B5: CM Nephro Exam 2 Final
1/242
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
243 Terms
glomerulonephritis
What is swelling and inflammation of the glomeruli (reduces ability of the kidneys to maintain balance of substances in blood)?
glomerulonephritis
What disorder:
-swelling and inflammation of the glomeruli
-reduces ability of kidneys to maintain balance of substances in blood
-hematuria and/or proteinuria
-renal insufficiency (azotemia, oliguria)
-HTN
-edema
-can be classified as focal or diffuse:
1. diffuse = all glomeruli are involved
2. focal = some glomeruli are involved
diffuse
(diffuse/focal) glomerulonephritis involves ALL glomeruli
focal
(diffuse/focal) glomerulonephritis involves SOME glomeruli
nephritic syndrome
What disorder:
-INFLAMMATORY
-presents hematuria, proteinuria, edema, HTN, azotemia, oliguria with possible other systemic Sx
-RBC casts
-cola/smoky urine
-<3.5 g/day
-HTN, edema
nephritic
(nephritic/nephrotic) syndrome:
-RBC casts
-cola/smoky urine
-<3.5 g/day
-hematuria
-HTN, edema
nephrotic
(nephritic/nephrotic) syndrome:
-fatty casts
->3.5 g/day
-+ hematuria
-generalized edema, periorbital edema, HTN
nephrotic
(nephritic/nephrotic) syndrome:
• Minimal Change Disease
• Focal Segmental Glomerulosclerosis
• Membranous Nephropathy ( Hepatitis, Lupus )
• Amyloidosis
• Diabetic Glomerulonephropathy
nephritic
(nephritic/nephrotic) syndrome:
• Post-streptococcal/Post infectious
• Subacute Bacterial Endocarditis
• IgA Nephritis ( Berger disease )
• Goodpasture Syndrome - (+) hemoptysis - pulm involvement
• Granulomatosis with polyangiitis
• Eosinophilic Granulomatosis with polyangiitis
• Microscopic polyangiitis
• Diffuse Proliferative Glomerulonephritis
• Alport Syndrome
• Membranoproliferative Glomerulonephritis
rapidly progressive glomerulonephritis
What disorder:
-nephritic syndrome that progresses to renal failure in weeks to months
3 categories
1. Anti-GBM Diseases: good pasture syndrome
2. Immune Complex Mediated Injury: IgA nephritis, post-strep glomerulonephritis, lupus nephritis, C3 glomerulopathy, Henoch Schonlein purpura
3. Pauci-Immune: microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis, granulomatosis with polyangiitis
Eval & Tx:
- HTN
- Other organ involvement & symptomatology
- Lab findings and Kidney Biopsy
post-streptococcal glomerulonephritis
What is the likely nephritic syndrome based on the following renal biopsy results:
Light Micro: enlarged and hypercellular glomeruli, leukocyte infiltration
Immunofluorescence: granular, "starry sky", lumpy/bumpy appearance
Electron Micro: subepithelial immune complex humps
IgA nephropathy
What is the likely nephritic syndrome based on the following renal biopsy results:
Light Micro: mesangial proliferation
Immunofluorescence: IgA immune complex deposits in mesangium
Electron Micro: mesangial immune complex deposition
rapidly progressive glomerulonephritis
What is the likely nephritic syndrome based on the following renal biopsy results:
Light Micro: crescent moon shape
Immunofluorescence: linear immunofluorescence/negative immunofluorescence
Electron Micro: no deposits, if there are deposits it is immune complex deposits
goodpasture syndrome
What is the likely nephritic syndrome based on the following renal biopsy results:
Light Micro: crescent moon shape
Immunofluorescence: linear immunofluorescence
Electron Micro: breaks in GBM, necrosis and crescent formation with no deposits
diffuse proliferative glomerulonephritis
What is the likely nephritic syndrome based on the following renal biopsy results:
Light Micro: wire looping of capillaries
Immunofluorescence: granular
Electron Micro: subendothelial, subepithelial or intramemranous IgG based immune complexes with C3 deposition
Ex: Lupus nephritis
alport syndrome
What is the likely nephritic syndrome based on the following renal biopsy results:
Light Micro: irregular thickening and thinning and splitting of GBM
Immunofluorescence: irregular deposits
Electron Micro: basket weave appearance due to irregular thickening and splitting of GBM
membranoproliferative glomerulonephritis
What is the likely nephritic syndrome based on the following renal biopsy results:
Light Micro: GBM splitting, tram track appearance on H&E, PAS
Immunofluorescence: granular
Electron Micro: Type 1 = subendothelial immune complex deposits, Type 2 = intramembranous deposits
Ex: Hepatitis
acute
(acute/chronic) treatment for glomerulonephritis involves:
-depends on the specific disease
-may be self-limiting
-in general: treat underlying disorder, ACEI/ARBs, corticosteroids, immunosuppressants, plasmapharesis, dialysis
chronic
(acute/chronic) treatment for glomerulonephritis involves:
-eat less protein, Na+ and K+
-control BP (ACEI/ARBs, other HTN meds)
-take diuretics to treat puffiness and swelling
-take Ca2+ supplements
-kidney transplant
infection related glomerulonephritis
What disorder (GENERAL):
-triggered by an infectious antigen
-typically affects children (2-14 y/o) in developing countries
-can affect adults, most commonly have chronic infections
-immune complexes in glomeruli will activate C'
post-streptococcal glomerulonephritis
What disorder:
-develops 1-6 WEEKS after infection with nephritogenic strains of group A strep (M1/M4/M12) --> can be strep pharyngitis OR strep skin infection (impetigo)
-most common cause of acute nephritis in children globally
-risk increased in pts >60 and children 5-12 y/o
-M > F
Pathogenesis:
-most likely strep antigens = NAPlr and SPEB
-Immune complex formation/deposition in glomeruli --> C' activation and PMN recruitment
-inflammation --> proliferative glomerulonephritis and variable decline in GFR
-M protein molecular mimicry: M protein protects GAS from phagocytosis, anti-M protein Abs can possibly cross react w/ heart antigens causing rheumatic fever
Histology:
-granular deposition of IgG and C3
-Humps: dome-shaped subepithelial electron-dense deposits along GBM
Diagnosis:
-clinical findings of acute nephritis
-(+) culture or serologic test w/ anti-streptolysin O or streptozyme test
S/Sx:
-most children asymptomatic
-may have Sx of hematuria, edema, and HTN
Tx: supportive to minimize fluid overload + control BP, Abx if infection still present
IgA nephropathy
(post-strep glomerulonephritis/IgA nephropathy):
-has a precedent URI with disorder occur DAYS after (or even with URI)
post-strep glomerulonephritis
(post-strep glomerulonephritis/IgA nephropathy):
-has precedent URI with disorder occurring a WEEK+ after infection
endocarditis
What disorder:
-inflammation of endocardium
-usually caused by an infection -- primarily bacterial, rarely fungal
-RFs = >60 y/o, M > F, IVDU, poor dental health, heart disease
Presentation:
-Sx of infection (not very specific) --> Fever, fatigue, flu-like Sx, petechiae or splinter hemorrhages
-Less common that are highly suggestive = janeway lesions (nonpainful), osler nodes (painful), roth spots
-cardiac murmurs common
Complications = glomerulonephritis, sepsis, arrhythmias, systemic embolization
subacute endocarditis
What disorder:
-Etiology = S. aureus (most commonly)
-RFs = IVDU, hepatitis C, DM, congenital heart disease/rheumatic heart disease
-Presentation = acute kidney injury, hematuria
-Pathogenesis = direct activation of C' by bacterial antigens, superantigens that directly injure endothelial cells, and/or C' regulatory protein dysfunction
-Diagnosis = Hx, cardiac exam, (+) blood culture, UA
-Biopsy = crescentic glomerulonephritis w/ evidence of tubular injury and interstitial inflammation
-Immunofluorescence = C3 predominantly
-Electron Micro = subepithelial humps in minority of pts
crescent formation
Pathogenesis of __________ involves:
-2+ layers of proliferating cells in Bowman's space
-can be circumferential or segmental
-histologic marker of severe glomerular injury
-typically associated w/ RPGN
-non-specific response to injury of glomerular capillary wall (movement of plasma products into bowman's space w/ subsequent fibrin formation)
-indicative that disease will not respond to immunosuppressive therapy
membranoproliferative glomerulonephritis
What disorder:
-caused by hepatitis B and C
-Light Microscopy = GBM has splitting with a "tram track" appearance due to GBM proliferation
-may have mixed cryoglobulinemia syndrome (hypocomplementemia and circulating cryoglobulins) when associated w/ HCV
Presentation:
-when associated with HCV, may have polyarteritis nodosa with systemic Sx and signs of multisystem involvement --> biopsy shows increased hyalinization around arteries. Gold standard = renal arteriogram
Labs:
-C' levels tend to be normal
-don't find cryoglobulins or RF
Treatment:
-antiviral therapy
-vasculitic manifestations treated w/ immunosuppressive therapy --> glucocorticoids + methotrexate or azathioprine
alport syndrome
What disorder:
-aka hereditary nephritis
-a glomerular disease that starts as asymptomatic persistent microscopic hematuria with eventual kidney failure between 16-35 y/o
-Ocular abnormalities = anterior lenticonus (pathogomonic), fleck retinopathy, corneal changes)
-Sensorineural hearing loss
-Pathogenesis: impaired integrity of BM due to abnormalities in collagen IV alpha chains
-Genetics: abnormalities in collagen IV genes (really any type of mutation)
Pathology:
-histologic changes typically increase in severity with age
-interstitial infiltrate with foam cells
-thinning of GBM, splitting of lamina densa, laminations
anti-glomerular basement membrane disease
What disorder:
-aka Goodpasture syndrome/disease
-RF = HLA-DR15
-Bimodal Distribution = 20-30 y/o (male, renopulmonary syndrome) & 60-70 y/o (female, renal involvement only)
Presentation:
-rapidly progressive glomerulonephritis (>90%) --> proteinuria, macroscopic hematuria
-+ alveolar hemorrhage (SOB, cough, hemoptysis)
-malaise, weight loss, fever, arthralgias
Diagnosis: Kidney biopsy
-LM = crescentic glomerulonephritis
-Immunofluorescence = linear deposition of IgG along glomerular capillaries
Pathogenesis:
-Anti-GBM Abs --> principal target is NC1 domain of alpha3 chain of type IV collagen
Histology:
-disruption of glomerular BM caused by fibrinoid necrosis and crescent formation
-crescents are usually widespread, of similar age, and activity b/c disease onset is acute (not ongoing process)
-immunofluorescence = strongly linear for IgG
ANCA-associated vasculitides
What is the most common cause of rapidly progressive glomerulonephritis in adults >60 y/o?
