Bioinorganic Chemistry
1/54
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
55 Terms
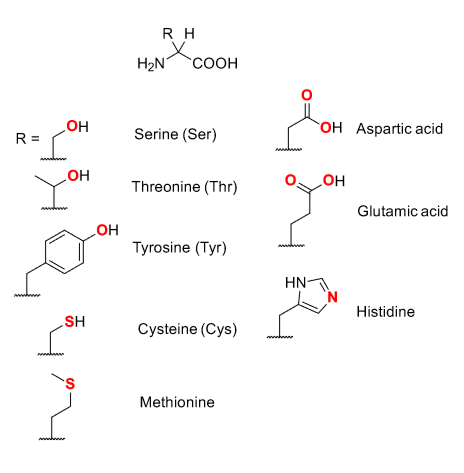
Major binding site for metal ions are provided by proteins - in red are where metals bind to.
Side chains usually provide selective coordination environments.
Proteins can enforce unusual coordination geometries on the metal.
Proteins can induce a strained geometry that resembles a transition site.
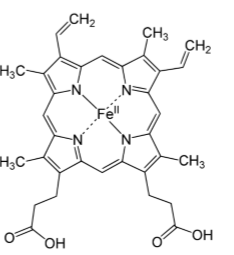
Fe(II)-porphyrin in heme.
Fe-porphyrins are used for: fast electron transfer (in cytochromes), reversible ligand binding, catalysis (particularly involving O2)
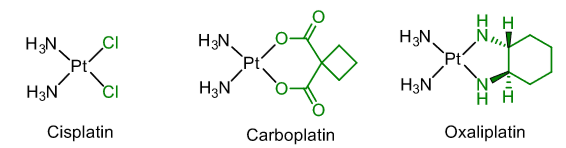
Anti-cancer agents
Therapy for lung, ovarian, testicular cancers.
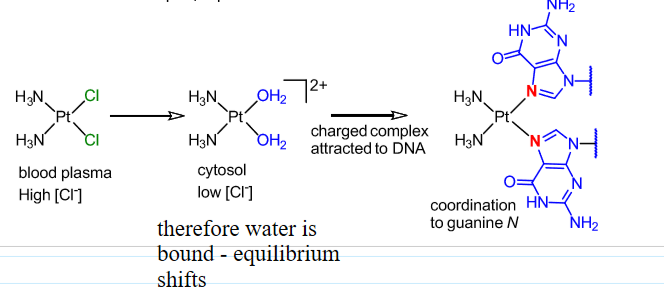
Mode of action thought to involve cross-linking DNA via coordination of guanine residues.
Interferes with cell repair/replication.
Mechanism of cisplatin
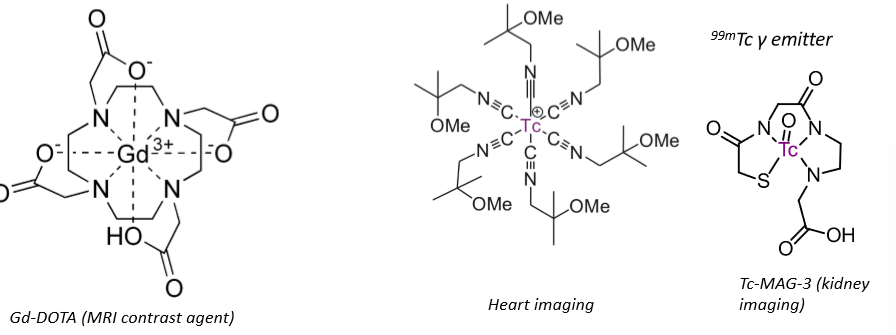
Imaging agents
Chelating ligands.
High thermodynamic and kinetic stability, therefore no ligand exchange.
Means that they don’t bind to anything, doesn’t interfere with the cell - essential for imaging.
Key points of group one metal cations, Na+ and K+
Weak interactions - only 1+.
Hard ligands.
Selectivity from size match and hydrophobicity.
No redox chemistry.
