occlusion of vessels
- ADAMTS13 is absent/inadequate and thus cannot shorten the vWF.
- platelets aggregate on the large vWF molecule and aggregates eventually break free and obstruct vessels. RBC become stressed and fragment
heparin induced thrombocytopenia (HIT)
immune mediated drug reaction
- antibodies are produced against heparin and platelet factor 4. this leads to over activation of platelets and widespread clotting
thrombocythemia
overproduction of platelets due to defect in megakaryocyte progenitor, occlusions
- platelets have abnormal TPO receptors on surface and TPO cannot bind to these receptors. this TPO continues stimulating bone marrow to produce megakaryocytes and ↑platelet production
- DVT and PE can occur
disseminated intravascular coagulation (DIC)
trauma event leads to widespread clotting and depletes the body of clotting factors, fibrinogen,& platelets which then leads to hemorrhage
factor v leiden
genetic mutation of factor V which alters the site of attachment of protein C.
- factor V cant be inactivated by protein C, levels of factor V remain↑ , and clotting persists (risk for thrombosis)
hemophilia A
classic __, most common cause of life threatening bleeding, X-linked recessive trait
- genetic mutation of gene causing Factor VIII deficiency. clotting factors ↓= reduced coagulation and higher risk of bleeding
hemophilia B
genetic mutation of gene causing factor IX, X-linked recessive traight
- clotting factors ↓= reduced coagulation and higher risk of bleeding