pauci-immune glomerular diseases
What disorder:
-aka ANCA-associated vasculitides
-necrotizing small vessel vasculitis (preference for kidneys, lungs, peripheral nervous system)
-sometimes limited to one organ
-Renal Biopsy: focal, necrotizing, and crescentic glomerulonephritis with immunofluorescence showing cytoplasmic staining of granulocytes OR perinuclear staining
Abs against cytoplasmic antigens:
-Myeloperoxidase (MPO) = p-ANCA
-Proteinase 3 (PR3) = c-ANCA
granulomatosis with polyangiitis
What disorder:
-type of pauci-immune glomerular disease
-most commonly in older adults
-necrotizing vasculitis predominantly involving small-medium sized vessels
-typically produces granulomatous inflammation of upper + lower respiratory tracts
-c-ANCA (+) (PR3)
microscopic polyangiitis
What disorder:
-type of pauci-immune glomerular disease
-most commonly in older adults
-necrotizing vasculitis that primarily affects capillaries, venules, or arterioles
-most commonly manifests as necrotizing glomerulonephritis and/or pulmonary capillaritis
-NO granulomatous inflammation
-50% have MPO-ANCA (p-ANCA), 40% have PR3-ANCA (c-ANCA)
eosinophilic granulomatosis with polyangiitis
What disorder:
-type of pauci-immune glomerular disease
-eosinophilia, asthma, small vessel vasculitis
-p-ANCA (+) (MPO)
lupus nephritis
What disorder:
-occurs in SLE pts
-proteinuria, microscopic hematuria, kidney function impairment, HTN
Pathogenesis:
-driven by auto-Ab against nuclear and non-nuclear antigens
-form immune complexes in circulation or in situ
-C' activation, leukocyte recruitment, and intrarenal cytokine signaling contribute to glomerular and tubulointerstitial injury
Histology = 6 histopathologic classifications
Treatment: depends on the class
-In general = corticosteroids, immunosuppressants, ACEIs/ARBs, diuretics, diet modification, maybe kidney transplant
-Immunosuppressive therapy for Class III, IV, or V
-NO immunosuppressive therapy for I, II, VI
-initial therapy w/ glucocorticoids in addition to mycophenolate or cyclophosphamide & monoclonal Abs can be used as well as initial Tx as an alternative
-subsequent therapy after response would include MMF or azathioprine could use cyclosporine or tacrolimus
Prognosis
-most pts do well
-for those w/ type III, IV, V --> any response to Tx will improve outcome, complete response will reveal improved renal function is usually 4 wks, partial response associated with unfortunate relapse
minimal mesangial (class I)
Which histologic classification of lupus nephritis:
-normal UA and serum creatinine
-no/minimal proteinuria
-no light microscopic abnormalities
-rarely, if ever diagnosed
-have only mesangial immune deposits ID by immunofluorescence alone or by both immunofluorescence and electron microscopy
-earliest and mildest form of glomerular involvement
mesangial proliferative (class II)
Which histologic classification of lupus nephritis:
-microscopic hematuria and/or proteinuria
-mesangial hypercellularity or mesangial matrix expansion
-prognosis is excellent
-no specific therapy indicated unless pt progresses to more advanced disease or has evidence of extensive podocyte effacement and nephrosis
focal (class III)
Which histologic classification of lupus nephritis:
-hematuria and proteinuria
-some pts have HTN, dec. GFR, and/or nephrotic syndrome
-<50% of glomeruli affected
-immunofluorescence shows IgG and C3 deposits -- almost uniform involvement
-active or inactive endocapillary or extracapillary glomerulonephritis almost always segmental (e.g., involves <50% of glomerular tuft)
-electron microscopy reveals immune deposits in subendothelial space of glomerular capillary wall + mesangium
diffuse (class IV)
Which histologic classification of lupus nephritis:
-most common and severe form
->50% of glomeruli affected
-diffuse wire loop deposits, little or no glomerular proliferation
-hematuria, proteinuria, nephrotic syndrome, HTN, dec. GFR
-significant hypocomplementemia (esp. C3), elevated anti-dsDNA levels, esp. during active disease
-electron micro reveals subendothelial deposits (at least during active phase)
-With active disease, hypercellular, necrotizing lesions, and crescent formation all may be present
-thickening of the glomerular capillary wall and a pattern on light microscopy that is similar to that in membranoproliferative glomerulonephritis. These lesions are characterized by the marked influx of proinflammatory cells (monocytes, suppressor/cytotoxic T cells), sometimes resulting in cellular crescents
membranous nephropathy (class V)
Which histologic classification of lupus nephritis:
-diffuse thickening of glomerular capillary wall on light microscopy and subepithelial immune deposits on immunofluorescence or electron micro
-present with nephrotic syndrome
-microscopic hematuria and HTN may also be seen
-[creatinine] either normal or slightly elevated
advanced sclerosing (class VI)
Which histologic classification of lupus nephritis:
-slowly progressive kidney dysfunction with proteinuria
-global sclerosis (>90%) of glomeruli
-immunosuppressives not going to help
IgA nephropathy
What disorder:
-most common cause of primary glomerulonephritis in industrialized countries
-more frequent in East Asian and Caucasian pts
-peak incidence 20-30 yrs old
-M > F in N. America
-IgA deposition occurs in many disease (lupus nephritis, minimal change disease, diabetic nephropathy, IgA vasculitis, linear IgA bullous dermatosis)
-may be associated with other conditions --> cirrhosis, celiac disease, HIV
Pts typically Present in 1-3 ways:
1. 1 or recurrent episodes of gross hematuria --> often with URI but may present like UTI or urolithiasis
2. Microscopic hematuria --> usually mild proteinuria, detected during routine exam
3. Nephrotic Syndrome or RPGN --> edema, HTN, renal insufficiency, hematuria
Pathogenesis:
-Galactose-deficient IgA1 (Gd-IgA1) and Gd-IgA1 auto-Abs form immune complexes
-C3 is co-deposited with IgA; alteernative and lectin pathways activated
-mesangial immune complex deposits activate mesangial cells and incite glomerular injury
Pathology: mesnagial and endocapillary hypercellularity, segmental glomerulosclerosis, tubular atrophy, crescents
IgA nephropathy
What disorder:
Pts typically Present in 1-3 ways:
1. 1 or recurrent episodes of gross hematuria --> often with URI but may present like UTI or urolithiasis
2. Microscopic hematuria --> usually mild proteinuria, detected during routine exam
3. Nephrotic Syndrome or RPGN --> edema, HTN, renal insufficiency, hematuria
Pathogenesis:
-Galactose-deficient IgA1 (Gd-IgA1) and Gd-IgA1 auto-Abs form immune complexes
-C3 is co-deposited with IgA; alteernative and lectin pathways activated
-mesangial immune complex deposits activate mesangial cells and incite glomerular injury
Pathology: mesnagial and endocapillary hypercellularity, segmental glomerulosclerosis, tubular atrophy, crescents
-glomerular deposition of C3 --> C1q almost always absent
-mesangial hypercellularity is major finding of light microscopy
Diagnosis confirmed by kidney biopsy: immunofluorescence shows mesangial deposits of IgA
membranoproliferative glomerulonephritis
What disorder:
-a pattern of glomerular injury on kidney biopsy
-hypercellularity and glomerular basement membrane thickening
-a histologic lesion (not specific disease) classified based on immunofluorescence microscopy
-immune complex/monoclonal Ig mediated = from Hep C infection (typically assoc. w/ mixed cryoglobulinemia, C3/C4/CH50 low), autoimmune diseases, monoclonal gammopathy (monoclonal Ig deposition, dense deposit disease, C3GN)
-immune complexed mediated caused by HCV, C3GN or DDD, due to chronic infections, autoimmune disease, plasma cell dyscrasias
2 primary mechanisms:
-deposits of immune complex or monoclonal Ig leading to C' activations
-dysregulaton and persistent activation of alternative C' pathways
without Ig or C' deposition: seen in healing phase of several disease
Underlying cause: endothelial injury, followed by reparative changes to repair that damage
Micro: no Ig or C' in glomeruli on immunofluorescence, no electron-dense deposits along capillary walls, electron microscopy shows subendothelial and mesangial deposits
C3 glomerulopathy
Which type of complement mediated membranoproliferative glomerulonephritis:
-C3 dense deposit disease
-may have persistently low C3 but normal C4
-C3 nephritic factor: auto-Ab to alternative C3 convertase
C4 glomerulopathy
Which type of complement mediated membranoproliferative glomerulonephritis:
-C4 dense deposit disease
-glomerular deposition of C4
-absence of C3, C1q, and Ig
ARBs (control BP and reduced proteinuria) and follow up to ensure proper kidney function maintained
How do you treat membranoproliferative glomerulonephritis in pts with normal kidney function, no active urinary sediment, and non-nephrotic range proteinuria?
nephrotic syndrome
What disorder is caused by a derangement in glomerular capillary walls resulting in increased permeability to plasma proteins?