Involved in controlling osmotic pressure in cells - transported in and out (requires relatively weak, reversible coordination chemistry).
Key points of group two metal cations, Mg2+ and Ca2+
More charge dense so stronger ionic interactions.
Hard ligands.
Selectivity from size match and hydrophobicity.
Ca has fast ligand exchange kinetics.
No redox chemistry.
Mg: ATP always coordinated to Mg2+. Structural role in enzymes.
Ca: used for signalling because fast ligand exchange with hard ligands. Structural role because Ca phosphates and carbonates are insoluble.
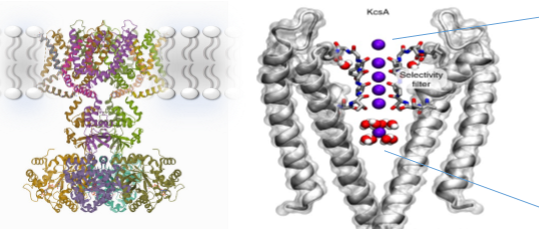
Key features of potassium channels
Helical ladder of peptide carbonyl O donors - provides 8-fold coordination of K+. By replacing the water interactions with coordination to carbonyl oxygens, there is no energetic barrier for moving into the protein channel. The binding is also fast and weak.
Correct size for eight bonds to carbonyl from potassium, but sodium is too small to form sufficient bonds to compensate for the dehydration energy, therefore it cannot move through the channel so it is selective for potassium.
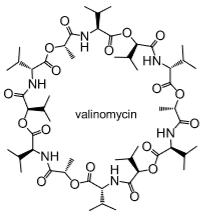
Ionophores
Naturally occurring antibiotic for K+ membrane transport.
Interior is polar to interact with K+, exterior is lipophilic so can move through phospholipid bilayer.
Disrupts ion gradients to destroy bacteria.
Anion-selective ionophores use H-bonds.
How is ATP used for active transport
ATP hydrolysis gives energy to change (rotate) the structure in order to ‘pump’ ions across the membrane.
Why is calcium suitable for signalling?
Large, flexible coordination geometry.
Intermediate binding constants.
Fast ligand exchange kinetics.
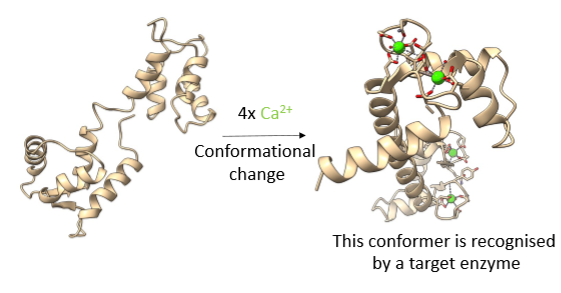
Calmodulin
Ca2+ sensor.
Has 4 6-coordinate Ca2+ binding sites.
Binding constant isn’t too strong - that’s why it’s suitable for signalling.
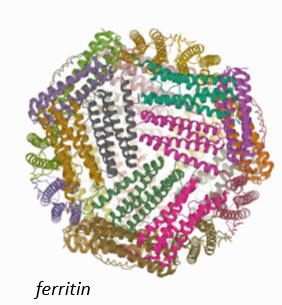
Ferritin
Mineral core stores Fe(III) and resembles ferrihydrate.
Protein shell.
Common features of Fe(III) transporters - transferrins
Hard donors.
Multi-dentate.
Chelate effect.
High binding constants (for scavenge Fe(III) at low concentrations).
No macrocycles - high kinetic stability so can release the iron.
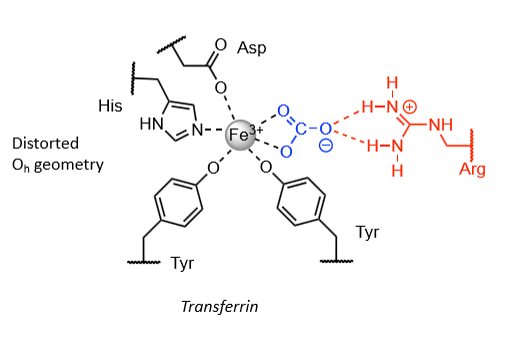
Low pH release mechanism
CO32- is protonated (synergistic ligand). Decreases charge and unbinds, releasing iron.