Hint: Manifestations include massive proteinuria (>3.5 g or more of protein), hypoalbuminemia (serum <3 g/dL), generalized edema, and hyperlipidemia/lipiduria
heavy proteinuria
____________ depletes serum albumin levels at a rate beyond the compensatory synthetic capacity of the liver, resulting in hypoalbuminemia
foamy
In pts with nephrotic syndrome, their urine can be described as "________" because of the increased protein in urine
decreased intravascular colloid osmotic pressure
generalized edema in pts with nephrotic syndrome is a direct consequence of _________. Water and sodium retention will also aggravate this
edema
__________, often found in nephrotic syndrome, is characteristically soft and pitting and is more marked in the periorbital regions and dependent portions of the body (very prone to gravity -- will collect in LE); if severe it may lead to pleural effusions and ascites.
nephrotic syndrome
Most pts with _________ have increased blood levels of cholesterol, TGs, VLDL, LDL, Lp lipoproteins and apoprotein. These defects seem to be due to a combination of increased synthesis of lipoproteins in the liver, abnormal transport of circulating lipid particles, and decreased lipid catabolism. Lipiduria follows hyperlipidemia because lipoproteins will also leak across the glomerular capillary wall
salt restriction (2g in adults) and loop diuretics
How do you treat the edema in nephrotic syndrome?
nephrotic syndrome (maltese cross formation in fat droplets under polarized light in urine)
ID

infection
Nephrotic pts are particularly vulnerable to _____________, esp. staph and pneumococcal, likely due to the loss of immunoglobulins in the urine
thrombotic and thromboembolic
______________ complications are common in nephrotic syndrome due in part to los of endogenous anticoagulants in urine.
nephrotic syndrome
What disorder:
S/Sx: increased swelling (periorbital, BLE, abdominal, genital), frothy/foamy urine, weight gain, fatigue, SOB
PE: HTN, edema (periorbital, LE, ascites, genital), leukonychia (white streaks on fingernails suggesting hypoalbuminemia)
Labs/Urine Studies:
-hypoalbuminemia (<3.5 g/dL)
-hyperlipidemia
-proteinuria (>3.5 g/day or >350 mg/mmol on spot urine protein to creatinine ratio) within 24 hrs
-fatty casts with maltese cross sign
Causes
-Primary Glomerular Diseases (membranous nephropathy, minimal change diseae, focal segmental glomerulosclerosis)
-Systemic Diseases (DM, amyloidosis, SLE, drugs, infections, malignancies, bee sting allergy, hereditary nephritis)
membranous nephropathy
What disorder:
-characterized by diffuse thickening of the glomerular capillary wall due to accumulation of deposits containing Ig along the subepithelial side of the BM
-75% are primary in origin (from kidney)
-25% are secondary to drugs, malignancy, SLE, infections, other autoimmune disorders
-form of chronic immune mediated diases
-Primary: autoimmune disease linked to HLA-DQA1 and other HLA alleles and caused in most cases by Abs to renal auto-antigen (most often phospholipase A2 R)
-Secondary: inciting antigens (endogenous or exogenous) can deposit complexes of self nuclear protein and auto-Abs (like in SLE) which can be identified in immune complexes
Clinical Course & Tx:
-need to rule out secondary causes b/c treating underlying disorder or d/c drug can stop/reverse injury
-complete or partial remissions may occur even without therapy (can observe but treat nephrotic syndrome)
-progression associated w/ sclerosis of glomeruli, rising SCr reflecting renal insufficiency, and developing HTN