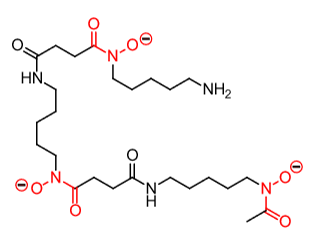
Which metal?
Fe(III)
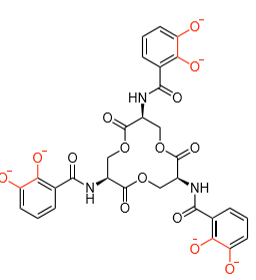
Which metal?
Fe(III)
Helix E and helix F in myoglobin
E - provides group to stabilise coordinated O2 (distal histidine)
F - provides fifth ligand (proximal histidine)
Which metal?
Fe(II) - porphyrin, haem
Explanation for change in structure of myoglobin upon O2 binding
When oxygen is unbound, Fe(II) is high spin d6, meaning electrons are in the eg (antibonding orbital). This causes a larger effective radius than if it was low spin, and so Fe(II) is too big to sit in the porphyrin plane.
When oxygen binds, it changes Fe(II) to low spin since it is a pi acceptor ligand (strong field), meaning there are no electrons in the eg, so the radius is small enough for Fe(II) to sit in the porphyrin plane.
Why does O2 bind in a bent fashion to myoglobin?
In order to get sigma and pi interactions.
(And also because it is forced to sterically due to the histidine).
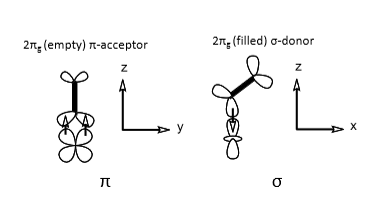
O2 binding curves for myoglobin and haemoglobin, and what they tell us
Myoglobin - simple 1:1 equilibrium, strong O2 binding, curve flattens off as fraction of available binding sites decreases.
Haemoglobin - sigmoidal binding curve (cooperativity), weaker O2 binding overall, optimised to bind O2 in lungs (high concentration) and release in tissue (low concentration, binding constant changes with partial pressure of oxygen indicating some structural change.
When thinking about effectiveness of O2 transport molecules, what two values should you compare?
Saturation in lungs vs saturation in tissues - gives you the percentage of binding sites that are actually used to pick up and release oxygen.
Explanation for the allosteric cooperativity of haemoglobin
Oxygen binding to the first Fe(II) porphyrin means it moves into the porphyrin plane. This pulls on the distal histidine (coordinated to O2), pushing on a different proximal histidine, pushing the second iron into the binding site. This increases the uptake of O2 for the second Fe(II) porphyrin, K2>K1.
‘Enhances the O2 binding at the other porphyrin centres’.
Hemocyanin
O2 transport in arthropods and molluscs.
O2 binds η2η2.
Colourless to blue.
Differences between hemocyanin and haemoglobin
Not cooperative.
A redox reaction - Cu(I) to Cu(II).
Electron transfer centres fall into three main classes:
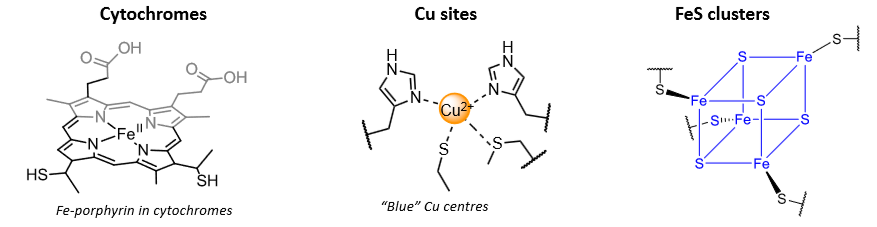
Factors that affect reduction potentials of metals in protein environments
Coordination chemistry factors: ionisation energy; ligands (pi acceptors stabilise lower oxidation states, strong sigma donors stabilise higher oxidation states).
Influence of active site: relative permittivity (stabilises centres with low overall charge); neighbouring charges (on amino acid residues or other bound metals); hydrogen bonding interactions that may stabilise reduced states.
From Marcus theory, rate of electron transfer depends on
Free reaction energy.
Free activation energy.
Reorganisation energy.
Electronic coupling of the electron donor-acceptor pair (HAD).
Temperature.
Fast electron transfer centres in proteins tend to exhibit
Redox active metal centres.
Minimal bond length changes between oxidised and reduced forms.
Sufficiently close donor and acceptor.
Ability to delocalise electrons (eg. extended pi systems, metal-ligand clusters).
Electron transfer in cytochromes
Fe porphyrins, using the Fe(II)/(III) couple.
Pi conjugation means a good HAD term.
Electrons conveyed between central Fe and porphyrin edge using porphyrin pi star orbital.
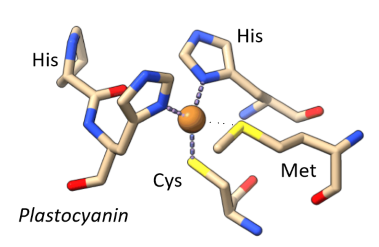
Which metal?
Copper - electron transfer centre
Explanation of the intense blue colour in the blue copper centre
Ligand to metal charge transfer transition - Cys-S to Cu(II)
Why is the blue copper centre fast and efficient at electron transfer
In an entatic state - rigid coordination sphere is enforced by the protein, so Cu(II) and Cu(I) are in virtually the same coordination environment.
Effectively holds the Cu ion close to the transition state for Cu(II) to Cu(I) interchange.
Minimising bond length changes meaning small reorganisation energy.
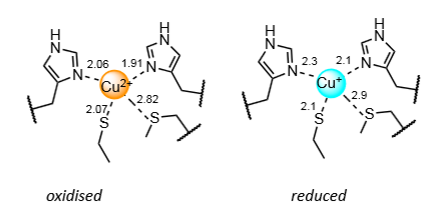
Entatic state
A state of an atom or group which, due to its binding in a protein, has its geometric or electronic condition adapted for function.
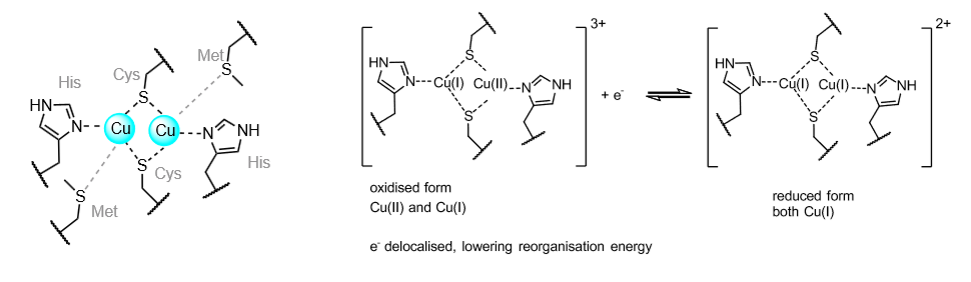
Binuclear copper electron transfer centres
Used in long range electron transfer in some enzymes, including cytochrome c oxidase.
Coordination environment is very similar to plastocyanin (blue copper).
Responsible for electron transfer to the active site used for O2 reduction.
What are metal-sulphido and metal-oxido centres used for?
Electron transfer, redox catalysis, acid-base catalysis and sensing.
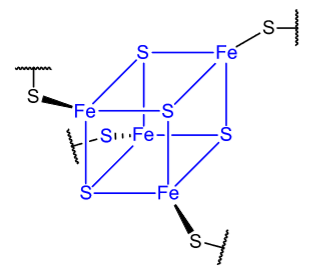
Why are FeS centres excellent fast ET transfer centres?
Ability to delocalise the extra electron(s).
Minimise bond length changes.
Decreased reorganisation energy.