-Proteinuria can be persistent, but only 10% die or progress to renal failure within 10 years and no more than 40% develop CKD/ESRD
-recurs in 40% who undergo transplant for ESRD
minimal change disease
What disorder:
-relatively benign disorder characterized by diffuse effacement of foot processes of visceral epithelial cells (podocytes), detectable only by electron microscopy in glomeruli that appear virtually normally by light microscopy
-most common cause of nephrotic syndrome in children (2-6 y/o)
-sometimes follows URTI or routine prophylactic immunization
-Light Micro: normal appearing glomerulus
-Electron Micro: marked degenerative changes of podocyte with diffuse retraction and effacement of foot processes ("fusion), microvillous degeneration of cell surface, and presence of vacuoles in cytoplasm
-Etiology: involves some immune dysfunction that results in elaboration of factors that damage visceral epithelial cells and cause proteinuria. NO immune deposits in glomerulus
-There are several factors that point to an immunologic basis: (1) clinical association w/ URTI and prophylactic immunizations, (2) response to corticosteroids and immunosuppressive therapies, (3) can have association w/ atopic disorders, (4) inc. prevalence of HLA haplotypes (suggesting genetic predisposition), (5) increased incidence in pts with Hodgkin lymphoma
minimal change disease
What disorder:
-relatively benign disorder characterized by diffuse effacement of foot processes of visceral epithelial cells (podocytes), detectable only by electron microscopy in glomeruli that appear virtually normally by light microscopy
-most common cause of nephrotic syndrome in children (2-6 y/o)
-sometimes follows URTI or routine prophylactic immunization
-Light Micro: normal appearing glomerulus
-Electron Micro: marked degenerative changes of podocyte with diffuse retraction and effacement of foot processes ("fusion), microvillous degeneration of cell surface, and presence of vacuoles in cytoplasm
-Etiology: involves some immune dysfunction that results in elaboration of factors that damage visceral epithelial cells and cause proteinuria. NO immune deposits in glomerulus
Clinical Course & Tx
-renal function maintains good w/o HTN/hematuria despite massive proteinuria
-proteinuria mostly made of albumin
-RESPONDS TO CORTICOSTEROIDS, most children respond rapidly to this!
-long-term prognosis excellent, disease usually resolves once puberty hits (also excellent for adults)
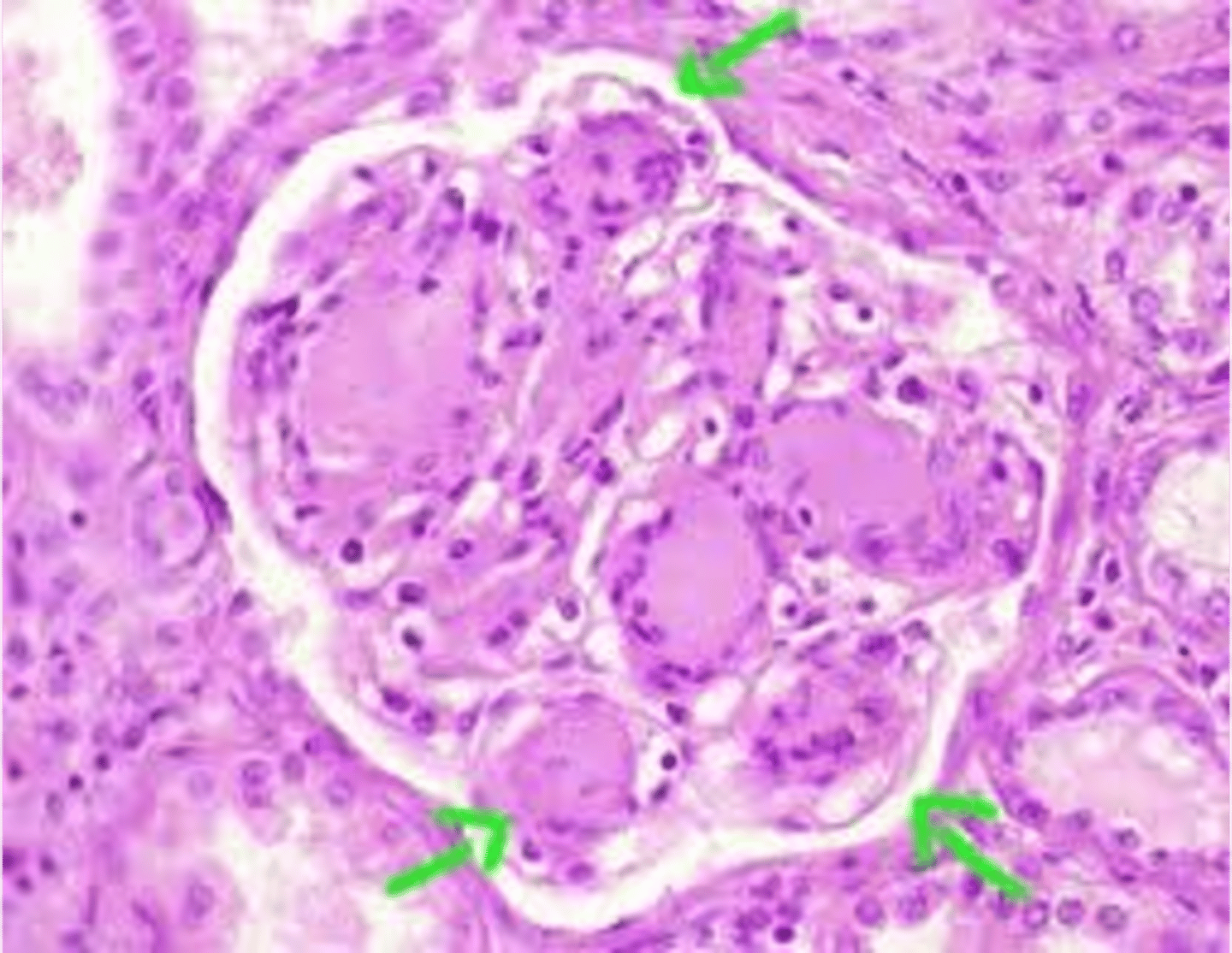
focal segmental glomerulosclerosis
What disorder:
-most common cause of nephrotic syndrome in adults in the US
-considered to be a primary disorder of podocytes
-characterized by sclerosis of some, but not all glomeruli
-only a portion of capillary tuft in the glomerulus is involved
-can be primary, idiopathic, or secondary to HIV, heroin use, sickle cell disease, massive obesity
S/Sx:
-acute or subacute onset of nephrotic syndrome or non-nephrotic proteinuria
-HTN, microscopic hematuria, and some degree of azotemia present
Pathogenesis:
-degeneration and focal disruption of podocytes w/ effacement of foot processes (similar to minimal change disease)
-epithelial damage!!!! = hallmark
-hyalinosis and sclerosis stem from entrapment of plasma proteins in extremely hyperpermeable foci and increased extracellular membrane deposition
-mutations in NPHS1 encodes nephrin, a component of slit diaphragm that controls glomerular permeability
-MPHS2, alpha actinin 4, and TRPC6 mutations may also be present
focal segmental glomerulosclerosis
ID
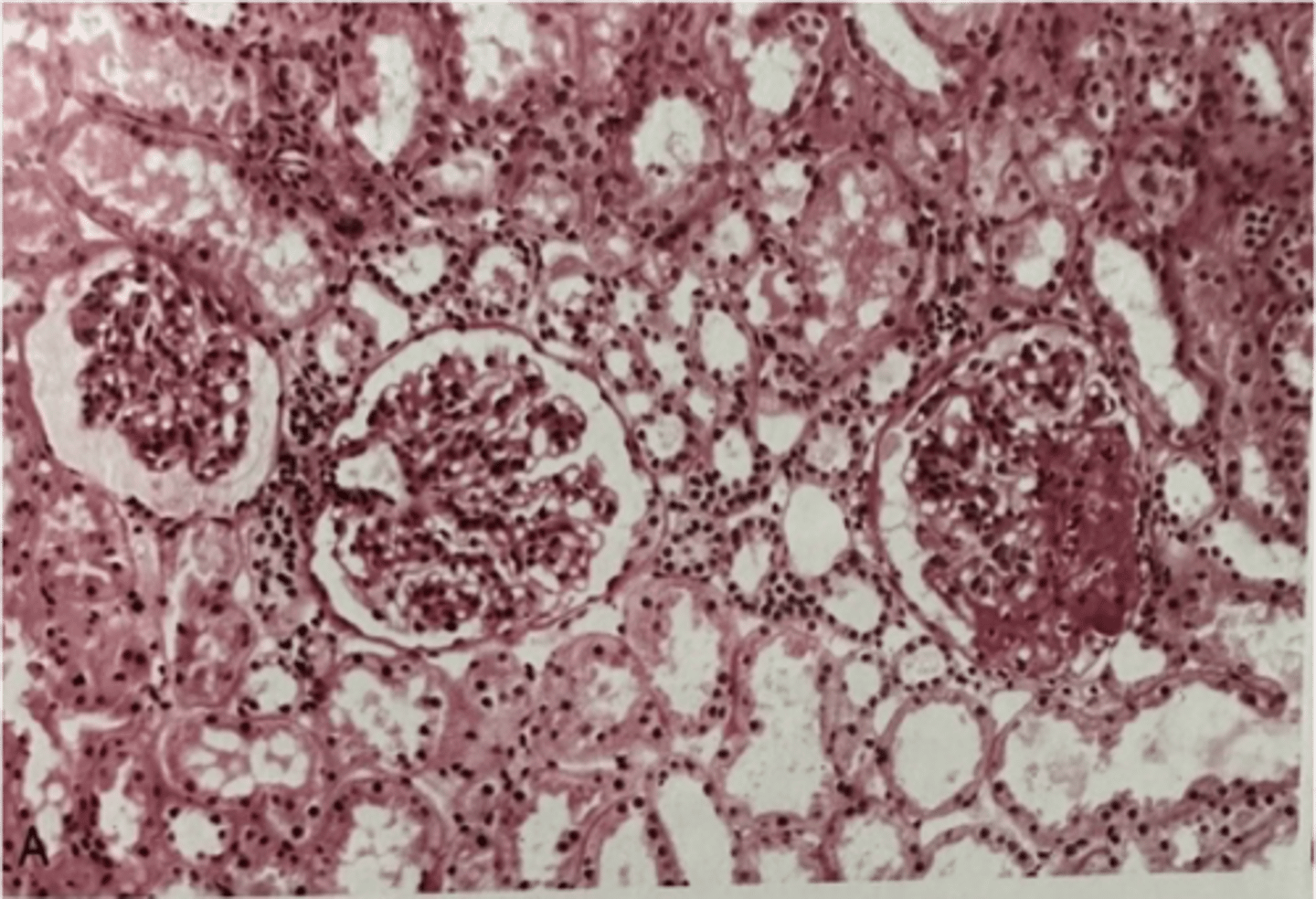
focal segmental glomerulosclerosis
What disorder:
-most common cause of nephrotic syndrome in adults in the US
-considered to be a primary disorder of podocytes
-characterized by sclerosis of some, but not all glomeruli
-only a portion of capillary tuft in the glomerulus is involved
-can be primary, idiopathic, or secondary to HIV, heroin use, sickle cell disease, massive obesity
S/Sx:
-acute or subacute onset of nephrotic syndrome or non-nephrotic proteinuria
-HTN, microscopic hematuria, and some degree of azotemia present
Electron Micro: diffuse effacement (fusion) of podocyte foot processes and early hyalin deposition
Clinical Course and Tx:
-little tendency for spontaneous remission in idiopathic and response to corticosteroids is variable
-children have better prognosis
-20% follow an unusually rapid course and end up with massive proteinuria and renal failure within 2 years
diabetic nephropathy
What disorder:
-largest cause of renal failure in US
-Hallmark = microalbuminuria on dipstick early on
-attempt to slow progression by controlling DM and decreasing proteinuria with ACEIs or ARBs
-glomerulosclerosis leads to hyperfiltration of other glomeruli and eventual burnout of the kidney
-Light Micro: mesangial expansion, GBM thickening, eosinophilic nodular glomerulosclerosis (Kimmelstiel-Wilson lesions)
-50% will have ESRD 7-10 years after onset of proteinuria
diabetic nephropathy
ID
multiple myeloma and light chain disease
What disorders:
-type of dysproteinemias
• Often have renal insufficiency at time of presentation
• Not usually a glomerular lesion but tubular disorders due to cast nephropathy
• Can see glomerularoclerosis
• Often leads to ESRD
• SPEP/UPEP: serum and urine protein electrophoresis looking for M-spikes
amyloidosis
What disorder:
-4 subtypes according to chemical deposition of fibrillar deposits
1. primary = light chains
2. secondary = chronic inflammatory or infectious states
3. hereditary = AA proteins
4. Dialysis associated = B2 macroglobulin
-kidney most commonly involved organ
-associated w/ chronic conditions that predispose to amyloid deposition
Light Micro: apple green birefringence of Congo Red stain due to amyloid deposition in mesangium
Electron Micro: mesangial expansion by amyloid fibrils
-80-85% of pts develop nephrotic syndrome
-cardiac failure most common cause of death
mixed essential cryoglobulinemia
What disorder:
-associated w/ HCV
-active sediment
-RF (+)
-low C' (hypocomplementemia)
-cryoprecipitate removed by plasmaphoresis and cytotoxic agents
-relapses common
SLE
What disorder:
-5-10% of these pts will develop nephrotic syndrome
-low C' (C4)
-clinical presentation and severity of glomerular disease classified by histopathology of biopsy
-classifications of system of proliferative lesions and membranous lesions (stages I-VI)
-Tx = cytotoxics and steroids and other underlying disorders/co-morbidities
hereditary
(hereditary/acquired) polycystic renal disease:
-AD (most common), AR
-medullary sponge kidney
-medullary cystic kidney
-VHL diseasee
-Tuberous clerosis
-AD liver disease
acquired
(hereditary/acquired) polycystic renal disease:
-simple cysts
-acquired cysts with dialysis
-medullary sponge kidney
-multicystic renal dysplasia
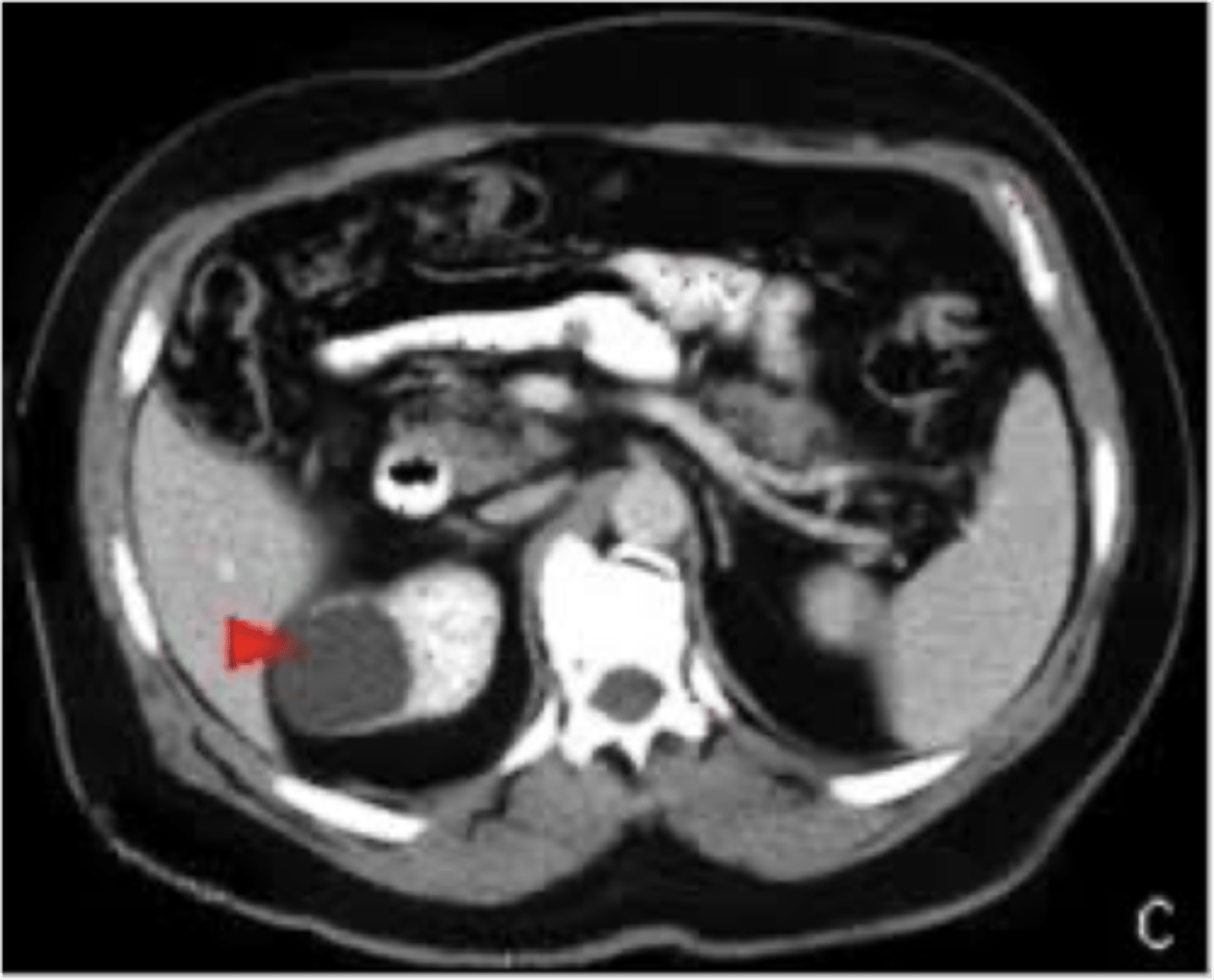
simple renal cyst
What disorder:
-most common renal mass
-solitary, multiple, and BL
-smooth appearing with translucent fluid per CT imaging, anechoic on US
-occurs most often in pts >50 y/o
-often seen on screening US
-range in size from <1 cm - 10 cm
-usually does not compromise renal function
-usually 1 epithelial layer without renal elements