What is NADH the biological equivalent of?
Hydride.
What are hydrogenases?
Metalloenzymes that catalyse interconversion of H2 and H+.
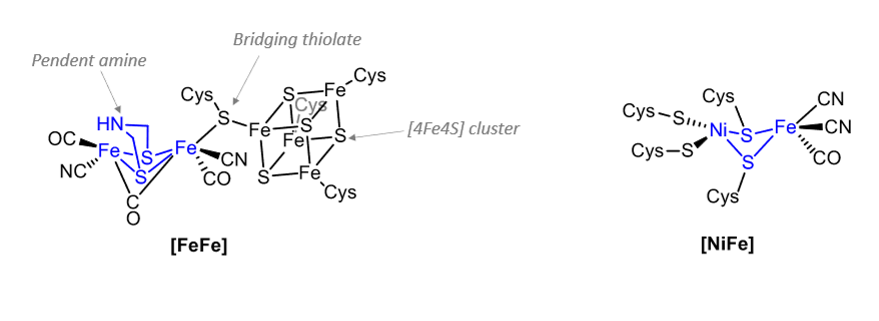
What are these part of?
Hydrogenase active sites.
General features of hydrogenase active sites
Bimetallic.
Coordinated to protein via Cysteine S ligands.
At least one CO and CN ligand.
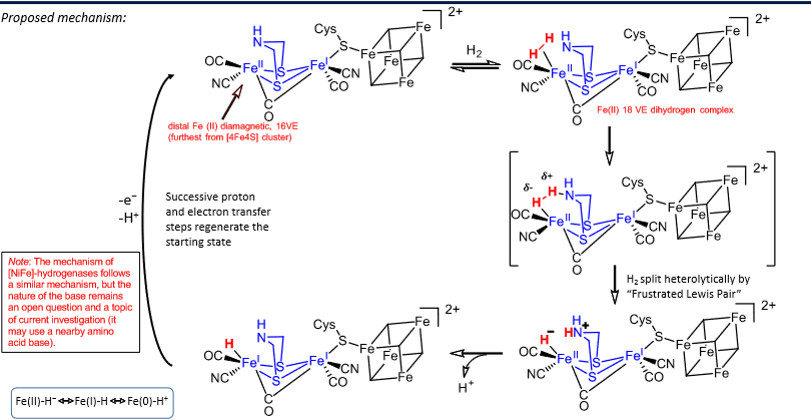
Steps for the oxidation of H2 by [FeFe] hydrogenase
H2 binds to distal Fe(II) which goes from 16 VE to 18 VE.
H2 split heterolytically by frustrated lewis pair mechanism - nitrogen on sulphur bridges acts as a lewis base whilst the iron acts as a lewis acid, causing the H-H bond to polarise until it breaks.
H+ lost from the nitrogen.
H+ and e- lost from iron, completing the cycle.
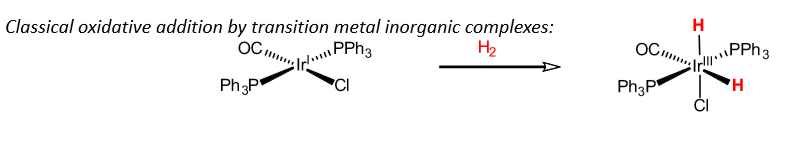
Synthetic catalysts for H2 oxidation
Frustrated lewis pairs - compounds which contain a lewis acid and base that cannot combine to form an adduct.
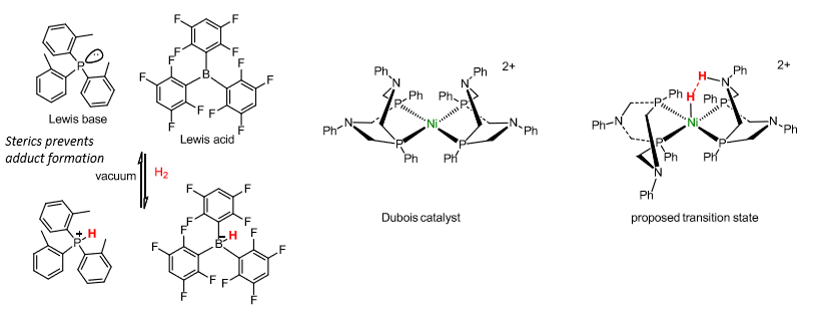
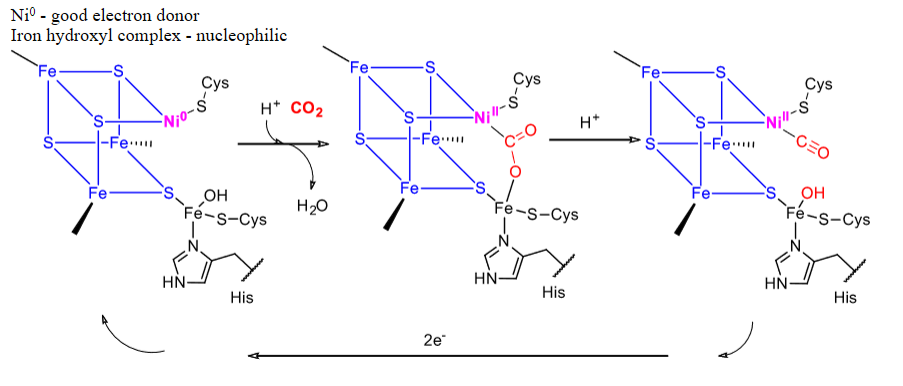
What is used for CO2 and CO interconversion?
Carbon monoxide dehydrogenase, which is an FeS cluster with Ni.
Mechanism for carbon monoxide dehydrogenase
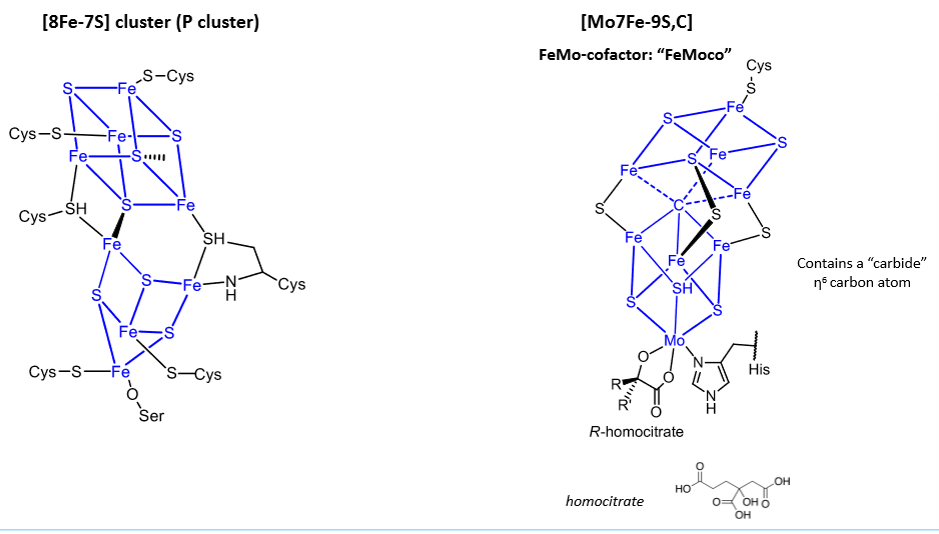
What are these used for?
Nitrogenases.
The second cluster is the site responsible for N2 to NH3 reduction, but the full mechanism is unresolved.
Iron-sulphur bonds, delocalised, gives fast electron transport because of good overlap and low reorganisation energy.
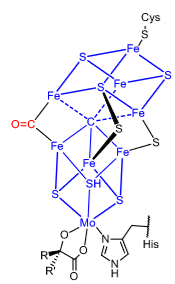
What does this show us about the nitrogenase mechanism?
CO can bind in place of the sulphur - shows that one of the sulphurs is labile so not necessarily required to hold the cluster together. Could mean one of the steps in the mechanism is removing a sulphur.