-need to rule out more serious disorders!
-Tx: usually none required
-pain is rare, acetaminophen/NSAIDs short course if renal function is preserved
-rare if ever aspiration and surgery
-infection is rare
simple renal cyst
ID
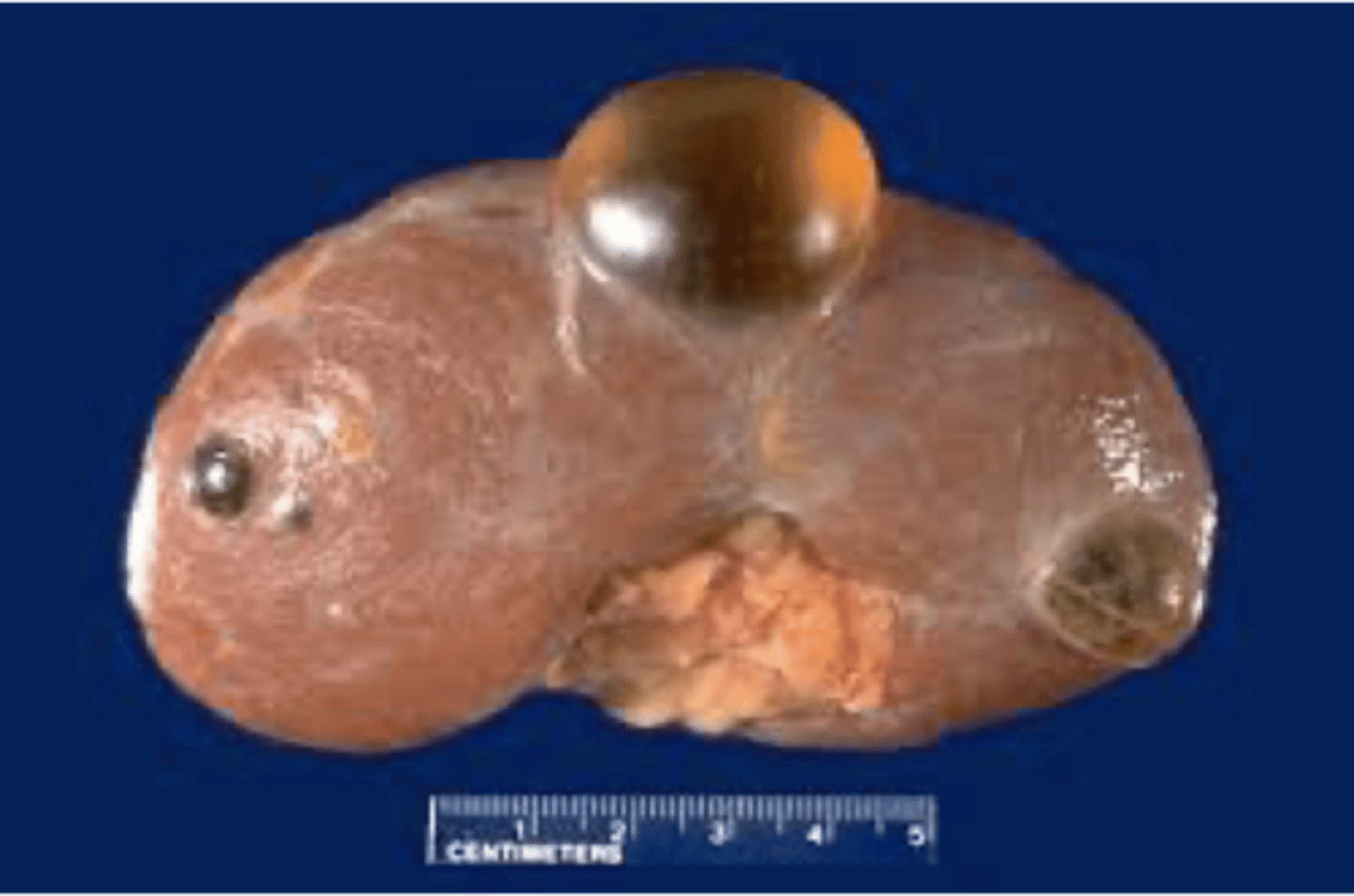
simple renal cyst
ID
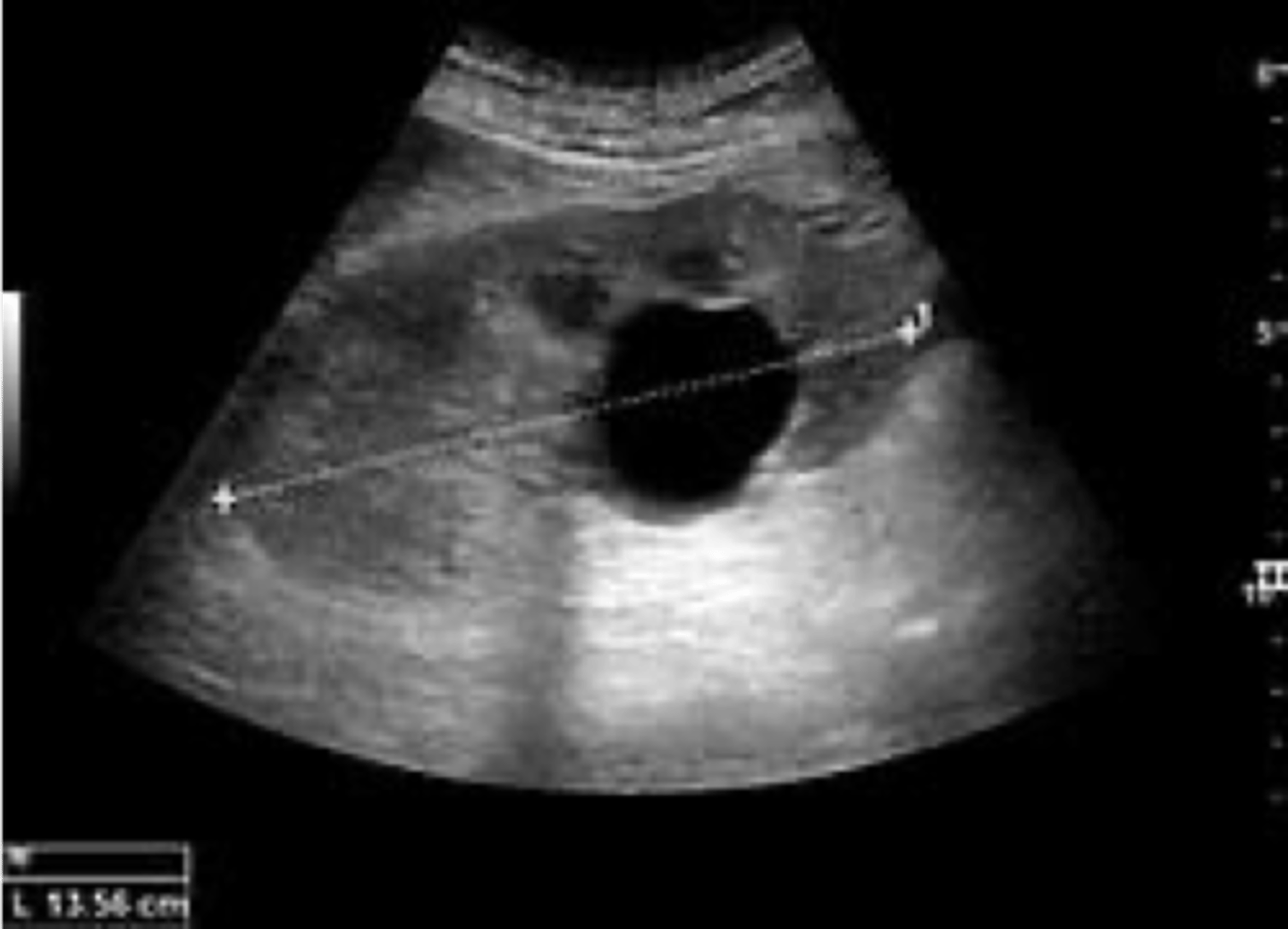
simple renal cyst
ID
AD polycystic kidney disease
What disorder:
-85% of families have abnormality on chromosome 16 (PKD1 locus)
-remaining pts have abnormalities on chromosome 4 (PKD2 locus) --> less severe and often seen in late presenting
-Dx = imaging + Hx
-numerous cysts in cortex and medulla causing BL enlarged kidneys ultimately destroy kidney parenchyma
-Presentation = combo of flank pain, hematuria, HTN, urinary infection, calculi, progressive renal failure
Screening
-Screening is NOT recommended in pts <18 y/o
-RFs = FMHx but may not know the phenotype
-Pts 40+ and 0-1 cyst --> excludes disease
-Pts 30-39 y/o w/o cysts --> excludes disease, may need follow up CT
-PTs <30 --> US limited in ability to exclude disease
Clinical Course:
-renal function starts to decline usually by 30 y/o (GFR decreases 4-6 mL/min/year)
-gradual progression to ESRD
-pain from cysts and enlarged kidneys is common manifestation
-RFs for progression to ESRD = genotype PKD1, HTN, early onset protienuria and hematuria, M gender, increasing kidney size and rate of growth, LV mass index, proteinuria alone, FMHx progression to ESRD
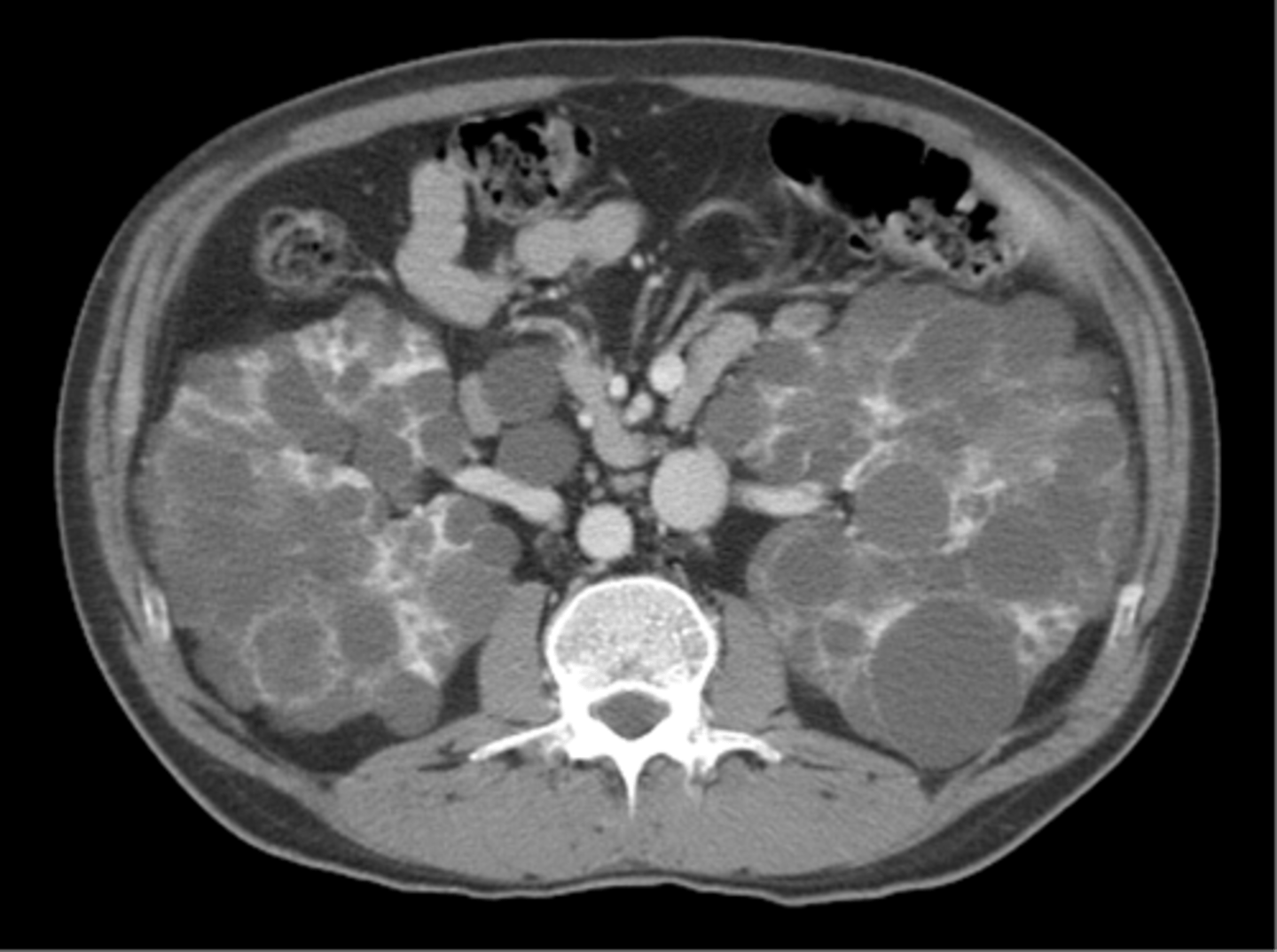
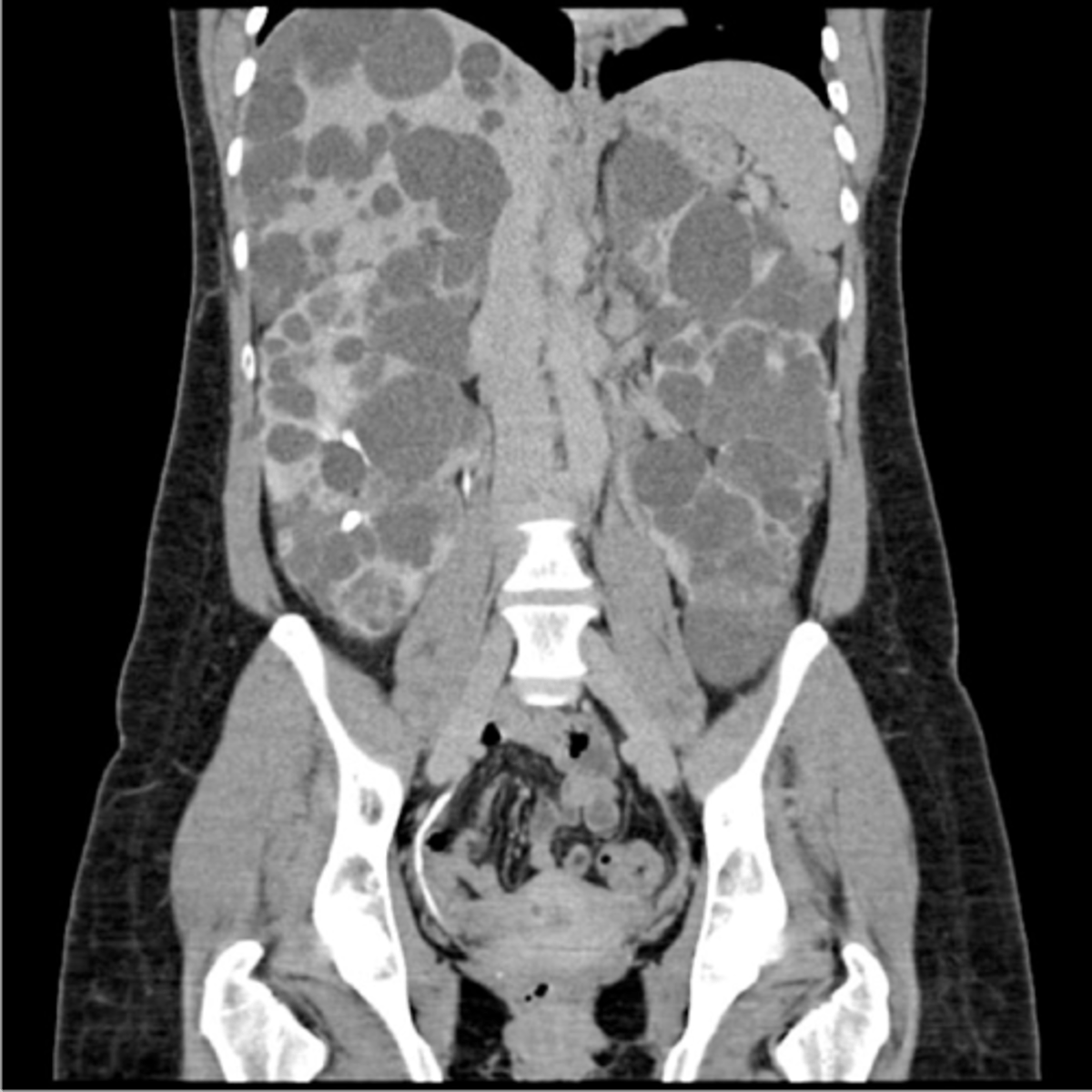
autosomal dominant polycystic kidney disease
What disorder:
-85% of families have abnormality on chromosome 16 (PKD1 locus)
-remaining pts have abnormalities on chromosome 4 (PKD2 locus) --> less severe and often seen in late presenting
-Dx = imaging + Hx
-numerous cysts in cortex and medulla causing BL enlarged kidneys ultimately destroy kidney parenchyma
-Presentation = combo of flank pain, hematuria, HTN, urinary infection, calculi, progressive renal failure
Clinical Course:
-renal function starts to decline usually by 30 y/o (GFR decreases 4-6 mL/min/year)
-gradual progression to ESRD
-pain from cysts and enlarged kidneys is common manifestation
-RFs for progression to ESRD = genotype PKD1, HTN, early onset protienuria and hematuria, M gender, increasing kidney size and rate of growth, LV mass index, proteinuria alone, FMHx progression to ESRD
-cause of death = cardiac (36%), infection (24%), neurologic (12% -- either ruptured intracranial (berry) aneurysm or hypertensive intracerebral hemorrhage)
-pregnant women with this disorder + hypertension have increased risk of fetal loss, no issue with fertility, though. Inc. risk ectopic preg
autosomal dominant polycystic kidney disease
What disorder:
-85% of families have abnormality on chromosome 16 (PKD1 locus)
-remaining pts have abnormalities on chromosome 4 (PKD2 locus) --> less severe and often seen in late presenting
-Presentation = combo of flank pain, hematuria, HTN, urinary infection, calculi, progressive renal failure
Extrarenal manifestations:
-cerebral aneurysms --> high risk in pts w/ FHx, require anticoagulants, pre-op, high risk occupation, symptomatic pts
-Hepatic cysts
-cardiac valve disease, mitral valve prolapse
-colonic diverticula (more in pts on dialysis)
-abdominal wall inguinal hernias
-berry aneurysms (most common around anterior communicating, then MCA, then posterior communicating, then PCA)
Treatment:
-no specific Tx proven to delay progression
-rigorous BP control may slow progression to ESRD and dec. risk of cardiovascular mortality
-if no CI --> ACEI!!!!! first line!!!!!
-statin therapy to dec. risk of CVD
-vasopressin receptor antagonists have some evidence of being beneficial
-increase fluid intake, suppress ADH, inhibit cyst growth
-dialysis
autosomal dominant polycystic kidney disease
ID
autosomal dominant polycystic kidney disease
ID
transplantation
____________ is indicated as a potential treatment for autosomal dominant polycystic kidney disease when:
-post-transplant erythrocytosis
-diverticulitis with perforation
-symptomatic aneurysms
-often require UL or BL --> avoid recurrent UTIs, need a place to put the graft, no benefit to survival
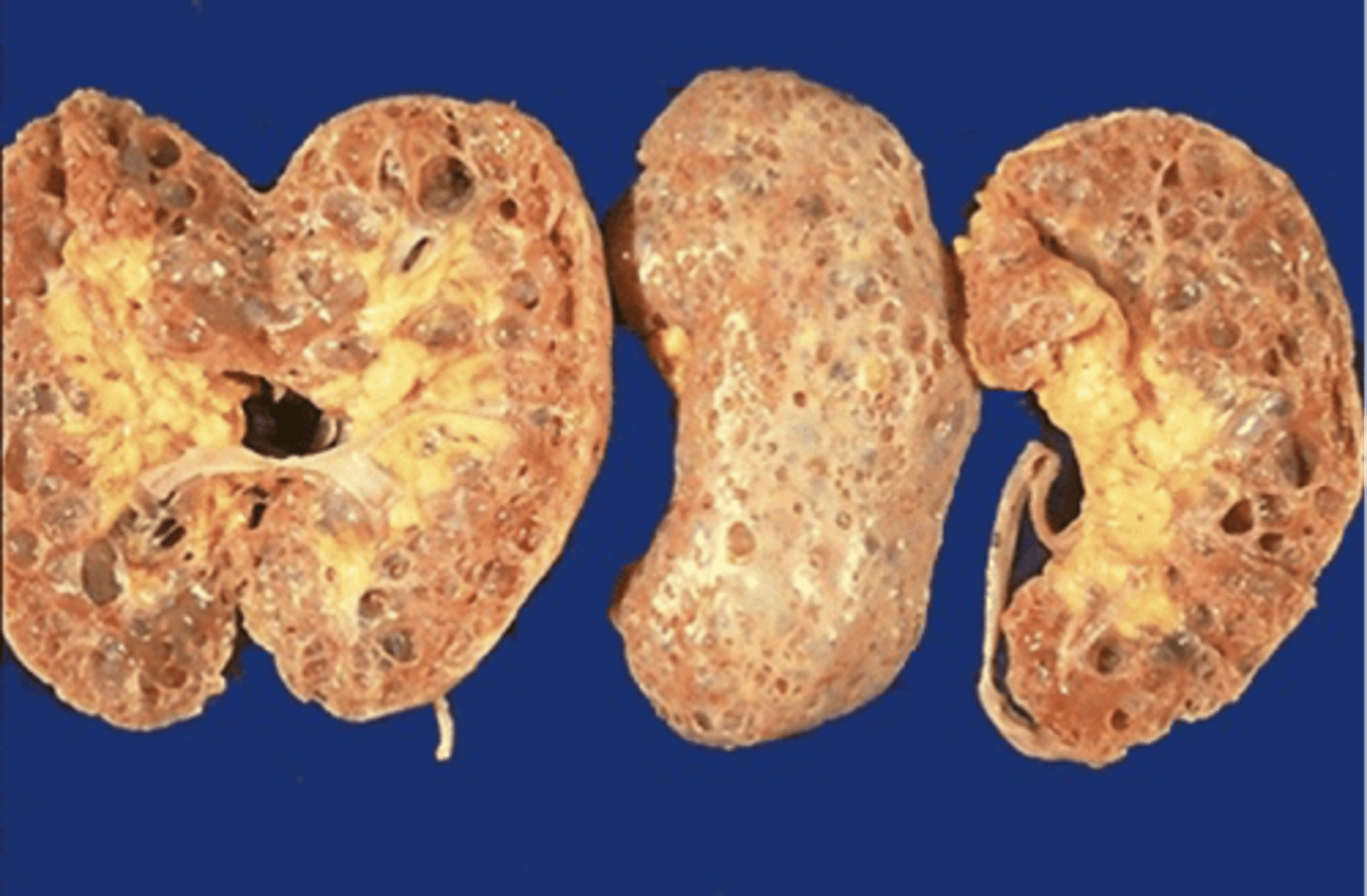
autosomal recessive polycystic kidney disease
What disorder:
-always associated with hepatic involvement (congenital hepatic fibrosis)
-most severe cases on ultrasound in utero
-less severe seen with enlarging kidneys after brith (pts alive after 1st month have 80% chance of survival to 15+ y/o)
-genetic defect on chromosome 6 of PKHD1 gene --> encodes fibrocystin which is found in cortical and medullary collecting ducts as well as epithelial cells of bile ducts
Diagnosis + Outcomes:
-Dx made by US --> kidneys diffusely hyperechogneic w/ multiple small cysts
-adults have slowly progressive kidney dysfunction
-systemic HTN in 75%
medullary sponge kidney
What disorder:
-congenital malformation of terminal CDs in renal pyramids --> urine collects here --> urinary stasis
-F > M
-formation of small and large medullary cysts that do not involve the cortex
-usually BL but can be UL
-benign disease w/ complications of nephrolithaisis and UTIs
-underlying defect unknown
-often incidental discovery by radiology (IVP) --> "brush like appearance"
-S/Sx = flank pain, hematuria, UTIs related to stones, nephrolithiasis
Cut surface = ductal dilatation (bubbles beneath cortex)
Prognosis:
-impaired urinary Ca2+ leads to stones, long-term prognosis is excellent
-rarely advances to ESRD
tuberous sclerosis
What disorder:
-AD genetic disorder, 33% familial
-neurocutaneous disorder involving many organ systems
-clinical diagnosis made by disease findings
2 gene mutations
• TSC1 chromosome 9q34, encodes hamartin protein, expressed in normal tissues
• TSC2 chromosome 16p13, encodes tuberin protein, participates in normal brain development
• The two proteins form a complex that is thought to function as a negative regulator of the cell cycle
Clinical Features:
-triad seizures, mental retardation, and facial angiofibromas
Diagnosis: 2 major features or 1 major feature with > 2 minor features
-Major features: hypomelanotic macules (>3, at least 5 mm diameter), angiofibromas (> 3) or fibrous cephalic plaque, ungual fibromas (> 2), shagreen patch, multiple retinal hamartoma, cortical dysplasias, ubependymal noduels, subependymal giant cell astrocytoma, cardiac rhabdomyoma, lymphangioleiomyomatosis, angiomyolipomas
-Minor Features: confetti skin lesions, dental enamel pits (>3), intraoral fibromas (>2), retinal achromatic patch, multiple renal cystsm non-renal hamartomas
von hippel lindau disease
What disorder:
-AD syndrome
-syndrome of benign and malignant tumors
-presents in childhood, adolescence, or adults (mean age 26 y/o)
-VHL gene mutation (type I or type II, type II at high risk for pheochromocytoma)
S/Sx:
-renal cysts
-retinal hemangiomas (most common)
-clear cell carcinomas of kidney
-cerebellar and spinal hemangioblastomas
-pheochromocytoma
-endocrine pancreatic tumors
-epididymal cystadenomas
Tx:
-genetic counseling
-early diagnosis of more invasive tumors
medullary cystic disease
What disorder:
-rare AD cystic disease
-renal cysts at corticomedullary junction
-small to normal size kidneys
-hyperuricemia and gout
-grouped w/ other hyperuricemic diseases (nephronophthisis/MCKD complex)
Diagnosis + Tx:
-strong FHx
-preponderance of gout
-bland urinary sediment, no protein
-US may show medullary cysts
-slow progressive CKD
-allopurinol for gout
-transplant is best option b/c disease does not recur
acquired cystic disease
What disorder:
-associated w/ hemo/peritoneal dialysis
-multiple, small, BL cysts
-easily distinguished from PCKD b/c no FHx, kidneys are small to normal size, and incidence rises with time on dialysis
-Pathogenesis not completely understood. Nephron loss leads to hypertrophy of normal nephrons, activates proto-oncogenes, growths factors and overtime tubular hyperplasia and cyst formation occurs
-Most pts are asymptomatic
-risk of RCC 4-7% over 10 years
dialysis associated (acquired) cystic disease
ID
multicystic renal dysplasia
What disorder:
-abnormal differentiation of renal parenchyma
-often UL, opposite kidney will undergo UL hyperplasia
-abnormal differentiation of metanephric parenchyma
-one of the most common forms of congenital cystic renal diseases AND abdominal masses in newborns
multicystic renal dysplasia
ID
renal cell carcinoma
What disorder:
-accounts for 85% of renal cancers in adults
-occurs mostly in older pts (50-60 y/o)
-M > F
-mostly sporadic but can be AD (esp. young)
-associated with VHL syndrome (associated w/ retinal/cranial hemangioblastomas, pheochromocytomas, pacnreatic neuroendocrine tumors/cysts, braod ligament/epididymal cystadenoma), hereditary leiomyomatosis and RCC syndrome, hereditary papillary carcinoma, birt-Hogg dube syndrome
RFs = SMOKING!!!!!, obesity, HTN, unopposed estrogen therapy, exposure to asbestos, petroleum products, heavy metals, ESRD, CKD, acquired cystic disease, tuberous sclerosis
PE = flank pain, abdominal mass, hematuria
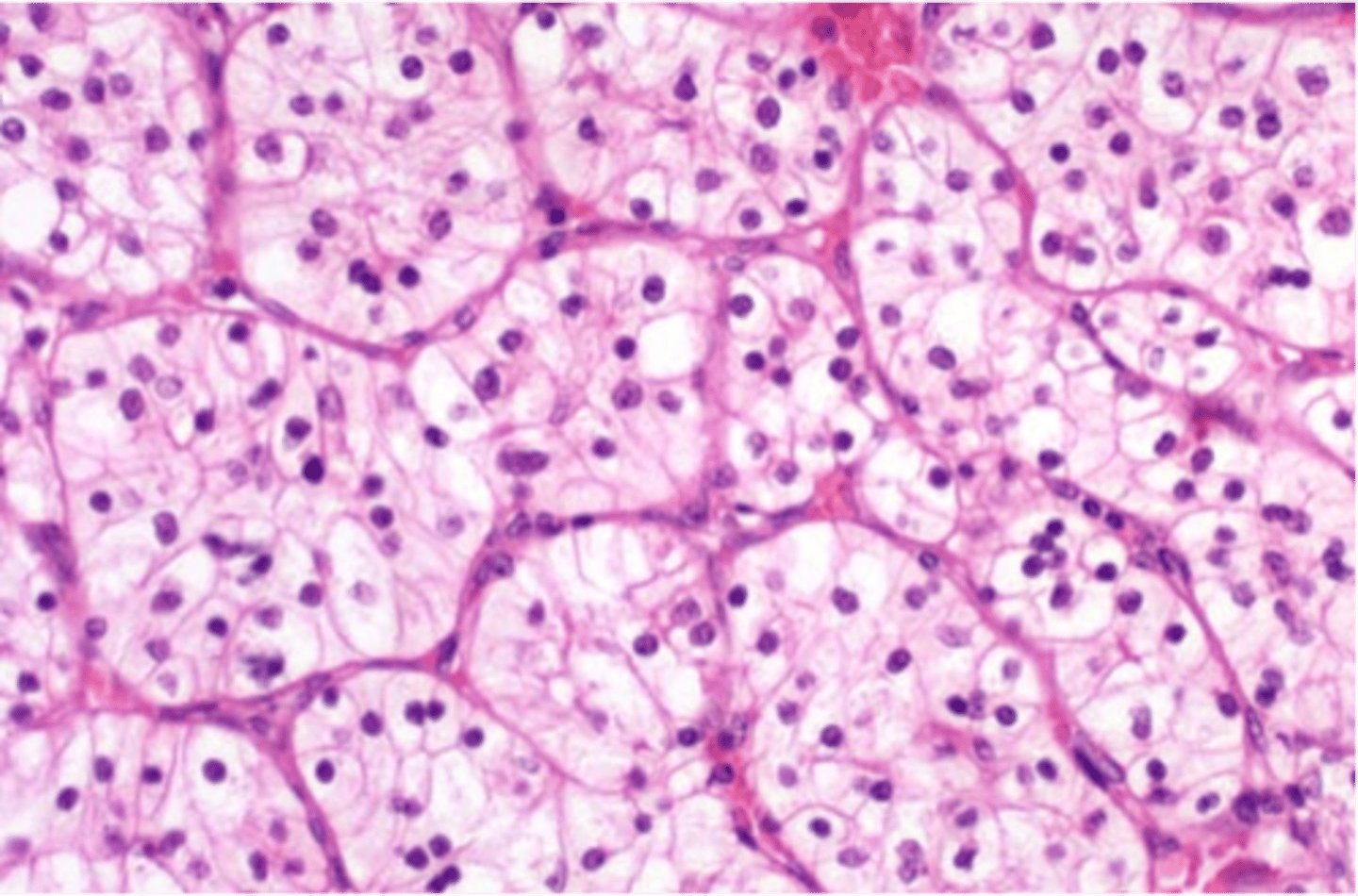
Histology = clear cell most commonly (cells w/ clear or granular cytoplasm and are non-papillary)
S/Sx = may develop as tumor progresses with hematuria, pain/lump in lower back or abdomen, fatigue, weight loss, fever, LE edema
-tumors are quite large and displace other organs and blood vessel before Sx, can invade IVC causing thrombus
Treatment:
-most agents target VEGF and mTOR
clear cell renal carcinoma
ID
renal medullary carcinoma
What disorder:
-malignant, almost always found in young pts (10-40 y/o, mean 22 y/o), M > F
-infiltrative, heterogeneous mass w/ medullary epicenter
-hemorrhage and necrosis contribute to heterogeneity
-associated w/ caliectasis
transitional cell carcinoma
What is the second most common kidney cancer?
renal cell carcinoma
What is the most common kidney cancer?
hydronephrosis
What disorder:
-occurs when ureter is obstructed
-in a pt w/ hematuria, can imply kidney stone or invasive bladder cancer
-can occur from intraluminal obstruction or from external compression of ureters (GU/GI cancers)
-flank pain = common presenting Sx
-renal failure doesn't occur if only one kidney affected but can die if pressure is not relieved
Tx = relieve pressure by placing stent in lumen of affected ureter and extract blockage if one is apparent OR place nephrostomy tube percutaneosuly into renal pevlis to relieve pressure
transitional cell carcinoma
What disorder:
-aka urothelial carcinoma of bladder
RFs = tobacco smoke, occupational exposure (dyes, metal working, paint industry), drinking water (arsenates, chlorination), rad/chemo, anti-diabetic meds, genetics
S/Sx = painless hematuria
-CT for staging (helps assess depth)
-often metastasizes to retroperitoneum, lungs, and bone
Tx = neoadjuvant therapy (giving chemo or chemo-rad prior to definitive therapy)
-50% in-situ and thus curable
-invasive cancers have worse prognosis based on size, LN spread, distant metastasis
true
T/F: an abdominal mass in a child is malignant until proven otherwise
neuroblastoma
What disorder:
-most common extra cranial solid tumor in children
-most commonly diagnosed malignancy in INFANTS
-M > F
-arises from neuroblast cells of SNS
-most cases in abdomen, can be noted in any site
-associated w/ many genetic syndromes and paraneoplastic syndromes
-Cytogenetics/Molecular Genetics = DNA ploidy, MYCN gene amplification, chromosome changes, neurotrophin receptor
-metastasis in children > 12 mos --> LN, long bones, skull, bone marrow, liver, and skin
-S/Sx = abdominal mass, periorbital ecchymosis, fever, irritability, bone pain, subq nodules, ataxia, swelling face/LE, weight loss
-Diagnosis = CT or MRI (mass contains calcification or hemorrhage), tumor markers elevated in 95% (HVA/VMA), Biopsy of tumor tissue or bone marrow (small round blue cell tumor)
-better prognosis in younger children
-higher ferritin + LDH levels have worse prognosis